
Conservation, Regulation, Synteny, and Introns in a Large-scale C. briggsae–C. elegans Genomic Alignment
Abstract
A new algorithm, WABA, was developed for doing large-scale alignments between genomic DNA of different species. WABA was used to align 8 million bases of Caenorhabditis briggsae genomic DNA against the entire 97-million-base Caenorhabditis elegansgenome. The alignment, including C. briggsae homologs of 154 genetically characterized C. elegans genes and many times this number of largely uncharacterized ORFs, can be browsed and searched on the Web (http://www.cse.ucsc.edu/∼kent/intronerator). The alignment confirms that patterns of conservation can be useful in identifying regulatory regions and rarely expressed coding regions. Conserved regulatory elements can be identified inside coding exons by examining the level of divergence at the wobble position of codons. The alignment reveals a bimodal size distribution of syntenic regions. Over 250 introns are present in one species but not the other. The 3′ and 5′ intron splice sites have more similarity to each other in introns unique to one species than in C. elegans introns as a whole, suggesting a possible mechanism for intron removal.
Biologists often compare DNA sequences to determine the relationships between species and to learn about the function of genes. As a result of the efforts of various sequencing projects, it is now becoming possible to do sequence comparisons between complete genomes. These comparisons can answer questions about how genomes change over time in more detail than is possible by chromosome painting (O'Brien et al. 1999). Comparing coding regions between species can locate functionally important parts of proteins, which are more highly conserved than other parts (Makalowski et al. 1996; Makalowski and Boguski 1998). Comparing noncoding regions of homologous genes between species separated by an appropriate evolutionary distance can locate promoters and other conserved regulatory elements (Hardison et al. 1997; Endrizzi et al. 1999;Jareborg et al. 1999). Comparisons between mouse and human, between maize and rice (Wilson et al. 1999), and between Fugu rubripes, Tetraodon fluviatilis, and Danio rerio(Boeddrich et al. 1999) have all revealed important functional regions of the genome.
This paper presents a genomic DNA comparison between the nematodesCaenorhabditis briggsae and Caenorhabditis elegans. C. elegans and C. briggsae are closely related nematodes in the same genus (Baldwin et al. 1997; Blaxter 1998; Blaxter et al. 1998; Voronov et al. 1998). They are estimated to have diverged 25–50 million years ago, although without a fossil record these estimates are highly dependent on assumptions about mutation rates that vary considerably between organisms and between genes (Ayala et al. 1996). In practical terms, C. elegans and C. briggsaeare separated by a nearly ideal distance for comparative genomics. In regions experiencing selective pressure, close to 80% base identity is preserved between species. In other regions base identity is close to 30% (Shabalina and Kondrashov 1999). The large-scale comparative genomic study we present here aligns 8 million bases of C. briggsae sequence from 229 cosmids with 97 million bases of C. elegans sequence covering essentially the entire genome ( C. elegans Sequencing Consortium 1998). The scale of this comparison presented unique challenges and resulted in the development of new algorithms that can cope with long insertions. Our algorithms are also able to recognize homologous regions at the DNA level despite the rapid divergence in the wobble position of most codons.
RESULTS AND DISCUSSION
An Algorithm for Interspecies Genomic Sequence Alignments
The alignment of 8 million bases of C. briggsae against 97 million bases of C. elegans involves, at least conceptually, almost 800 trillion nucleotide comparisons. This requires a relatively fast algorithm. Genomic alignment also requires handling both small and large insertions and deletions and a high degree of divergence in the third, wobble, position of codons. We developed a three-pass algorithm—the Wobble Aware Bulk Aligner (WABA)—that meets these requirements and is described in some detail in Methods. Homologous regions were identified with the first pass of WABA, which took 20 hr of run time on a Pentium III 450 mHz. The second pass, which did a detailed alignment of overlapping 2000 × 5000 base regions, took 11 days to run on the same machine. The third pass joined the overlapping alignments in 15 min on a comparable machine. Although the first and especially the second passes are slow on a single workstation, it is easy to distribute them on many machines. Fortunately, it is in the first pass that the run time varies with the product of the genome sizes that are being compared, where the run times of the other passes are close to linear with respect to the shorter genome size. It should thus be possible in the future to apply WABA to aligning large vertebrate genomes.
To help assess the utility of WABA, we performed the same alignment with BLAST (Altschul et al. 1997). We ran the BLASTN program with two different settings: the default and the slower, but more sensitive, settings. The results are summarized in Table 1. WABA aligned over twice as many bases as BLAST. The average length of a single WABA alignment was >10 times as long as the average BLAST alignment. WABA alignments with inserts of >5 bases or with sections of poor conservation >20 bases long tended to appear as several separate BLAST alignments. Blast found 80% of the regions WABA classified as coding or highly conserved (see below). Table2 shows an alignment that only WABA found. Running BLAST with a window size of 8 and an extension threshold of 8 rather than the default 11 and 11 resulted in BLAST aligning 6% more bases, but this also increased the run time by a factor of 41. WABA was also slower than BLAST at default settings but only by a factor of 24.
Comparison of BLASTN and WABA Alignments
An Alignment WABA Finds but BLAST Misses Between the C. briggsae Cosmid G47H01 and the C. elegans Cosmid T23C6
Web-based Browser and Database
The results of the genomic sequence alignment of C. briggsae with C. elegans are available through a Web-based display called the Intronerator. The alignments can be viewed both as a graphical display indicating the degree of sequence conservation and as a base-by-base alignment in the context of gene predictions and mRNA alignments (Kent and Zahler 2000) athttp://www.cse.ucsc.edu/∼kent/intronerator. A sample Intronerator high-level view is shown in Figure 1. This high-level view displays the region of the C. elegans genome containing the unc-47 gene. Regions containing homology to C. briggsae are represented by a bar immediately under the gene predictions. Areas of stronger homology are represented in a darker shading of the bar than regions of lower homology. Clicking on the bar brings up a base-by-base view of the alignment. The browser is entered via the Tracks Display link on the Intronerator home page. It is possible to search as well as browse the alignments by following the Extract Sequences link. C. elegans genes can be searched for ORFs, for sequenced genes, for the presence of aligning mRNA, and for the presence of aligning C. briggsae genomic sequence. The various search criteria can be combined in a flexible manner. The names of genetically characterized and sequenced C. elegans genes with C. briggsae sequence homology are shown in Table3. The program can also restrict the returned sequences to introns only, exons only, or windows relative to the translational start or finish. This last feature can be used to extract likely promoter regions. A Web interface to the alignment algorithm itself is available at http://www.cse.ucsc.edu/∼kent/xenoAli/. From this page the user can align sequences against the C. elegansgenome or align two sequences to each other. The page also links to a list of all homologous C. briggsae and C. elegansregions sorted by either alignment score, location in C. briggsae, or location in C. elegans.
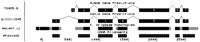
An Intronerator tracks display of the region aroundunc-47. The top track shows the gene prediction of the C. elegans Sequencing Consortium (1998) that is used in AceDB. The second track shows a prediction by the Genie gene finding program (Kulp et al. 1996, 1997; Reese et al. 1997). The third track gives a high level view of the C. briggsae homolog. Regions of high homology (classified by the seven state aligner as highly conserved or coding) appear darker. Regions of lower homology appear in a lighter shade. Nonaligning regions are white. The fourth track shows a cDNA alignment. Note that in this case the Genie prediction agrees with the mRNA, whereas the AceDB prediction misplaces the first exon. Note too the highly conserved region upstream of the first exon containing known promoter elements for this gene (Eastman et al. 1999).
A List of Genetically Characterized and Sequence C. elegans Genes for Which There Is Homology in the Washington Univeristy C. briggsaeSequence Data
Alignment Statistics
Overall, 59% of the C. briggsae sequence is homologous toC. elegans. The alignment algorithm developed here has at its core a pair–hidden Markov model (HMM) (Durbin et al. 1998) that, along with the alignment itself, classifies regions into weakly conserved regions, strongly conserved regions, and conserved coding regions (see Methods). Within weakly conserved regions, an average of 49% of nucleotides are an identical match. Within strongly conserved regions, 89% of nucleotides match. In the first two nucleotide positions inside codons in conserved coding regions, 92% of nucleotides match. In the third, wobble, position 53% of the nucleotides match. Because, as discussed below, the discrimination between coding and highly conserved regions is not robust, for purposes of further analysis we lump the coding regions in with the highly conserved regions. Of the 8,074,467 bases of C. briggsae sequence, 30.5% are in strongly conserved regions, 28.5% are in weakly conserved regions, and 41.0% are in nonaligning regions. Fifty-five percent of the highly conserved regions are coding, 20% are in introns, and 25% are in intergenic regions according to the gene predictions of the C. elegansSequencing Consortium (1998). It is likely that many of the conserved non-coding regions are regulatory in nature.
Compared with mammalian genomes in which ∼50% of the bases are part of repeating elements (Smit 1999), there are relatively few repeating elements in C. elegans and C. briggsae.Nineteen percent of the strongly conserved regions and 8% of the weakly conserved C. briggsae regions align to more than one place in the C. elegans genome. Table 4presents a breakdown of the highly conserved multiply aligning regions. The highly conserved intergenic and intronic sequences are less likely to align to multiple places than the coding bases, indicating that the repeating regions are dominated by duplicated genes. The region that aligned in the most places, 32 in all, was a coding region of the major sperm protein. The transposable element Tc1 aligned in 25 places. The majority of repeating regions, however, only align to two or three places.
Distribution of Highly Conserved Regions Comparing the Number of Places a Region Aligns vs. Whether the Region is Coding, Intronic, or Intergenic
In the current implementation, WABA discriminates weakly between strongly conserved and coding regions. In the regions of alignment, the gene predictions of the C. elegans Sequencing Consortium (1998) classify 1,364,947 bases as coding, WABA classifies 1,845,169 bases as coding, and 1,192,602 bases are classified as coding by both WABA and the gene predictions. If the gene predictions are correct, then WABA has a false-negative rate of 13% and a false-positive rate of 55%, which is not as good as the current state of the art in gene prediction software. The goal of this project was merely to create a sensitive alignment tool, not a gene-finding program. However, in the future it would be interesting to extend the pair–HMM in such a way that it can model codon bias in general and stop codons in particular as well as account for wobble position variations. It is possible such extensions would yield a program that would produce both sensitive genomic alignments and reliable predictions of coding regions.
Conservation of Promoters
Figure 1 shows an Intronerator display of the unc-47 gene. This is a fairly typical example of the patterns of conservation in protein-coding genes between C. elegans and C. briggsae. Most of the coding regions are heavily conserved as is the promoter region. Beyond the splice consensus sequences, most introns are lightly conserved if at all. In this case the gene has been cloned and sequenced (McIntire et al. 1997) and the promoter region studied (Eastman et al. 1999).The conserved region upstream of the first exon covers almost exactly the minimum promoter needed to drive reporter constructs in the same expression pattern as unc-47(Eastman et al. 1999). Identification of conserved regions in promoters will help biologists identify the minimal promoter sequence necessary to drive the proper expression of a gene in functional studies.
Cross-species alignments will be important for dissection of regulatory networks of transcription factors and their binding sites. Two other very powerful techniques for dissecting promoter logic are clustering expression patterns from DNA microarrays (Eisen et al. 1998) and searching for motifs with tools like MEME (Bailey and Elkan 1995) athttp://www.sdsc.edu/MEME/meme/website and Improbizer athttp://www.cse.ucsc.edu/∼kent/improbizer/index.html. Together, these techniques can find cis-acting regions in the promoters of coexpressed genes. Areas in the promoter region conserved in cross-species alignments can serve as an independent confirmation that these regions are biologically relevant or, alternatively, can serve to reduce the size of the region that the motif searching algorithms must consider. As more microarray data is publicly released, it will become possible to apply these powerful bioinformatic approaches to characterizing transcription factor binding sites in C. elegans.
Conservation of Splicing Regulatory Elements
In genes that are alternatively spliced, such as let-2(Sibley et al. 1993), it is not unusual to find highly conserved areas in introns (Fig. 2). Presumably, these conserved areas may be involved in the regulation of alternative splicing. There are several blocks of conserved intron sequence in the alternatively spliced region of let-2. These are seen in the intron just upstream of exon 9, in the intron sequence between exons 9 and 10, and in several blocks between exons 10 and 11. A poly-GT motif conserved in the intron between exons 10 and 11 is rare in C. elegansintrons as a whole. Searching the Intronerator's Alternative Splicing Catalog (Kent and Zahler 2000) shows that this motif does occur frequently in introns near alternative splice sites (data not shown). This poly-GT motif is also found near the region of alternative splicing of bli-4 and is conserved between C. briggsaeand C. elegans in that gene as well (Thacker et al. 1999).
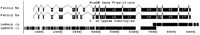
Tracks display of let-2, a gene that is alternatively spliced in a developmentally regulated manner. The C. briggsaesequence is split between two overlapping cosmids. Note the relatively high level of conservation in the introns compared withunc-47. (The exons numbered 8 on the top and bottom splicing diagrams are numbered 9 and 10 in the text and in Table 5. This difference is due to Tracks display not including exons in the UTR regions and numbering the exons in each isoform separately.)
Splicing regulatory elements are often found in exons as well. Because exons tend to be highly conserved, these regulatory elements do not stand out well in the high-level view. However, the base-by-base alignment view can be used to look for sequence conservation in the wobble positions inside coding regions, which only match 53% of the time in the alignment as a whole. Table 5 shows a base-by-base alignment of the let-2 mutually exclusive exons 9 and 10. Sixty-eight bases at the end of exon 10 are perfectly conserved. This includes 22 wobble-position nucleotides. The last 19 bases of this conserved region are all purines. Some members of the SR protein splicing factors have been shown to bind directly to purine-rich elements in exons to promote regulated splicing events (Nagel et al. 1998)
Alternatively Sliced Region of let-2
Intron/Exon Structure of Rarely Expressed Genes
Even though there are > 100,000 C. elegans EST sequences in GenBank, only 9313 of 19,722 (47%) of the predicted C. elegans ORFs align with any ESTs. However, rarely expressed genes as well as commonly expressed genes can be identified by their patterns of conservation, as illustrated in Figure 3. Here, a single homologous region of C. briggsae covers two predictedC. elegans genes. One prediction is supported by seven EST sequences derived from four different cDNA clones. The other gene prediction has no supporting EST sequences at all. However, both genes are strongly supported by the pattern of conservation of predicted exons in the homologous C. briggsae region.

C. elegans predicted genes K04F10.1 and K04F10.7. The K04F10.7 gene prediction is supported by a number of ESTs. K04F10.1 appears by lack of EST coverage to be rarely expressed. The pattern of conservation of K04F10.1 nonetheless provides strong support for this gene prediction.
Introns Unique to One Species
Long insertions and deletions (indels) are common in the alignments. For indels from 3 to 30 nucleotides long, indels of an integral multiple of three were more common than other indels (data not shown). As indicated in Table 6, 95% of the longer indels occurred in regions of weak homology. Of the indels in regions of strong homology, 72% were in coding regions. Seventy-three percent of the indels in coding regions of strong homology, 263 in all, aligned with GT … AG ends on the region unique to one species. These indels appear to correspond to introns present in one species but not the other (Table 7). 263 unique introns is a conservative number. There are many additional cases where it appears that a codon or two were inserted or deleted along with the intron. In addition, there are cases where the insert resembles an intron that has since undergone a few point mutations in the flanking region. There also may be introns inserted or deleted in weakly conserved regions and noncoding regions as well. The figure of 263 unique introns does not include any of these cases.
Summary of Insertions and Introns Unique to One Species
Examples of Introns Unique to One Species
From an alignment between just two sequences it is not possible to tell whether the introns are being gained, lost, or a mixture of both. There is only a small amount of genomic sequence data available for other members of the genus Caenorhabditis in GenBank and only a single instance (Hansen and Pilgrim 1998) where this data covers a coding indel in C. elegans and C. briggsae. In this case C. briggsae is missing a stretch of DNA that is present in C. elegans and Caenorhabditis remanei. C. remanei is considered to be a closer relative of C. briggsae than of C. elegans (Blaxter et al. 1998). Thus, this indel appears to represent an intron that was lost along with a single additional codon sometime since C. briggsae and C. remanei diverged. In a previous study comparing sequences of two large families of chemoreceptor genes in C. elegans andC. briggsae (Robertson 1998), Robertson found a pervasive pattern of intron loss over recent evolutionary time. Therefore, in all, it seems most likely that the majority of the introns unique to one species represent intron loss in the other species. Robertson noted a bias toward introns being lost in C. briggsae rather thanC. elegans. Assuming the unique introns we observe in this larger study represent intron loss, 65% of the introns were lost fromC. briggsae (Table 6).
We tested the model that flanking the unique introns may be some sequences associated with intron loss. The consensus sequence near the splice site of unique introns is different from the consensus sequence near the splice site of C. elegans introns as a whole (Table8). In particular, the consensus of the AG before the 5′ splice site is much stronger in unique introns, particularly inC. briggsae. This and other changes to the consensus have the effect of strengthening the homology between the 3′ and 5′ splice sites. However, the homology is only strengthened significantly in the two bases before, and the one base after, the splice site (Table9). This homology is too short for homologous recombination to be the mechanism for intron loss. However, the Ku mechanism for repairing double-stranded breaks in DNA (Jeggo 1998) joins broken ends at regions of microhomology (≈3 bp). The Ku mechanism deletes bases between the broken ends and the microhomology region as well. Thus, it seems possible that introns may be lost during repair of double-stranded breaks in DNA.
Profiles of Nucleotide Frequencies Adjacent to 5′ and 3′ Splice Sites
Percent Homology Between 3′ and 5′ Splice Sites
Synteny
The large scale of this alignment also permitted us to observe the level of short-range synteny between the two genomes. On average, aC. briggsae sequence would only align for 8553 bases to one part of the C. elegans genome before the best alignment shifted to another part of the C. elegans genome. Because the average size of a C. briggsae clone was only 35,722 bases, approximately one-fourth of the alignments ended because the C. briggsae sequence ended. Thus, the average size of an aligning segment is actually larger. Figure 4 depicts the number of fragments clones are broken into by the alignment. Curiously, this quantity exhibits a bimodal distribution, with peaks at one fragment per clone and three fragments per clone. The first peak indicates that ∼40% of the genome is resistant to rearrangment, going longer between rearrangement events than the 35,722-bp size of the C. elegans clones. The other 60% of the genome appears to be susceptible to rearrangements, with rearrangement events occurring approximately every 4000 bases. Because the average length between genes in C. elegans is ∼5000 nucleotides, this is consistent with arrangement occurring freely in regions between genes in the 60% of the genome that is susceptible to rearrangement.
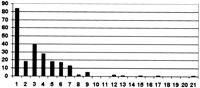
Synteny summary graph. The horizontal axis is how many pieces a C. briggsae clone needed to be broken into for maximal alignment with the C. elegans genome. The vertical axis is the number of clones broken into that many pieces. There were 229 clones, and the average clone size was 34,722 bases. Clones were broken up if the best aligning region of one 2000-base region of the C. briggsaeclone aligned to a spot 50,000 bases or more away or on a different chromosome from where the previous 2000-base region of the C. briggsae clone aligned best. The 2000 base regions overlapped by 1000 bases. The average length of a broken-up region was 8553 bases. The bimodal distribution suggests that ∼40% of the genome is resistant to rearrangements and the rest is not.
The 40% of the genome resistant to rearrangement may reflect regions that are transcriptionally coregulated. The largest single alignment is part of the homeobox complex. Genes that are part of operons (Spieth et al. 1993) or that share an enhancer would also seem likely to be resistant to rearrangement. In part, the length of an alignment reflects the position on the chromosome. Eight of the 10 longest alignments, 15 of the 20 longest alignments, 36 of the 50 longest alignments, and 63 of the 100 longest alignments are found in the gene-rich clusters near the middle of chromosomes. The chromosome arms appear to be more susceptible to rearrangement. One cosmid, G46J06, aligns in nine widely separated pieces on chromosomes I, II, III, and IV. None of the G46J06 alignments are in the gene-rich cluster regions.
Conclusions
We developed a new algorithm for aligning genomic DNA. We used the algorithm to perform a large-scale alignment between C. briggsae and C. elegans genomic sequences. The alignment confirms that patterns of conservation can be useful in identifying regulatory regions and rarely expressed coding regions. The alignment, including C. briggsae homologs of 154 cloned C. elegans genes and many times this number of largely uncharacterized ORFs, can be viewed and searched in the Intronerator. The detailed alignment display can help identify conserved regulatory elements even inside exons. There is a bimodal distribution on the size of syntenic regions. Over 250 introns are present in one species but not the other, within the ∼10% of the C. briggsae genome sequenced.
METHODS
Sources of Sequence Data
The genomic sequence data for C. elegans came from the C. elegans Sequencing Consortium (1998) downloaded from http://www.sanger.co.uk/Projects/C_elegans/chromosomes.shtml on August 4, 1999. The C. briggsae genomic sequence data came from the Washington University siteftp://genome.wustl.edu/pub/gsc1/sequence/st.louis/briggsae/finish/ on October 6, 1999. Genomic sequences from other organisms and all the mRNA sequences were obtained from GenBank (Benson et al. 1999) athttp://www.ncbi.nlm.nih_.gov/Entrez/ on December 4, 1999.
Cross-species DNA Alignment Algorithms
The large scale of the C. briggsae and C. elegansgenomic alignment required a relatively fast algorithm. Yet the algorithm also needed to handle both small and large insertions and deletions and deal with the statistical challenges presented by the AT-richness of the Caenorhabditis genome. To cope with these somewhat conflicting concerns, we broke the alignment problem into three passes: a fast program that took overlapping 2000 base regions ofC. briggsae and scanned the C. elegans genome for homologous regions, a program that aligned the homologous regions in a very detailed manner, and a program that stitched together the overlapping alignments.
The fastest DNA alignment algorithms, such as BLAST (Altschul et al. 1990), demand areas that match perfectly to serve as seeds for the alignment. Although such approaches work well for searching EST databases, they break down when comparing DNA sequences between species. Although coding regions of DNA are fairly well conserved in the first and second codon positions, the third, wobble, position is largely redundant in the genetic code and tends to diverge freely. To cope with this we developed an algorithm that worked on the general principles of gapped BLAST (Altschul et al. 1997) but that, rather than requiring six consecutive nucleotides to match perfectly to seed an alignment, required six nucleotides spread out in the pattern XXoXXoXX (where the X's must match but the o's need not) to serve as the seed. We call this algorithm WABA. There are three passes to the WABA algorithm.
The first pass of WABA works in the following fashion: The C. elegans DNA (the target sequence) is packed so that each 16-bit word contains 8 nucleotides. This serves two purposes—to reduce the amount of RAM required to store the genome and to allow multiple nucleotides to be compared in a single computer operation. For each section of the C. briggsae sequence (the query sequence) we do the following: An array that contains an entry for all possible 8-nucleotide sequences (8-mers) is allocated. The query sequence is then scanned, and each 8-mer is checked and possibly added to the array. Note that these 8-mers in the query sequence overlap each other by 7 bases. Eight-mers are rejected if they contain ambiguous nucleotides such as N or if they could match degenerate repeating DNA sequences of period 4 or less. (This eliminates 8-mers such as AAAAAAAA, ACACACAC, ACGACGAC, and ACGTACGT.) The 8-mers are packed into a 16-bit word, which is then binary logical anded with the wobble mask (hexadecimal 0xF3CF) so that the third and sixth nucleotides are ignored. The packed, masked 8-mer is then added to the corresponding entry in the array that stores the position of the 8-mer in the query sequence as well. Then, the packed genome is scanned. Each packed word of the genome is “anded” with the wobble mask, and the result is used to index the array. If the corresponding array entry is nonempty, then the current genome position and the position of the corresponding query 8-mer are noted in a hit list. Because the hit list grows quite quickly, it is periodically scanned and nonpromising hits are eliminated. If a hit occurs within 1000 bases on the target sequence of another hit and the positions of the hits in both query and target indicate that they could be part of a homologous region lacking inserts (which is simply checked by subtracting from query offsets of both hits to get a “diagonal” offset and then checking that the diagonal offsets are identical), it is considered promising. In this way, WABA scans the entire genome and assembles a list of hits. The hits are then clumped together. A clump starts with a single hit. Each hit within 48 nucleotides in both target and query and with the same diagonal offset is added to a clump. As hits are added to a clump, the boundaries of the clump are extended to include the new hits. The clumps are then scored by the square of the number of hits in the clump, and the clumps scoring <25% of the highest score are eliminated. Because the distribution of scores is highly peaked, this saves processing in later steps in the common scenario where there are one or a few areas of strong homology and many areas of weak homology. The remaining clumps are scored for the best local alignment without inserts between the full query sequence and the corresponding part of the target sequence. Because the worm genome is AT rich, AT matches are scored as 2, whereas GC matches are scored as 3. Mismatches are scored as 4. The bottom scoring 25% of the alignments without inserts are dropped, and the remaining alignments are stored for the next pass.
Though the first pass is quite fast, it has no provision for insertions and deletions. The first pass merely identifies regions of the target homologous to the 2000-base query sequence. A 5000-base window of the target sequence centered around the first pass aligment is then aligned with a slower algorithm that can cope with insertions and deletions. The Smith–Waterman algorithm (Smith and Waterman 1981) and extensions of it to allow affine gap scores (Pearson and Miller 1992) would serve this purpose reasonably well. However, even affine gap penalties tend to penalize long gaps excessively in genomic and cDNA alignments. Furthermore, we wanted our slow, detailed aligner to be at least as wobble-aware as the fast front end. HMMs have proven to be very useful in predicting gene structure (Kulp et al. 1996). Pair HMMs of three (hidden) states can provide a conceptually simple and effective implementation of alignment with affine gap costs (Durbin et al. 1998). This motivated us to design a seven state pair HMM to do the detailed alignment (Fig. 5). One state captures long inserts in the target sequence, one state captures long inserts in the query sequence, one state captures highly conserved regions (∼90% base similarity), one state captures lightly conserved regions (∼50% base similarity), and three states capture coding regions. As the HMM matches a nucleotide pair in one coding state, it shifts to the next coding state. Match and mismatch scores for the first two coding states are very similar to match and mismatch scores for the highly conserved regions. Match and mismatch scores for the third coding state are lower and favor mismatches between C and T or G and A over other mismatches. The heavily and lightly conserved and coding states all allow inserts within the state with a gap penalty that is approximately equivalent to eight mismatches. To transition into a long gap state invokes a penalty equivalent to roughly 12 mismatches. To continue a gap in a long gap state costs a penalty that is only about one-fifth of a mismatch cost. The result is an algorithm that penalizes 1-base gaps by 8, 2-base gaps by 12, and longer gaps by 12 plus one-fifth of their length. A 60-bp gap is only penalized twice as much as a 2-bp gap. This is a fair approximation of the distribution of gap sizes in genomic alignments (data not shown) that is not as computationally expensive as more elaborate gap penalty schemes (Gotoh 1990). The matches are also somewhat more sensitive than the usual practice of treating all nucleotide matches equivalently. Because the worm genome is 61% A/T, we weighed G/C matches more heavily than A/T matches. The various match, mismatch, and gap scores were chosen first by an educated guess of the equivalent probabilities. The resulting HMM was used to align a substantial fraction of the available data. The distributions of matches, mismatches, and gaps in this first alignment were used to refine the scores for a second alignment over the entire data set. This provided one step of parameter optimization by expectation maximization (Durbin et al. 1998). The result is a robust and sensitive alignment algorithm. It also produces predictions as to whether a region is coding or not and a score that can be used to compare the significance of different alignments. We empirically set the scoring threshold for an alignment to pass onto the final pass of WABA to a number that just excluded a large number of ≈40-bp alignments that match primarily in poly-A and poly-T regions. This is necessary because the worm genome is very rich in these regions. The oligonucleotide AAAA occurs 7.5 times as often as would be expected from a random distribution of nucleotides.
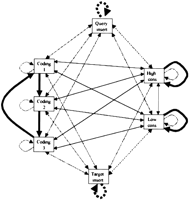
Finite state machine depiction of seven state aligner. This is a pairwise HMM. Transitions between states are associated either with aligning a pair of nucleotides (solid arrows) or aligning a single nucleotide with a gap (broken line arrows). The most likely transition out of each state is shown by a thick arrow. Thin arrows represent possible, but unlikely, transitions. In coding regions, the most likely path will typically cycle between coding states 1, 2, and 3. In strongly conserved regions, the most likely path will typically stay in the high conservation state. In weakly conserved regions, the most likely path will typically stay in the low conservation state. Short inserts follow the broken line arrows in the coding and conserved states. Long inserts transition into the query or target insert states.
The pairwise HMM that is the second pass of WABA is implemented in optimized C code. Nonetheless, because the run time increases with the product of the length of the two sequences that it is aligning, it is not efficient to use it for long alignments. This is the primary motivation for breaking the query sequence into 2000-bp blocks that overlap each other by 1000 bp. This necessitates a third pass to scan the resulting short alignments and, when possible, merge them into longer alignments. This last pass works by looking for alignments that overlap in both query and target. Overlapping areas are scanned for regions of at least 15 nucleotides where the alignments are identical and are joined if such a region is found. After this joining, the alignment is complete.
Acknowledgments
We thank David Haussler for his sage advice on HMMs, David Kulp for making Genie predictions for the Intronerator, and members of the Zahler, Jin, and Chisholm labs and Manny Ares for helpful discussions. Thanks to David Haussler, David Hoffman, and Jane Silverthorne for critical reading of this manuscript. We are indebted to all the various sequencing projects for providing sequence data. This work was supported by grant 1R01GM52848 from the National Institutes of Health to A.M.Z. and a training grant from the University of California Biotechnology Program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL kent{at}biology.ucsc.edu; FAX (831) 459–3737.
-
- Received January 27, 2000.
- Accepted June 2, 2000.
- Cold Spring Harbor Laboratory Press








