
Benchmarking bulk and single-cell variant-calling approaches on Chromium scRNA-seq and scATAC-seq libraries
- Matthew Wiens1,
- Hossein Farahani1,
- R. Wilder Scott1,
- T. Michael Underhill1,2,4 and
- Ali Bashashati1,3,4
- 1School of Biomedical Engineering, University of British Columbia, Vancouver, British Columbia V6T 2B9, Canada;
- 2Department of Cellular & Physiological Sciences, University of British Columbia, Vancouver, British Columbia V6T 2A1, Canada;
- 3Department of Pathology & Laboratory Medicine, University of British Columbia, Vancouver, British Columbia V6T 1Z7, Canada
-
↵4 These authors contributed equally to this work.
Abstract
Single-cell sequencing methodologies such as scRNA-seq and scATAC-seq have become widespread and effective tools to interrogate tissue composition. Increasingly, variant callers are being applied to these methodologies to resolve the genetic heterogeneity of a sample, especially in the case of detecting the clonal architecture of a tumor. Typically, traditional bulk DNA variant callers are applied to the pooled reads of a single-cell library to detect candidate mutations. Recently, multiple studies have applied such callers on reads from individual cells, with some citing the ability to detect rare variants with higher sensitivity. Many studies apply these two approaches to the Chromium (10x Genomics) scRNA-seq and scATAC-seq methodologies. However, Chromium-based libraries may offer additional challenges to variant calling compared with existing single-cell methodologies, raising questions regarding the validity of variants obtained from such a workflow. To determine the merits and challenges of various variant-calling approaches on Chromium scRNA-seq and scATAC-seq libraries, we use sample libraries with matched bulk whole-genome sequencing to evaluate the performance of callers. We review caller performance, finding that bulk callers applied on pooled reads significantly outperform individual-cell approaches. We also evaluate variants unique to scRNA-seq and scATAC-seq methodologies, finding patterns of noise but also potential capture of RNA-editing events. Finally, we review the notion that variant calling at the single-cell level can detect rare somatic variants, providing empirical results that suggest resolving such variants is infeasible in single-cell Chromium libraries.
Single-cell sequencing technologies have emerged as one of the most powerful interrogative tools currently available for studying cell populations in a variety of biological scenarios. Single-cell DNA sequencing (scDNA-seq) tools revealed new insights into the ancestral hierarchy and subclonal composition of various cancers (Navin et al. 2011; Gawad et al. 2016). Additionally, other single-cell approaches, such as single-cell RNA sequencing (scRNA-seq) and single-cell ATAC sequencing (scATAC-seq), have resulted in major advances in our understanding of intercellular heterogeneity, cellular differentiation, and cell–cell interactions (Jaitin et al. 2014; Buenrostro et al. 2015). These single-cell sequencing technologies are increasingly being paired with short variant-calling approaches to interrogate the unique haplotype state of individual cells (Fan et al. 2018; Poirion et al. 2018; Ding et al. 2019; Petti et al. 2019; Vu et al. 2019; Zhou et al. 2020; Lu et al. 2021). This pairing enables investigators to extract additional information from these libraries regarding the single-cell genetic state, thus saving the time and money otherwise required to obtain scDNA-seq libraries. For instance, using scRNA-seq, the clonal composition of a tumor sample and expression phenotypes of individual subclones have been determined independently of scDNA-seq (Ding et al. 2019; Zhou et al. 2020; Lu et al. 2021). An additional benefit is that modality-specific sequence-altering effects such as RNA editing can be investigated at the single-cell level (Picardi et al. 2017).
The most popular single-cell methodology is the Chromium platform provided by 10x Genomics, which employs a droplet-encapsulation-based approach to sequence many thousands of cells in parallel (Zheng et al. 2017). Single-cell alternative sequencing modalities, especially libraries generated by Chromium pipelines, pose new challenges for existing variant callers owing to unique biological and technical limitations. To start, cell loadings in Chromium libraries range anywhere from 500 to more than 50,000 cells, and at these high loadings, most cells have low coverage so that they may be sequenced in a single run. As a result, alternative allele visibility can be poor at the single-cell level, at which cells will often only have a single read at a given site. Additionally, the sparse coverage provided by approaches such as scATAC-seq and scRNA-seq limits the regions in which variants can be detected. ATAC-seq is sparse because of its targeted sampling approach, whereas Chromium scRNA-seq only sequences one end of a mRNA transcript, significantly reducing overall gene coverage. Modality-specific effects, such as allelic dropout, can further impact the accuracy of the allele called in each individual cell. Finally, at higher cell loadings, beads may contain more than one cell, which can complicate the analysis. Very few variant callers have been designed to address these unique challenges.
Nonetheless, multiple studies have performed variant calling on libraries generated by the Chromium 3′/5′ scRNA-seq and scATAC-seq chemistries (Fan et al. 2018; Petti et al. 2019; Lu et al. 2021; Massarat et al. 2021; Prashant et al. 2021; Dou et al. 2024). A wide variety of variant-calling approaches have been applied. These fall into one of two categories: (1) treating the reads as having been called in bulk (i.e., applied to the pooled reads) and disregarding cell-level information or (2) leveraging cell level information and intercell statistics to improve variant calling. The latter approach is of interest because of its potential to discover rare variants visible only at the level of small groups of cells. Indeed, studies have claimed that cell-level variant calling reveals a wealth of variants not visible in whole-genome and whole-exome sequencing (Prashant et al. 2021). However, the extent to which such observed variants occur owing to biological variations versus false positives resulting from artifactual sources requires further investigation. Additionally, although one recent study has performed benchmarking of pooled callers as part of a comparison with their purpose-built single-cell variant caller (Dou et al. 2024), there are no publications that focus on comparing the performance of pooled and individual-cell variant-calling approaches which have been used in studies with Chromium-derived single-cell sequencing libraries (Dou et al. 2024).
In this study, we aim to provide a comprehensive comparison of commonly used single-cell and pooled variant-calling approaches on matched whole-genome sequencing (WGS) and 10x Chromium scRNA-seq and scATAC-seq libraries derived from mouse tumor samples. We additionally aim to investigate rare variants that might be detected only in single-cell modalities and only at the individual-cell level to determine to what extent real rare variants may be extracted from these modalities.
Results
Benchmark data sets
Three tumor samples derived from a genetically engineered mouse model of epithelioid sarcoma (EPSRC) featuring an induced adulthood deletion of Smarcb1 were acquired, and each sample underwent Chromium scRNA-seq and scATAC-seq, as well as Illumina WGS, library generation (see Supplemental Table S1). Additionally, a control sample underwent Chromium scRNA-seq and scATAC-seq. Single-cell sequencing was performed at loadings of around 7000–15,000 cells per sample. This loading is typical for Chromium pipelines and serves to reproduce the low per-cell coverage typical of this single-cell methodology.
Benchmark metrics
Our unique experimental setting allowed us to capture both real variants that would be present in many cells as well as rare variants that would be present in a small fraction of cells. To identify real variants, the consensus of multiple variant callers (HaplotypeCaller [Poplin et al. 2018], Strelka2 [Kim et al. 2018], SAMtools [Li 2011], and VarScan [Koboldt et al. 2009]) on the matched WGS libraries was utilized (referred to as common ground-truth variants) (Table 1). Given that WGS is limited in its capability to detect variants present in a small number of cells, compared with other modalities, we would expect that such rare variants would most often be undetected or indistinguishable from noise in WGS data. As such, variant calls identified in scRNA-seq or scATAC-seq data without significant evidence in WGS were treated as potential rare variants (referred to as rare positive variants). We define a number of variant categories and metrics (Table 1; Supplemental Fig. S1) to facilitate comparison of various variant-calling algorithms. These are described in more detail in Methods (see subsection “Variant categories and metrics”).
Quick reference for definitions of variant categories defined and used frequently throughout this study
As our primary metric to compare various variant-calling approaches on single-cell sequencing, we use a precision-recall measure that evaluates missed variants (pooled false negatives) and extra variants that have been called (rare positives) for each caller. We used the quality value for each variant generated by each caller as the score threshold to generate precision and recall across different levels. We further computed the area under the precision-recall curve (AUPRC) to compare the overall performance of each caller. Precision and recall were computed for variants across multiple samples to provide an overall score. We additionally use metrics termed the common true-positive rate (cTPR) and common false-discovery rate (cFDR) to evaluate the performance of variant callers with different filtering approaches. For more details on these metrics, see Methods (subsection “Variant categories and metrics”).
Benchmarking variant callers
We consider variant-calling approaches for single-cell data sets as falling into two categories: pooled and individual-cell methods. Bulk callers have been tested previously on Chromium scRNA-seq and scATAC-seq libraries at the pooled read level (Liu et al. 2019; Massarat et al. 2021). However, no studies have tested the application of bulk callers to individual cells for Chromium libraries. Given that such approaches are increasingly being used (Ding et al. 2019; Lu et al. 2021), it is essential to benchmark their performance against bulk methods and confirm their utility.
Typically, for individual-cell calling, the intersection of variants called across multiple cells is used to derive a consensus list that can be filtered by quality. To investigate the potential of these approaches, we apply Strelka2 (Kim et al. 2018) and GATK HaplotypeCaller (Poplin et al. 2018) on individual BAM files for reads corresponding to a single cell. We then take the intersection of variants from individual cells and define the quality of each variant as the maximum quality value of the variant over the cells in which it was found.
Based on previous benchmarking studies applying bulk callers to Chromium single-cell pooled reads (Liu et al. 2019; Massarat et al. 2021), we test Strelka2, SAMtools (Danecek et al. 2021), GATK HaplotypeCaller, and FreeBayes (Garrison and Marth 2012) as the most promising options, given their previously tested performance. We run them on the pooled reads for each single-cell library according to their recommended best-practice approach for each modality.
It is notable that there are many purpose-built options for individual-cell variant calling, such as Monovar (Zafar et al. 2016), SCcaller (Dong et al. 2017), SciPhi (Singer et al. 2018), ProSolo (Lähnemann et al. 2021), ScanSNV (Luquette et al. 2019), scAllele (Quinones-Valdez et al. 2022), and Monopogen (Dou et al. 2024). Many of these callers were developed to address allelic bias owing to multiple-displacement amplification used in scDNA-seq methodologies. As a result of their tailored statistical modeling approaches and traditional application to high-coverage single cells, they have been dismissed as inapplicable to Chromium single-cell methodologies (Lu et al. 2021). However, the ability of these tools to adaptively model changing allele frequency might be helpful to correct for allelic imbalance owing to preferential visibility of one chromosome in scRNA-seq and scATAC-seq, thus improving variant detection. The major question is whether they will transfer to the low read coverage per cell of Chromium-based sequencing. To verify this viability, we benchmark SCcaller, which among the caller options has a varying window allelic modeling approach suitable for RNA sequencing.
It should be mentioned that there are recently developed callers developed specifically for improving sensitivity in single-cell modalities through the use of additional population-level information to overcome the limitations of single-cell sequencing. Examples include Monopogen (Dou et al. 2024), which leverages population-level statistics such as linkage disequilibrium to help differentiate real variants from noise, and scAllele (Quinones-Valdez et al. 2022), which leverages splicing patterns to help inform variant confidence. Although we do not benchmark them in this paper, approaches like these may have significant promise for Chromium-based single-cell modalities.
Given that single-cell data have the potential to capture variants that show evidence in a low number of cells and thus have a low presentation frequency, we first investigated the performance of variant callers at different variant allele frequencies (VAFs; the fraction of alternate-allele reads over total reads at a variant site). We can see from Figure 1, C and D, that, for both scATAC-seq and scRNA-seq, the majority of variants have either very low VAF (less than 0.01) or very high VAF (equal to 1.0). As such, a failure to detect low-VAF variants will miss many WGS variant sites being lost. All callers suffer a drop in sensitivity at low VAF, especially below VAF of 0.01 (Fig. 1A,B). However, two of the individual-cell callers, SCcaller and Strelka, perform quite well at low VAF compared with all other callers, possibly highlighting the potential of individual-cell callers to pick up lower-frequency variants. Based on the results of Figure 1, in our further analyses, we kept only those variants with VAF of 0.01 or greater to filter out the large number of very-low-frequency variants missed by all callers.
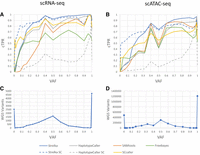
Common true-positive rate (cTPR) averaged over samples for different variant-calling approaches, for scRNA-seq (A) and scATAC-seq (B), binned by VAF (e.g., the point VAF = 0.5 would include variants from 0.5 to the next bin, which is 0.6). The number of WGS variants with coverage in the single-cell sequencing modality, for scRNA-seq (C) and scATAC-seq (D), binned by VAF.
Figure 2, A and B, demonstrates the performance of variant-calling approaches on scRNA-seq and scATAC-seq, respectively, based on precision-recall curves. We can see that pooled read calling can achieve good sensitivity in both cases, reaching up to 90.5% and 76.9% recall in each modality, respectively. Based on AUPRC, we find that Strelka demonstrates the best performance on scATAC-seq, whereas SAMtools demonstrates the best performance on scRNA-seq (see Table 2). Notably, in scRNA-seq, most callers have lower precision than in scATAC-seq, indicating that extra variants are consistently called in scRNA-seq that are not present in the common ground-truth set. This is especially notable when variants are called in nonexonic and exonic regions (see Supplemental Fig. S2) and raises the importance of limiting scRNA-seq variant calling to exonic regions. In later sections, we investigate what proportion of these rare positive variants are noise/artifacts, biological factors unique to sampling RNA, or true biological variants. The fact that callers perform well at detecting common ground-truth variants indicates that the WGS variants are well represented at the pooled read level in both modalities.
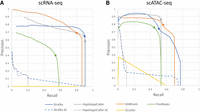
Precision-recall curves averaged over samples for different variant-calling approaches, for scRNA-seq (A) and scATAC-seq (B). Individual-cell calling (Strelka SC, HaplotypeCaller SC) shown with dashed line; dots signify the recommended quality filter level for each caller. QUAL > 30 for HaplotypeCaller, QUAL > 20 for others; no dot for single-cell callers that do not have a recommended filter level.
Area under precision recall curve (AUPRC) for bulk callers on pooled reads, in exonic regions for the case of scRNA-seq
Contrary to bulk callers applied to the pooled reads, the same callers applied at the individual-cell level do not effectively resolve common ground-truth variants. Both Strelka SC and HaplotypeCaller SC demonstrate low recall at most filtering thresholds, only detecting common ground-truth variants when quality filters are so stringent that only a handful of variants remain (corresponding to the left side of the graph in Figs. 2A,B). On scATAC-seq data sets, the single-cell callers do manage to match pooled read calling for precision at the very extremum of their quality threshold, indicating that variant calling at the single-cell level is able to resolve common ground-truth variants but at the cost of extremely low recall. HaplotypeCaller, on the other hand, performs poorly at any filtering threshold, indicating a very low sensitivity to common ground-truth variants.
A common expectation of variant callers applied at the individual-cell level is the ability to resolve rare variants that have low alternate allele evidence. To determine the ability of variant callers to detect low-read-support variants, we look at the variants called on the single-cell libraries without applying any filtering to the variant sites determined by each caller. As we can see in Figure 3, A and B, certain bulk callers considerably outperform others, with Strelka consistently demonstrating the highest sensitivity for both scRNA-seq and scATAC-seq. Additionally, the individual-cell application of Strelka (Strelka SC) appears to match the performance of Strelka when called in bulk. On the other hand, HaplotypeCaller calling on individual cells (HaplotypeCaller SC) demonstrates poor performance at detecting common ground-truth variants across all read coverage levels, indicating that the caller has decreased sensitivity when applied at the single-cell level.
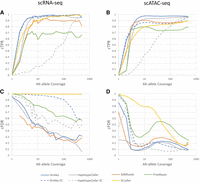
Average over samples of cTPR (A,B) and common false-discovery rate (cFDR; C,D) versus alternate allele coverage for variants binned by total (e.g., pooled) alt-allele coverage (e.g., the point coverage = 400 would include variants from 400 to the next bin, which is 500), for each of the variant-calling approaches used, for both scRNA-seq (A,C) and scATAC-seq (B,D).
Individual-cell application of bulk callers demonstrate extremely high numbers of rare positive variants, as is evidenced by the cFDR statistics in Figure 3, C and D. HaplotypeCaller SC demonstrates lower FDR than Strelka SC, possibly because of its decreased sensitivity. Pooled read callers (with the exception of FreeBayes) demonstrate similar performance in cFDR across allele coverage levels, indicating more rare positives for variants with low alternate allele coverage.
By comparison to other calling approaches, SCcaller significantly underperforms, mostly owing to a high cFDR, especially in scRNA-seq. Although the cTPR of the caller is decent, being outperformed only by Strelka, it appears that SCcaller is indiscriminate in its sensitivity. Given that SCcaller has been shown to obtain good results on MDA scDNA-seq data sets, this may be primarily owing to the low coverage present in the cells. Additionally, Strelka SC also generates a very high cFDR across different allele coverages in the case of scRNA-seq. Overall, this result supports the suspicion of others that existing single-cell variant callers are not suitable for Chromium data, although this may not be the case for all such callers.
Overall, these results agree with previous studies demonstrating that a wide range of variants present in a sample are visible in libraries generated by Chromium scRNA-seq and scATAC-seq modalities (Liu et al. 2019; Massarat et al. 2021). This enables such modalities to supplement, or act independent of, DNA sequencing to discover biologically relevant variant sites. The choice of caller has a significant impact on the ability to resolve these variants. Applying bulk callers to individual cells results in extremely high rare positive rates likely owing to a large number of false-positive calls, making such approaches impractical for calling biologically relevant variants. The potential veracity of such rare positives is discussed in later sections. Applying bulk callers to pooled reads can provide quite useful results, even at standard-quality levels (see dots on Fig. 2). Strelka, HaplotypeCaller, and SAMtools perform well on both scRNA-seq and scATAC-seq, with small differences such that it is difficult to say which one is best. Depending on whether precision or recall is more important and depending on which modality is used, Strelka or SAMtools might give optimal performance. There appear to be trade-offs between callers, and we hope the data we have presented here will help inform researchers in the field as to which approach they would prefer.
Investigating rare positive variants in pooled reads
Previously, we noted that bulk callers applied to pooled reads detect variants in addition to those found in the common ground-truth set. It is possible that some of these rare positive variants are either real and not detected in WGS or owing to other biological factors. ATAC-seq and RNA-seq modalities frequently sample specific regions to a depth much higher than the 40× coverage we obtained for our WGS. This deeper coverage may enable higher alternate allele visibility and greater confidence in variant calling at a given locus. However, rare positive variants may just as well be the result of artifacts owing to noise/error or modality-specific sequence alterations. Considering that explicit verification of these variants would likely require high-depth sequencing to confirm, we use indirect approaches to draw conclusions about the portion of rare positive variants that are potentially real.
To examine the composition of rare positive variants in general, we defined union rare positive (URP) variants as the union of rare positive variant calls for all callers performed on pooled scRNA-seq or scATAC-seq reads. We quantified the number of URP variants without any filtering except that which demonstrate evidence (at least one alt-allele read) in WGS (Table 3). This is to account for any variants that did not pass filters for calling in our common ground-truth set (i.e., WGS) but that gave sufficient evidence in scRNA-seq or scATAC-seq to be called. We find that, on average, 9.8% of URP variants called in ATAC-seq (scATAC-RPs) are present in at least five reads in WGS, with 6.8% present in at least 10 reads. On the other hand, only 2.2% and 1.2% of scRNA-seq URP variants (scRNA-RPs) show evidence of being present in WGS at depths greater than five or 10 reads, respectively. Thus, it appears that a portion of scATAC-RPs may be real and possibly missed in the common ground-truth set, whereas this is significantly less so for scRNA-RP variants. This raises a concern that scRNA-RPs are enriched for noisy or artifactual variants.
Counts of variants found in each modality
It should be noted that scRNA-seq appears to generate more URPs than scATAC-seq. If we normalize by the total coverage of each modality (see Table 3), we find that scRNA-seq has 2.9× more URP variants per megabase than scATAC-seq (P = 0.087 by two-tailed paired t-test, n = 3), despite scATAC-seq detecting similar or more (∼1.8×) common ground-truth variants than scRNA-seq (P = 0.068 by two-tailed paired t-test, n = 3). The extra variants we see may either be real and owing to differences in coverage location or owing to higher error rates in scRNA-seq.
To determine if rare positive variants tend to have a lower read coverage or lower VAF distribution, potentially indicating noise as a cause for these variants, we broke down scRNA-RP and scATAC-RP variants by VAF (Fig. 4A,C) and read depth (Fig. 4B,D) in comparison with pooled TP variants for control. Notably, although URP variants in both modalities appear to be slightly more distributed toward lower VAFs compared with TP variants, there appears to be no meaningful difference that would indicate VAF as a major correlating factor for rare positive variants. Likewise, the read depth distribution of URP for both modalities is relatively similar to that of TP variants, with TP variants presenting in a higher portion of low-read-coverage sites than URP variants, especially for scATAC-seq. The tendency of URPs to occur in regions of higher coverage for scATAC-seq may be indicative of a systematic source for their generation, which we attempt to explore more in the next section.
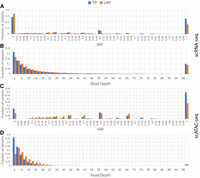
Comparing the composition of URP and TP variants by VAF and read depth for scRNA-seq (A,B) and scATAC-seq (C,D). The number of variants with a read depth of more than 100 are summed in the rightmost bin.
Analysis of the mutational signatures of variants, specifically the base sequence in the context of a SNV or indel locus, is a common approach for resolving the mechanisms underlying a particular variant (Omichessan et al. 2019). Such analyses have been used to identify signatures corresponding to mutations resulting from artifacts in sequencing and variant calling (Alexandrov et al. 2020). Here we used the SigProfiler tool developed by Bergstrom et al. (2019) to extract and fit known signatures for scATAC-seq and scRNA-RP variants, as well as common ground-truth variants. The COSMIC (Alexandrov et al. 2020) signatures serve as a reference of well-known mutational signatures that might compose some part of the variants seen in our samples.
We find that scATAC-RP variants feature a trinucleotide mutational breakdown similar to that seen in WGS (Fig. 5A), namely, with a high proportion of variants that may be explained as a composition of the COSMIC signature SBS5 (Fig. 6A). scATAC-RPs in EPSRC1 also appear to match the SBS45 and SBS46 signatures, which have been linked to sequencing errors. scATAC-seq also features a prominent CTG→CCG rate (Fig. 5A), which does not match any known signatures that we have reviewed and may be because of a preparation/sequencing error. As such, it appears that, although scATAC-RPs may contain real variants, there is a substantial risk of including noise based on the signatures observed.
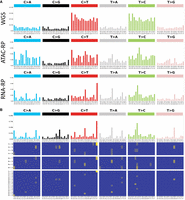
SBS 96 trinucleotide mutation breakdown for common ground-truth, scATAC-RP, and scRNA-RP variants as seen in EPSRC1 (A), which is representative of the group trend, and the SBS1536 breakdown of scRNA-RP variants in exonic regions only (B), demonstrating that the signature is maintained in exonic regions and is characterized by repeat extensions of all base patterns. Images were generated using the SigProfiler suite.
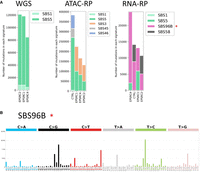
Identification of a novel SBS mutational signature presenting in scRNA-RP variants through (A) SigProfilerExtractor breakdown of mutational signatures by COSMIC SBS signatures (red star next to novel signature) and (B) the SBS 96 trinucleotide breakdown of the novel signature. Images were generated using the SigProfiler suite.
On the other hand, the trinucleotide mutational breakdown of scRNA-RPs variants differs substantially from that of WGS, demonstrating an extremely high incidence of repeat extension mutations of the form YY[X]YY > YY[Y]YY, especially in poly(T)/poly(C) regions (Fig. 5A,B). These mutations are present even if we reduce the list of variants to those found in exonic regions, which we would expect to have higher quality owing to increased coverage by scRNA-seq (Fig. 5B). According to SigProfiler, the signature of these mutations does not match any yet established signature, although they are somewhat similar to SBS58, another COSMIC potential sequencing artifact signature (Fig. 6A,B).
These previous results suggest that many URP variants are caused by noise. One major advantage of our data set is the matched scRNA-seq and scATAC-seq, which allows rare variants to be validated by confirming their presence in both modalities. We use the intersection of scRNA-RP and scATAC-RP variants as a validation set for rare positive variants, which we term multimodal rare positive (MRP) variants. We use estimated MRP and URP variant statistics (for definition, see Methods, subsection “Variant categories and metrics”; a minimum threshold of five supporting reads) (Table 4) alongside pileup overlap statistics to extrapolate the number of valid URP variants we would expect to see in each modality. We find that the rate of MRP variants per megabase is extremely small. If we extrapolate this rate to the URP variants unique to each modality, we find that they would make up ∼2% and 14% of unique URP variants in scRNA-seq and scATAC-seq, respectively. This agrees well with the portion of rare positive variants seen with evidence of the alternate allele in WGS discussed previously. It should be noted that the true number of overlapping rare variants may be larger owing to regions where one modality has very low coverage and thus does not sample the rare alternate allele. Overall, these results suggest that rare variants detectable through pooled-read variant calling in scRNA-seq and scATAC-seq make up only a small portion of rare positive variants.
scRNA-seq and scATAC-seq overlapping URP variants
Overall, it appears that many of the URP variants may be explained as owing to noise. Some of the URP variants show evidence of being real rare mutations, indicating that scATAC-seq and scRNA-seq de novo calling may yield variants not visible in typical WGS experiments. However, they make up such a small portion of the URP variant set that they would be infeasible to separate out from the majority of noisy variants in any practical scenario. The increased incidence of scRNA-RPs compared with scATAC-seq, as well as characteristic repeat extension mutations in scRNA-seq but not in scATAC-seq, strongly suggests that most of the scRNA-RPs are false positives generated by RNA-modality-specific effects. These effects may be the result of scRNA-seq-specific qualities, such as high coverage in low-complexity regions (3′ UTR, 5′ UTR and intronic regions) and poly-adenylation, although further investigation would be required to confirm this. In scRNA-seq, although these false positives are frequent, it may be possible to reduce their incidence. We recommend others consider removing repeat-extension variants of the forms TT[C]TT > TT[T]TT, GG[C]NG > GG[G]NG, and CC[T]CN > CC[C]CN. On the other hand, scATAC-RPs seem to contain a more prominent mixture of real variants among the noisy variants. As we have not discovered an easy way to separate these, it appears that noise must simply be accepted.
Single-cell-unique variants
As described previously, there is an abundance of variants that are unique to single-cell sequencing modalities, which tend to present in a low number of reads and hence a low number of cells. Previous studies have attributed these variants to real mutations unique to small-cell populations caused by high mutation rates in cancer. Given the incidence of systematic errors in regions of high coverage or motif repeats (Li 2014; Tørresen et al. 2019; Stoler and Nekrutenko 2021), variant loci called on loci with a low number of supporting reads should be treated with caution. Before any claims of rare single-cell-unique (scu) variants may be made, it is essential to determine what portion of these variants may be the result of noise or error.
To estimate the potential of rare single-cell-unique variants in our single-cell libraries, we quantify scu-variants in scRNA-seq and scATAC-seq. In line with previous results, we find abundance of scu-variants (average of 1180 per sample at this filtering level) for scRNA-seq (see Table 3).
To evaluate whether these scu-variants are the result of rapid mutations characteristic of cancer, we extract the scu-variants from an unmatched control for which we have scRNA-seq and scATAC-seq, but no WGS. To account for the lack of matched WGS, we filter out all variants seen in the intersection of the common ground-truth set from each of the sarcoma samples. We find considerably more scu-variants are generated (6600 for CTRL vs. average of 1170 for EPSRC). Repeating the analysis performed by Prashant et al. (2021), if we examine the breakdown of scu-variant locations and impact annotated by SnpEff (Cingolani et al. 2012), as seen in Figure 7, we find a breakdown very similar to that of the original paper, with 3′ UTR variants being extremely common and an abundance of missense and intergenic variants (see Fig. 7A). The rare positive variants called on pooled reads (referred to as URP variants in Table 1) also demonstrate breakdowns similar to those of Prashant et al. (2021; see Fig. 7B). Although our breakdowns are not directly comparable to previous results owing to a difference in the annotation tool used (SnpEff rather than SeattleSeq), these similarities give confidence that we have reproduced the scu-variants seen in previous studies.
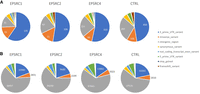
Breakdown of variant annotations for scu-variants (A) and rare positive variants (B) derived from scRNA-seq for each sample.
Notably, the distribution of both scu-variants and rare positive variants in our wild-type control closely follows that of the sarcomas. A chi-squared test of the category counts for scu-variants across samples results in a P-value of 0.0004 (see Table 5), which is mostly owing to the decreased incidence of intergenic and noncoding variants seen in EPSRC1. If intergenic and noncoding categories are removed, or EPSRC1 is removed from comparison, the P-value shifts to nonsignificant values of 0.357 and 0.359, respectively, indicating strong agreement. Because intergenic and noncoding regions typically have poor coverage in scRNA-seq, we are inclined to conclude these extra variants in EPSRC1 are caused by noise, possibly resulting from differences in the coverage profile of the scRNA-seq library sampled from EPSRC1. This overall result demonstrates that scu-variants are found in both cancerous and healthy tissue consistently across the same genomic locations and therefore cannot be easily attributed to low frequency somatic mutations associated with cancer development as previously described.
Breakdown of scRNA-seq scu-variants by predicted location/impact
The abundance of scu-variants seen in Chromium scRNA-seq appears to be modality specific, as performing the same analysis on scATAC-seq returns, on average, ∼230× fewer variants for the same filtering levels (average, 27 scu-variants) (see Table 3). This is surprising considering the significantly higher breadth of coverage seen in scATAC-seq versus scRNA-seq. Although the two modalities cover different genomic regions, with scRNA-seq focusing on the exome, we would not expect the background somatic mutation rate to vary so drastically by region. This discrepancy further implies that the abundant scu-variants seen in scRNA-seq are the result of RNA-specific artifacts.
Overall, although we do not dismiss the possibility that scu-variants may capture rare cancerous somatic mutations, the abundance of scu-variants in scRNA-seq seen in both normal and cancerous tissue suggests that isolating rare somatic variants from background noise in scRNA-seq would be a considerable challenge. The lack of scu-variants in scATAC-seq is further evidence that those seen in scRNA-seq are because of noise. As such, we conclude that the utility of scu-variants as candidate rare somatic variants is questionable at best.
RNA-editing events
RNA-editing events are a type of post-transcriptional modification that can alter the sequence of an mRNA transcript to modify the function or localization of the resulting protein (Nishikura 2010). RNA editing is widespread among eukaryotic organisms, and specific modifications are often tissue specific (Song et al. 2004; Picardi et al. 2015). There is a considerable interest in furthering our understanding of editing sites, such as the resulting impact on function and the context in which it occurs, as RNA editing has been suggested as a safer method of genomic alteration for cancer therapy (Reardon 2020). As a result, there is a demand for approaches that can resolve RNA editing at the cell type level and link this to cell activity.
RNA sequencing can capture RNA-editing events that occurs on mRNA strands as they are reverse-transcribed to cDNA for tagging and amplification. Moreover, scRNA-seq methods may resolve this at the single-cell level, enabling cell type–specific characterization of editing sites at a much finer level of detail than can be achieved with many bulk-RNA-seq libraries. Thus, scRNA-seq is uniquely suited to provide additional insight into RNA editing. However, some studies have questioned the ability to resolve RNA-editing events in tag-based scRNA-seq approaches such as Chromium (Chen et al. 2019) We set out to determine if RNA-editing events are visible in Chromium scRNA-seq libraries and to determine if they may resolve cell type–specific effects.
Prior work has amalgamated libraries of common RNA-editing sites in mice (Mansi et al. 2021). Using overlap between scRNA-RP variants derived from our scRNA-seq data sets and the known RNA-editing sites, we attempted to characterize the visibility of RNA edits in Chromium 3′ scRNA-seq. Across our four scRNA-seq samples, we find a total of 4942 unique variant sites (average, 1870 per sample) matching RNA-editing events in the REDIportal database, out of a total of 107,095 variants (average, 1.75% detection rate). For context, in scATAC-seq-derived common ground-truth and scATAC-RP variants, only two and 31 matches were discovered, respectively. The coverage of editing sites seen in scRNA-seq can vary, with most demonstrating low coverage. However, many editing sites appear in a sufficient quantity of reads such that many cells within a sample demonstrate visibility of the editing site (Fig. 8A). Given that editing alleles often appear on a fraction of reads (Fig. 8A), it is possible that a considerable number of editing sites may have relevance in cell type–specific analysis of editing events.
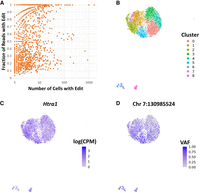
Evidence of potential RNA-editing events in scRNA-seq variants, demonstrated by (A) a high incidence of editing sites and the presence in both low and high portions of reads. (B–D) UMAP embedding of cells in EPSRC1 scRNA-seq sample based on per-cell expression profile, with clusters derived from Louvain clustering of normalized log counts per million for each gene after regressing cell-cycle effects to demonstrate the heterogeneity of cells (B); expression level of the gene on which the RNA edit of interest is located (LOG(CPM) indicates logarithm of counts per million), demonstrating a uniform expression profile (C); RNA-edit allele presentation frequency (VAF) differing across cells in a pattern similar to expression-based cell heterogeneity (D).
Most editing sites did not present in an adequate number of cells to infer cell type–specific editing. However, we found one site at Chr 7: 130,985,524 presenting as an A-to-G base substitution that demonstrated differential presentation of the RNA edit based on the expression profile of the cells in the sample. This was found in EPSRC1, and we display the presentation of the RNA-edit allele presentation frequency per cell, termed VAF, on the UMAP embedding of expression generated using the Seurat R package (Fig. 8D). For comparison, we used the expression profile of cells to perform unsupervised Louvain clustering for any cell type heterogeneity within our tumor sample (Fig. 8B). As we can see, the VAF varies considerably across the sample, with clusters 5 and 2 being enriched for the RNA edit and clusters 3 and 4 showing an absence of the edit (P = 4.0 × 10−14 by Fisher's exact test using alternate allele and reference allele counts across cluster categorization). As can be seen in Figure 8C, this is not because of differences in expression levels, as the gene on which the edit is located (Htra1) is expressed in all involved clusters.
Overall, this constitutes evidence that RNA-editing events may be seen in Chromium scRNA-seq libraries and used to infer heterogeneous presentation of RNA editing. Although only one differentially presented site was found in our samples, there are many other sites with high coverage. Given that our samples are relatively homogeneous with respect to cell type compared with other studies owing to all being sampled from a mesenchymal tumor phenotype, it is possible that performing the same analysis on Chromium scRNA-seq libraries composed of multiple distinct cell types would yield additional cell type–specific presentation of RNA edits. However, we realize that a fair number of the variants found to match known PTM sites may be false positives owing to noise, as discussed in previous sections. Furthermore, without a ground-truth data set of PTMs to compare, we cannot measure the accuracy of the approach. Thus, significant further study is required to confirm whether the results we have found represent real measurable RNA-editing events in Chromium scRNA-seq.
Discussion
Previous studies have applied various variant-calling approaches to Chromium-based scRNA-seq and scATAC-seq libraries without full consideration of the potential false-positives and artifacts generated by such approaches. In particular, traditional callers applied to pooled and individual cells are frequently used. Our analysis shows that a false-discovery rate of around 0.4 and 0.2 for Chromium scRNA-seq and scATAC-seq, respectively, may be expected for most pooled read callers on these modalities. Of the pooled read calling approaches, it appears that Strelka2, GATK HaplotypeCaller, and SAMtools Call appear to offer the best performance in both modalities.
Variant calling in Chromium single-cell sequencing modalities such as scRNA-seq and scATAC-seq is increasingly being done with the application of bulk variant callers at the individual-cell level. The major perceived advantage of variant calling at the individual cell is the ability to resolve rare variants not otherwise detected in variant calling on pooled reads. However, based on the results of this study, such approaches do not exceed the performance of pooled read calling for the detection of infrequently presented variants and are limited by their high false-discovery rates. Additionally, the excess of variants unique to individual-cell callers appear to mostly consist of noise or artifacts specific to the sequencing modality. We provide evidence of this fact in contrast to previous studies claiming that such variants might be rare somatic variants present in a small subgroup of cells. Although we do not refute the presence of rare somatic variants detected with the single-cell approach, we reason that it would be difficult to separate these from the number of false-positive variants generated without additional validation with high-depth DNA sequencing. As a result, we conclude that variant calling at the single-cell level offers no benefits over calling on the pooled reads in Chromium scRNA-seq and scATAC-seq, and we recommend others rely on variant callers such as Strelka, SAMtools, and HaplotypeCaller, which demonstrate good performance in both modalities.
Through further investigation of variants discovered by variant calling on pooled reads, we find that scRNA-seq generates a significant number of novel variants that are likely because of noise. We track this down to repeat-extension variants, possibly owing to the poly(A) tail or increased coverage in 3′ and 5′ low complexity regions characteristic of the Chromium scRNA-seq modality. We recommend others performing variant calling in Chromium scRNA-seq remove variants with XX[Y]XX > XX[X]XX signatures, especially if there is evidence that these compose a significant portion of the variants detected.
Finally, we find that Chromium scRNA-seq appears to detect instances of RNA editing on the sequenced transcripts. The incidence of these events is extensive and is found in many cells. We show that such a signature can be investigated at the single-cell level to resolve cell type–dependent RNA editing for a particular locus, provided there is sufficient coverage in cells. As such, we conclude that Chromium scRNA-seq may be a useful tool for investigating RNA-editing events at the single-cell and cell type level.
It should be noted that we are not discounting the possibility of rare somatic mutations in our data. However, based on the number of likely artifactual variants we see both in tumors and wild-type samples, it would be very difficult to resolve such variants if they exist without high-depth DNA sequencing for confirmation.
Of novel variants called on scRNA-seq, many of them appear to be owing to repeat-extension artifacts characteristic of Illumina sequencing. We believe this is because of higher coverage in the low complexity regions found in the 3′ and 5′ untranslated regions of genes. However, a certain portion of these variants may be associated with RNA-editing events seen in other scRNA-seq methodologies. Our preliminary results suggest that Chromium 3′ scRNA-seq may be used to detect and characterize intercell variations in RNA editing, which could significantly accelerate our understanding of this phenomenon.
It is worth mentioning that it is possible that any of the variant-calling approaches applied herein may be considerably improved by a different application of filtering approaches. For ease of comparison, we did not test filtering approaches exhaustively for all callers. Additionally, the quality metric used for individual-cell callers, namely, maximum overall variant quality, may not be optimal but is most similar to what is typically used. Of alternative approaches we tested, such as minimum number of cells in which the variant was visible or average quality, we found it to perform the best. More work to investigate other filtering techniques may improve results.
Overall, variant calling in Chromium scRNA-seq and scATAC-seq has great promise, and good results can be achieved using basic variant-calling approaches. We offer a broad review and empirical evidence of the performance of such approaches, which should be helpful to anyone wishing to apply them to their own sequencing libraries.
Methods
Mouse model and tumor samples
All samples used in this study were selected from an inbred mouse line derived from the C57BL/6J line. Smarcb1 (Smarcb1f/f, control Smarcb1f/+) deletion was induced in the Hic1CreERT2 background (Scott et al. 2019), including a B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Jax stock number 007914) reporter allele, following administration of tamoxifen (TAM) at ∼8 weeks of age. Epithelioid-like tumors appeared in various regions of the mouse with a latency of 10–14 months post-TAM weeks. From this study, three tumor samples from different animals labeled as EPSRC1, EPSRC2, and EPSRC4 were taken for further processing. Tumor resections were gently minced using scissors into 4 mm pieces and enzymatically dissociated using a cocktail of 1.5 U/mL collagenase D (Roche 11088882001) and 2.4 U/mL Dispase II (Roche 04942078001) for 60 min. The resultant cell suspension was passed through a 40 μm filter and centrifuged at 500g for 5 min. Cells were then incubated for 30 min on ice with the following lineage antibody cocktail (anti-CD45-Alx647 [Ablab 67-0047-01, 1:400], anti-CD31-APC [BD Biosciences 551262, 1:400], anti-CD11b-Alx647 [Ablab 67-0055-01, 1:500], anti-F4/80-Alx647 [Ablab 67-0035-05, 1:500], anti-CD117-APC [eBioscience 17-1172-82, 1:500], and anti-Ter119-647 [Ablab 67-0031-01, 1:200] dissolved in FACS buffer [PBS with 2% FBS and 2 mM EDTA]) before fluorescence-activated cell sorting (FACS) against lineage markers to reduce the number of contaminating cell types and for tdTomato-positive cells on an Influx (BD) sorter. Samples were then prepared and sequenced using Chromium-based scRNA-seq and scATAC-seq workflows (10x Genomics), as well as WGS (Illumina). This resulted in a highly enriched collection of tumor cells for each sample.
Sample preparation and sequencing
For scRNA-seq, sorted cells were prepared using the Chromium Next GEM Single Cell 3′ v3 Kit (10x Genomics). Cells were sequenced on the NextSeq 500 platform (Illumina). Reads were processed first using CellRanger v3.0.2 (10x Genomics) on default settings. CellRanger performs a variety of preprocessing steps, including unique molecular identifier (UMI) and cell barcode (CB) identification and denoising, adapter trimming, and duplicate marking, as well as alignment with STAR (Dobin et al. 2013). On average, there were 7900 cells per sample and 62,000 reads per cell.
For scATAC-seq, sorted cells were prepared using the Chromium Single Cell ATAC v1 Kit (10x Genomics). Cells were again sequenced on the NextSeq 500 platform (Illumina). Reads were processed using CellRanger ATAC v1.1.0 (10x Genomics) on default settings, which implement BWA-MEM for alignment (Li 2013) and custom cell barcode identification and cell filtering steps. There was an average of 12,400 cells per sample and between 5000 and 22,000 median fragments per cell.
Concomitantly to single-cell capture, additional cells were centrifuged, flash-frozen, and banked. Thawed cells were further processed for WGS. Control tissue was obtained from age-matched Hic1CreERT2; R26tdTomato, Smarbcb1+/+ controls and processed for WGS. Samples were prepared using the DNA PCR-free library tagmentation prep kit (Illumina). The libraries were sequenced on the HiSeq X platform (Illumina). Reads were aligned using BWA-MEM (Li 2013).
Variant categories and metrics
The full set of variant category definitions can be seen in Table 1. As a gold standard of common or high-allele-frequency variants that should be called in a single cell, we use the consensus of multiple variant callers on the matched WGS libraries for each sample. This gives a high-confidence list of biological common variants present in each sample. Given that WGS is limited in its capability to detect variants present in a small number of cells, compared with other modalities, we would expect that such rare variants would most often be undetected or indistinguishable from noise in WGS data. As such, we use WGS to generate what we call the common ground-truth set given that it should accurately represent the content of high-allele-frequency variants in the sample.
If a variant in the common ground-truth set is not detected in single cells, but the reads in that sample show sufficient evidence for an alternate allele, it is defined as a pooled false negative and used to calculate precision. Furthermore, we define a rare positive as a variant that was detected in single-cell sequencing that was not present in the common ground-truth set. It is possible that rare positives are rare variants detected owing to the high depth and allelic imbalance present in scRNA-seq and scATAC-seq. However, it is also likely that many are false positives because of noise or artifacts. Despite the potential veracity of rare positives, we use them to calculate recall out of convenience.
We define union rare positive (URP) variants as the union of rare positive variant calls for all callers performed on pooled scRNA-seq or scATAC-seq reads. We additionally required them to pass a weak quality filter (QUAL > 10) to remove low-quality variants. To ensure equivalent comparison of modalities, variants in all regions were considered except when otherwise noted.
To determine the performance of callers in regions of low versus high alternate allele read coverage, we define a common true-positive rate (cTPR) based on the portion of pooled true-positive and pooled false-negative variants (Table 6). Correspondingly, we define a common false-discovery rate (cFDR) using the portion of rare-positive and pooled true-positive variants.
Quick reference for definitions of metrics used frequently throughout this study
We cannot confirm rare-positive variants with our WGS libraries as they are not of high enough coverage to resolve low-frequency variants with confidence. However, there is a potential approach to estimate the rare variants present in our samples. A notable advantage of our data set is the availability of matched scRNA-seq and scATAC-seq for a specific sample. Given the much higher coverage and potential for visibility of rare alleles in these modalities, variants seen in both scRNA-seq and scATAC-seq but not in WGS are arguably very likely to be real rare variants. Thus, to provide an estimate of the incidence of rare variants in the absence of a high-coverage WGS or WES resource for verification, we can leverage the overlap of URP variants in scRNA-seq and scATAC-seq. We use the URP variant seen in scRNA-seq and scATAC-seq, keeping those that have at least five supporting reads in each modality and an alternate allele frequency of at least 0.5% to remove sequencing noise. We then count the number of base pairs with at least five reads of coverage in scRNA-seq, scATAC-seq, and overlapping between the two to provide an estimate for the rare mutation rate per megabase. We term this the estimated rare variant rate.
For comparison with existing studies (Prashant et al. 2021), we define a scu-variant as any variant that is called at the single-cell level in scRNA-seq or scATAC-seq but is not called on pooled reads or found in WGS. To keep in line with previous studies, we restrict this definition to the intersection of scu-variants called by both GATK HaplotypeCaller and Strelka2 with a quality greater than 100 and 40, respectively. We further restrict this to variants that show no evidence of being called at a statistically significant level in WGS or by bulk callers on pooled reads.
WGS common ground-truth variant calling
To ensure high quality of called variants, WGS-derived common ground-truth variants were taken as the intersection of multiple callers. We used GATK HaplotypeCaller (Poplin et al. 2018), Strelka2 (Kim et al. 2018), SAMtools (Li 2011), and VarScan (Koboldt et al. 2009) in their germline calling modes on default settings. For GATK, we performed recommended best-practices steps prior to variant calling, including Picard MarkDuplicates and GATK base quality score recalibration. Filtering was then done for all caller results to keep high-quality variants. For Strelka2 and VarScan, this involved keeping variants that passed all filtering steps. For GATK HaplotypeCaller, variants were filtered using the recommended hard filtering approach: for SNVs, QD > 2.0, QUAL > 30.0, SOR < 3.0, FS < 60.0, MQ > 40.0, MQRankSum > −12.5, and ReadPosRankSum > −8.0; for indels, QD > 2.0, QUAL > 30.0, FS < 200.0 and ReadPosRankSum > −20.0. For SAMtools, variants were filtered using the recommended hard filtering approach of QUAL ≥ 20.0. Variants were added to the common ground-truth set for a sample if they passed filters for at least three of the four callers.
Variant calling on pooled reads from single-cell libraries
Variant calling was performed on pooled reads from scRNA-seq and scATAC-seq libraries using Strelka2, SAMtools, GATK HaplotypeCaller, and FreeBayes. For HaplotypeCaller, pooled reads were preprocessed according to modality-specific recommended best practices. For scRNA-seq, this included SplitNCigarReads and Base Quality Score Recalibration, whereas for scATAC-seq, the DNA-seq variant-calling best practices approach was assumed and thus only Base Quality Score Recalibration was performed. SAMtools and HaplotypeCaller were run in their default configuration. Strelka2 was run using its new ‐‐rna mode, in which many filters are loosened, and a new scoring model is used. This was applied for both scRNA-seq and scATAC-seq, as it was reasoned that these new filters, especially relating to allele frequencies, would enable better sensitivity in both modalities.
Variant calling on individual-cell reads from single-cell libraries
To prepare reads to be called individually at the single-cell level, a custom Python script was used to split SAM/BAM read files by cell barcode into individual files for each cell. Libraries were split using the whitelist of cell barcodes generated by the CellRanger pipeline on its default settings for scRNA-seq and scATAC-seq, respectively, which uses a set of custom criteria for filtering out cells that are deemed to be low quality. Any reads with a cell barcode not within the whitelist were discarded. After splitting, variant callers were run on each individual-cell BAM file to generate a list of variants, which were then annotated with SnpEff (Cingolani et al. 2012). A custom Python script was used to collect variants called in each individual cell into one file and summarize collective statistics for each variant. A quality score was assigned to each variant, defined as the maximum quality seen for that variant across all cells in the sample.
Two bulk variant callers, Strelka2 and GATK HaplotypeCaller, were applied at the single-cell level. For HaplotypeCaller, reads were preprocessed according to recommended best practices prior to read splitting. Strelka2 was run in its ‐‐rna mode.
Additionally, SCcaller (Dong et al. 2017) was run as a purpose-built single-cell variant caller on each individual cell. SCcaller requires a list of known variants ideally called on matched DNA-seq for the sample in question to estimate allele frequencies in each region. For this purpose, the list of variants called on WGS was used for each sample. Additionally, SCcaller averages local allele frequencies following a moving window of length specified by the user. To keep this window on the same order of length as that of a protein and thereby model the allelic bias associated with each protein, a window size of 5 kb was used.
Defining scu-variants
Following previous studies, a single-cell-unique variant was defined as a variant that was detected in two independent individual-cell callers with sufficient quality score in at least one cell to pass a filtering threshold. The two callers used were Strelka and HaplotypeCaller, both applied at the single-cell level. Additionally, scu-variants had to be unique to single-cell calling, and therefore, any variants detected in WGS or by the application of variant callers on pooled reads were removed. We defined a bulk variant as the union of variants called on the pooled single-cell reads using Strelka, HaplotypeCaller, and SAMtools. The quality filters for scu-variants generated by Strelka and HaplotypeCaller were set to 40 and 100, respectively, and these were found to be the quality past which the number of variants stopped decreasing significantly for any further increase in quality.
Coverage and false-negative quantification
To determine if a variant found in WGS was visible in single-cell libraries, Vartrix by 10x Genomics was used to count the number of reads per cell containing a variant of interest. Possorted BAM files from CellRanger were run on Vartrix using the consensus variants from matched WGS. Reads with a mapping quality of less than 10 were removed. For scRNA-seq, reads have molecular barcodes and were thus deduplicated using the umi mode; in scATAC-seq, reads were deduplicated with the no-duplicates option. Variant loci were counted by number of alternate/reference allele reads per cell. Missing alleles were defined as a locus that did not show evidence of the allele seen in WGS but had coverage. False-negative calls were defined as loci that showed evidence of the alternate allele but were not called as such by the variant caller.
For situations in which GATK HaplotypeCaller was used for variant calling, all the above steps were run to generate a count on the BAM file after preprocessing according to GATK-recommended best practices for each modality.
To check variants found in single-cell libraries were visible in WGS libraries, GATK ASEReadCounter was used. Variants were treated as heterozygous and used to count the presentation of alternate and reference alleles on reads passing default quality filters.
To determine the regions and depth of coverage of the genome for BAM files, BEDTools (Quinlan and Hall 2010) was used. From this toolkit, the functions genomecov, merge, and intersect were used to determine regions of genome coverage, combine regions with coverage to contiguous sets, and determine overlapping regions of coverage between two BAM files, respectively.
Benchmark scoring
We use quality filtering measures at a variety of thresholds to generate precision-recall curves. For bulk-calling methods, the default variant quality returned is used. For individual-cell calling, a list of variants is available for each cell. To merge these into one list, the union of variants across all cells was taken, and any variants appearing in multiple cells were counted once. The maximum of variant call qualities over all cells in which the variant was detected was used as an aggregate quality measure.
For scATAC-seq, variants in any region are considered. As the validity of scRNA-seq results may extend only to exonic regions, we include the results for scRNA-seq limited to exonic regions in Figures 1, 2, and 3 but also include the results for scRNA-seq in all regions in Supplemental Figure S2, which demonstrate a substantially increased incidence of false positives.
Expression level analysis and plotting
Expression-level analysis was done in R version 4.0.3 (R Core Team 2022) using the Seurat package version 4.1.0 (Hao et al. 2021) to perform loading and manipulation of expression counts generated by CellRanger for each sample.
Some plots demonstrating cell–cell similarity based on expression were generated using the Seurat package. Cells were filtered based on percentage-mitochondrial RNA, number of total visible genes, and number of RNA reads according to Seurat recommended practices. The 2000 most variable features were kept. Features were reduced to the first 50 components found by principal component analysis. The cell-cycle impact on gene expression was regressed out by identifying cell-cycle genes based on the Harvard Chan Bioinformatics Core tinyatlas annotations followed by repeated PCA analysis. Cells were clustered using Louvain clustering at 0.8 resolution and plotted using UMAP (Sainburg et al. 2021) on all 50 PCA components.
Data access
The scRNA-seq and scATAC-seq data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE213338 and GSE213503, respectively. The WGS data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1099909. The source code, including all helper scripts, is available at GitHub (https://github.com/mvjw/variant_stats) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the following grants: Terry Fox New Frontiers program project grant 1021 (T.M.U.), National Institutes of Health grant CA231652 (T.M.U.), and Natural Sciences and Engineering Research Council of Canada discovery grant RGPIN-2019-04896 (A.B.). We also thank the following facilities for technical assistance: BRC-seq, ubcFLOW cytometry, AbLab, BRC genotyping, and the BRC transgenic unit.
Author contributions: Study conception and design were by M.W., A.B., H.F., R.W.S., and T.M.U. Data collection was by R.W.S. and T.M.U. Analysis and interpretation of results were by M.W., A.B., H.F., R.W.S., and T.M.U. Draft manuscript preparation was by M.W. Supervision of the study was by T.M.U. and A.B. All authors reviewed the results and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277066.122.
- Received August 5, 2023.
- Accepted August 12, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








