
Semiconservative transmission of DNA N6-adenine methylation in a unicellular eukaryote
- Yalan Sheng1,2,6,7,
- Yuanyuan Wang1,2,6,
- Wentao Yang3,6,
- Xue Qing Wang3,
- Jiuwei Lu4,
- Bo Pan1,2,
- Bei Nan1,2,
- Yongqiang Liu1,2,
- Fei Ye1,2,
- Chun Li5,
- Jikui Song4,
- Yali Dou3,
- Shan Gao1,2 and
- Yifan Liu3
- 1MOE Key Laboratory of Evolution and Marine Biodiversity and Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China;
- 2Laboratory for Marine Biology and Biotechnology, Qingdao Marine Science and Technology Center, Qingdao 266237, China;
- 3Department of Biochemistry and Molecular Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California 90033, USA;
- 4Department of Biochemistry, University of California Riverside, Riverside, California 92521, USA;
- 5Division of Biostatistics, Department of Preventive Medicine, University of Southern California, Los Angeles, California 90033, USA
-
↵6 These authors contributed equally to this work.
Abstract
Although DNA N6-adenine methylation (6mA) is best known in prokaryotes, its presence in eukaryotes has recently generated great interest. Biochemical and genetic evidence supports that AMT1, an MT-A70 family methyltransferase (MTase), is crucial for 6mA deposition in unicellular eukaryotes. Nonetheless, the 6mA transmission mechanism remains to be elucidated. Taking advantage of single-molecule real-time circular consensus sequencing (SMRT CCS), here we provide definitive evidence for semiconservative transmission of 6mA in Tetrahymena thermophila. In wild-type (WT) cells, 6mA occurs at the self-complementary ApT dinucleotide, mostly in full methylation (full-6mApT); after DNA replication, hemi-methylation (hemi-6mApT) is transiently present on the parental strand, opposite to the daughter strand readily labeled by 5-bromo-2′-deoxyuridine (BrdU). In ΔAMT1 cells, 6mA predominantly occurs as hemi-6mApT. Hemi-to-full conversion in WT cells is fast, robust, and processive, whereas de novo methylation in ΔAMT1 cells is slow and sporadic. In Tetrahymena, regularly spaced 6mA clusters coincide with the linker DNA of nucleosomes arrayed in the gene body. Importantly, in vitro methylation of human chromatin by the reconstituted AMT1 complex recapitulates preferential targeting of hemi-6mApT sites in linker DNA, supporting AMT1's intrinsic and autonomous role in maintenance methylation. We conclude that 6mA is transmitted by a semiconservative mechanism: full-6mApT is split by DNA replication into hemi-6mApT, which is restored to full-6mApT by AMT1-dependent maintenance methylation. Our study dissects AMT1-dependent maintenance methylation and AMT1-independent de novo methylation, reveals a 6mA transmission pathway with a striking similarity to 5-methylcytosine (5mC) transmission at the CpG dinucleotide, and establishes 6mA as a bona fide eukaryotic epigenetic mark.
As a base modification, N6-adenine methylation can occur in both RNA (referred to as m6A) and DNA (6mA). 6mA in eukaryotes has long been known, but its widespread presence is only lately realized (Fu et al. 2015; Mondo et al. 2017; Wang et al. 2017; Bochtler and Fernandes 2021). 6mA studies in eukaryotes are complicated by varying abundance and divergent functions across species. In protists, green algae, and basal fungi, 6mA is abundant, enriched at the ApT dinucleotide, and associated with genes, all of which are consistent with its role as an epigenetic mark (Fu et al. 2015; Mondo et al. 2017; Wang et al. 2017, 2019; Luo et al. 2018; Beh et al. 2019). In animals, plants, and higher fungi, 6mA is scarce, promiscuous in its sequence context, and associated with silenced genomic regions (Greer et al. 2015; Zhang et al. 2015; Koziol et al. 2016; Liu et al. 2016; Wu et al. 2016; Liang et al. 2018; Wang et al. 2018; Xiao et al. 2018; Zhou et al. 2018; Lyu et al. 2022). In these organisms, it remains controversial whether 6mA is an enzymatically deposited epigenetic mark; alternatively, the modified base, as an RNA breakdown product, is merely misincorporated into DNA (Schiffers et al. 2017; Musheev et al. 2020; Bochtler and Fernandes 2021; Liu et al. 2021a; Kong et al. 2022; Lyu et al. 2022). More rigorous application of cutting-edge technologies, like single-molecule real-time circular consensus sequencing (SMRT CCS) mentioned below, has the potential to further clarify the issue (Kong et al. 2022).
Methyltransferases (MTases) of the MT-A70 family are involved in N6-adenine methylation in eukaryotes (Iyer et al. 2011, 2016). They are classified into several clades with distinct structures and functions (Iyer et al. 2016; Wang et al. 2019). Two of these clades are represented by AMT1 (also known as MTA1) and AMT6/AMT7 (MTA9-B/MTA9), which are part of the eukaryotic 6mA-MTase complex first identified in the protist Tetrahymena thermophila (Beh et al. 2019; Wang et al. 2019). METTL4/DAMT-1 are members of another clade (Greer et al. 2015), but their status as bona fide 6mA-MTases is still not supported by biochemical evidence (Iyer et al. 2011, 2016; Beh et al. 2019; Wang et al. 2019). Critically, AMT1 and AMT6/AMT7 homologs are only found in protists, green algae, and basal fungi, whereas METTL4/DAMT-1 homologs are mostly found in animals, plants, and higher fungi (Beh et al. 2019; Wang et al. 2019). Phylogenetic distributions of these two deep branches of MT-A70 family members, therefore, closely match that of the two alternative modes of 6mA in eukaryotes (Beh et al. 2019; Wang et al. 2019). However, even in the best-characterized Tetrahymena system, molecular mechanisms of 6mA transmission still need to be elucidated.
Tetrahymena thermophila, a ciliated protist, is the first eukaryote with 6mA identified in its nuclear DNA (Gorovsky et al. 1973), and more recently, with AMT1, the eukaryotic 6mA-specific MTase, identified and characterized (Beh et al. 2019; Wang et al. 2019). Tetrahymena contains within the same cytoplasmic compartment two types of nuclei, the somatic macronucleus (MAC) and the germline micronucleus (MIC) (Karrer 2012). Although missing in the transcriptionally silent MIC, 6mA is abundantly present in the transcriptionally active MAC and associated with RNA polymerase II-transcribed genes, consistent with its role as a euchromatic mark (Gorovsky et al. 1973; Wang et al. 2017).
6mA is readily detected by single-molecule real-time (SMRT) sequencing via its perturbation to DNA polymerase kinetics—specifically, increase in the time between nucleotide incorporation, referred to as the interpulse duration (IPD) (Fig. 1A; Eid et al. 2009; Flusberg et al. 2010). Genome-wide mapping of endogenous 6mA in eukaryotes has previously been achieved only at the ensemble level, by combining different DNA molecules covering the same genomic position to overcome random fluctuations in IPD—an approach referred to as continuous long reads (CLR) (Wang et al. 2017, 2019; Beh et al. 2019). Effective implementation of circular consensus sequencing (CCS; also known as Pacific Biosciences [PacBio] HiFi sequencing), by combining reads from multiple passes of the same DNA template (Fig. 1B; Eid et al. 2009; Flusberg et al. 2010; Wenger et al. 2019; Kong et al. 2022), allows us to accurately map 6mA distribution in the Tetrahymena MAC genome at the single-molecule level and rigorously establish AMT1-dependent semiconservative transmission of 6mA.
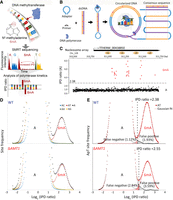
Exclusive methylation at the ApT dinucleotide in Tetrahymena. (A) Overview of 6mA detection by SMRT sequencing. (B) A schematic diagram for SMRT CCS. (C) IPD ratios (IPDr) for all A sites in a typical SMRT CCS read mapped to the Tetrahymena MAC reference genome. The IPDr threshold was set at 2.38, separating 6mA from unmodified A. Note the localization of 6mA clusters in linker DNA between the canonical nucleosome array within the gene body. (D) IPDr distributions (log2) of all A sites in wild-type (WT) (top) and ΔAMT1 cells (bottom). Also plotted were distributions for A sites at the ApA, ApC, ApG, and ApT dinucleotide, respectively. (E) Deconvolution of the 6mA peak and the unmodified A peak for IPDr distributions (log2) at the ApT dinucleotide. Note the low false positive and false negative rates of 6mA calling in WT (top) and ΔAMT1 cells (bottom).
Results
6mA detection at the single-molecule level
Recent development of SMRT CCS has allowed highly accurate and sensitive 6mA calling on individual DNA molecules. SMRT CCS has been applied to detect endogenous 6mA in several eukaryotes (Kong et al. 2022). However, very short inserts/reads (a few hundred bp) are used to maximize 6mA calling accuracy (high number of passes of the DNA template), but at the cost of genome-wide coverage. SMRT CCS has also been applied to detect exogenously introduced 6mA in chromatin fiber sequencing, which exploits in vitro methylation by 6mA-specific MTases (e.g., M.EcoGII and Hia5) to probe the chromatin organization (Abdulhay et al. 2020; Stergachis et al. 2020). Long inserts/reads (∼10 kb) are used to increase sequencing coverage of large genomes of higher eukaryotes, but at the cost of reduced 6mA calling accuracy at individual DNA molecules (low number of passes of the DNA template). Here, we developed an SMRT CCS-based pipeline to map 6mA on individual native DNA molecules from Tetrahymena (Supplemental Fig. S1A). Our analyses showed that at ≥30 passes, 6mA calling accuracy reached a plateau (Supplemental Fig. S1B). We, therefore, used intermediate-sized inserts (3–5 kb, enabling most CCS reads to have ≥30 passes, given >100 kb raw read length) to balance 6mA calling accuracy and sequencing coverage. We used the CCS read of a DNA molecule as its own reference sequence in the IPD analysis, yielding averaged and standardized IPD ratios (IPDr) for each site, relative to the in silico reference for its unmodified counterpart (Supplemental Fig. S1A; Kong et al. 2022). A typical DNA molecule from WT Tetrahymena cells showed low IPDr for most adenine (A) sites and a few clusters with high IPDr (Fig. 1C). As most A sites are presumably unmodified, they formed a baseline of IPDr around 1, with low dispersion across the read length (Fig. 1C). As exceptions, we found DNA molecules with global anomalies in IPDr, whose baseline dispersed or deviated from 1 (Supplemental Fig. S1C,D), possibly due to a compromised DNA polymerase. We also found DNA molecules with local anomalies in IPDr, which contained one or more clusters of G/C/T sites with high IPDr as well as A sites (Supplemental Fig. S1E), attributable to DNA damage (Clark et al. 2011). We removed reads with global or local anomalies in IPDr, to further improve 6mA calling accuracy.
We next mapped CCS reads back to the Tetrahymena MAC, MIC, and mitochondrion reference genomes (Supplemental Fig. S2A–D). Most were aligned across the entire read to a single genomic locus (Supplemental Fig. S2B). There were some chimeric reads with different parts aligned to separate genomic loci (Supplemental Fig. S2B), attributable to concatenation during sequencing library preparation. Their constituent DNA molecules were resolved before
further analysis (Supplemental Fig. S2C). For DNA molecules fully mapped to the MAC genome (Supplemental Fig. S2D), their IPDr for A sites exhibited a bimodal distribution: a large peak with low IPDr corresponding to unmodified A and a
small peak with high IPDr corresponding to 6mA (Fig. 1D, top). A similar bimodal distribution was observed when we focused on A sites at the ApT dinucleotide (Fig. 1D, top). The 6mA peaks of these two distributions were almost superimposable (Fig. 1D, top; Supplemental Fig. S2E, left). In contrast, IPDr distributions of A sites within the ApA/ApC/ApG dinucleotides all exhibited a single peak with
low IPDr (Fig. 1D, top). These analyses indicate that 6mA is exclusively associated with the ApT dinucleotide 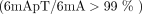 (Supplemental Fig. S2E, left).
(Supplemental Fig. S2E, left).
We deconvoluted the 6mA peak and unmodified A peak in the IPDr distribution for the ApT dinucleotide: The 6mA peak was closely fitted by a Gaussian distribution curve, whereas the unmodified A peak was deduced as the differential between the original data and the Gaussian fit (Fig. 1E, top). We set the threshold for 6mA calling at the intersection of the two peaks (IPDr = 2.38) and estimated that the false positive and false negative rates of 6mApT calling were 1.93% and 1.12%, respectively (Fig. 1E, top). We calculated that 6mApT represented 1.89% of all ApT sites (and 6mA represented 0.66% of all A sites) in DNA molecules fully mapped to the MAC genome.
To validate our bioinformatic pipeline, we reanalyzed a published data set of plasmid DNA sequenced by SMRT CCS (Abdulhay et al. 2020). We found that adenines in GATC sites, uniformly methylated by Escherichia coli Dam MTase, were distributed in a single peak with high IPDr, whereas all other adenines were distributed in a single peak with low IPDr; the combined bimodal distribution is readily deconvoluted by our method, with low false positive and false negative rates (Supplemental Fig. S2F). Using the same IPDr threshold for reads mapped to the MAC, 6mApT was called only at low levels for DNA molecules specifically mapped to the MIC (0.017%) or mitochondrion (0.014%) (Supplemental Fig. S2G; Supplemental Table S1). 6mA was called at low levels at the ApC/ApG/ApA dinucleotides regardless of their mapping (Supplemental Table S1), which were close to the background level observed in the plasmid DNA negative control (Supplemental Table S2). We also sequenced a negative control sample generated by whole genome amplification (WGA) of Tetrahymena DNA (Supplemental Fig. S3), effectively removing all base modifications while preserving the sequence information. Using the same bioinformatic pipeline, we found that all A sites, or A sites at the ApT dinucleotide, were distributed in a single peak with low IPDr (Supplemental Fig. S3A,B). We calculated the background level for 6mApT calling in Tetrahymena WGA DNA (Supplemental Fig. S3C), which was very low (0.030%) and comparable to the background level observed in the MIC and mitochondrion. Based on the accurate calling of 6mA, we conclude that 6mA occurs exclusively at the ApT dinucleotide in the MAC.
Our conclusion disagrees with previous estimates of substantial 6mA in non-ApT dinucleotides (12%) based on SMRT CLR (Wang et al. 2017, 2019; Beh et al. 2019). We examined whether 6mApT genomic positions called by CCS were also called by CLR in our previous study (Wang et al. 2017). Whereas SMRT CCS can call 6mA on individual reads/DNA molecules, SMRT CLR only calls 6mA at the ensemble level, by combining different reads covering the same genomic position, and is, therefore, affected by both sequencing coverage and 6mA homogeneity. We found that at high 6mApT coverage (i.e., the number of reads in which 6mApT has been called in a genomic position), CLR calls converged with CCS calls (Supplemental Fig. S4A,B); in contrast, they diverged at low 6mApT coverage (Supplemental Fig. S4A). Also, as 6mA homogeneity decreased (from high 6mA fraction [>80%], and intermediate [20%–80%], to low [<20%]), the percentage of 6mA called by CLR in non-ApT dinucleotides (regarded as false positive, based on our CCS analysis) increased (0.6%, 9.8%, and 82.7%, respectively). We performed additional analyses to attribute the difference between CCS and CLR calls to the poor performance of CLR, showing high rates for both false positive and false negative (Supplemental Methods; Supplemental Fig. S4C–G).
Distinguishing four methylation states of ApT duplexes
SMRT CCS makes strand-specific 6mA calls, as the DNA polymerase alternately passes through the Watson strand (W, defined as the forward strand in the reference genome) and the Crick strand (C, reverse) of a DNA template (Fig. 1B; Flusberg et al. 2010). For self-complementary ApT duplexes, we plotted their distribution according to IPDr values of A sites on W and C, respectively, and found four groups with diagonal symmetry, corresponding to four methylation states: full methylation, methylation only on W (hemi-W), methylation only on C (hemi-C), and no methylation (Fig. 2A–C). We demarcated these four groups and estimated that 89.3% methylated ApT duplexes were full methylation (full-6mApT), whereas 10.7% were hemi-methylation (hemi-6mApT) (Table 1; Fig. 2C, left, D, top; Supplemental Fig. S5). Importantly, consistent evaluation of the full- and hemi-6mApT percentages was obtained in duplicate experiments (Supplemental Fig. S5) and with varying numbers of CCS passes (Supplemental Fig. S7). Our results establish the predominance of full-6mApT over hemi-6mApT in WT Tetrahymena cells, in contrast to the near parity assessment (54% and 46%, respectively) based on CLR (Wang et al. 2019). Note that only SMRT CCS can distinguish between hemi- and full-6mApT at the single-molecule level, whereas CLR must extrapolate from the ensemble level.
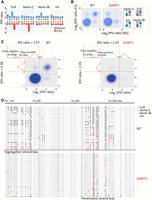
Distinguishing hemi- and full-6mApT. (A) Four states of ApT duplexes: full methylation, hemi-W, hemi-C, and unmethylated, distinguished by IPDr of adenine sites on W and C, respectively. (B) Distribution of ApT duplexes according to IPDr of adenine sites on W and C, respectively. Note the abundance of the full methylation state in WT and its absence in ΔAMT1 cells. (C) Demarcation of the four methylation states of ApT duplexes in WT (left) and ΔAMT1 cells (right) by their IPDr on W and C, respectively. (Left) For bulk ApT duplexes, the IPDr threshold for 6mA calling was set at 2.38, according to deconvolution based on Gaussian fitting of the small 6mA peak. For ApT duplexes with one 6mA as defined above, the IPDr threshold for calling 6mA on the opposite strand was set at 1.57, according to deconvolution based on Gaussian fitting of the small unmodified A peak. (Right) For bulk ApT duplexes, the IPDr threshold for 6mA calling was set at 2.55, according to deconvolution based on Gaussian fitting of the small 6mA peak. For ApT duplexes with one 6mA as defined above, the IPDr threshold for calling 6mA on the opposite strand was also set at 2.55, according to deconvolution based on Gaussian fitting of the small 6mA peak. (D) Typical DNA molecules from Tetrahymena WT (top) and ΔAMT1 cells (bottom). Note ApT duplexes with distinct methylation states (colored dots) distributed along individual DNA molecules (gray line). A DNA molecule with strong segregation strand bias in WT cells and a genomic position with strong penetrance strand bias in ΔAMT1 cells were marked.
6mA statistics in WT and ΔAMT1 cells
We compared genomic positions to which hemi-6mApT and full-6mApT sites were mapped back and found a very strong overlap between them (>99.9%) (Supplemental Fig. S6A). Furthermore, most genomic positions with multiple full-6mApT calls also contained multiple hemi-6mApT calls (Supplemental Fig. S6B). These observations support the interconversion between the hemi and full methylation states at most genomic positions. We define 6mA penetrance for each genomic position as the ratio between the number of 6mA sites and all adenine sites (with or without the modification) mapped to it. For WT cells, 6mA penetrance for most ApT positions showed no significant bias for either W or C (Fig. 7D, top); with increasing sequencing coverage, 6mA penetrance from both strands tended to converge (Fig. 7D, middle). In other words, at the ensemble level, most ApT positions in the genome were methylated at similar levels on W or C. We did not observe biased 6mA penetrance in most asymmetrically methylated ApT positions reported previously (Wang et al. 2019), and attributed them as a CLR artifact (for exceptions, see AMT1-independent de novo methylation). Our result is consistent with DNA replication splitting a full-6mApT into a hemi-W and a hemi-C.
Segregation of hemi-6mApT to the parental strand after DNA replication
We next investigated the segregation of hemi-W and hemi-C at the single-molecule level. We focused on DNA molecules with multiple hemi-6mApT, henceforth referred to as hemi+ molecules (Fig. 2D, top; Supplemental Fig. S6C,D). Their levels oscillated with cell cycle progression, starting low for cells synchronized in the G1 phase, climbing to the peak for cells in the S phase, and declining for postreplicative and dividing cells (Fig. 4A). In the vast majority of hemi+ molecules, hemi-6mApT were not randomly distributed across both strands; instead, their constituent 6mA sites were segregated with a strong bias for one strand (Figs. 2D, top and 4B,C; Supplemental Fig. S6D). Also, segregation strand bias was consistently observed with varying numbers of CCS passes (Supplemental Fig. S6). These robust results support hemi+ molecules as the product of DNA replication. Segregation was not always absolute: A minority of hemi-6mApT were occasionally detected on the opposite strand (Supplemental Fig. S6D). This is most likely due to de novo methylation, either AMT1-dependent (see AMT1-dependent maintenance methylation) or AMT1-independent (see AMT1-independent de novo methylation).
We used 5-bromo-2′-deoxyuridine (BrdU) labeling to determine whether hemi-6mApT are segregated to the parental (old) strand or the daughter (newly synthesized) strand after DNA replication. We first set up an in vitro BrdU labeling system (Fig. 3). Using a plasmid DNA fragment as the template, we performed specific labeling of either strand by primer extension, as well as total labeling of both strands by PCR (Fig. 3A). It is important to note that in the original plasmid DNA, there are three fully methylated GATC sites, which are readily converted to the hemi-methylated or unmethylated state as either or both DNA strands are replaced during BrdU labeling (Fig. 3A,B). We then performed SMRT CCS of these BrdU-labeled samples, as well as the unlabeled plasmid DNA as the negative control. We found that BrdU substitution of thymidine (T) resulted in IPDr increases, allowing us to adapt the 6mA calling pipeline for BrdU calling (Fig. 3B,C; Supplemental Fig. S8A,B). Although there were a few T positions with large shifts in IPDr, most showed only small changes (Supplemental Fig. S8C–E). We applied a single high IPDr threshold for BrdU calling (IPDr = 2.5) to achieve relatively low false positive rates (∼5%–10%; Fig. 3C), at the cost of a high false negative rate (estimated ≫50% overall). To further increase the chance to correctly identify BrdU-labeled DNA molecules, we focused on those with multiple BrdU calls, henceforth referred to as BrdU+ molecules (Supplemental Fig. S8F). There were very few BrdU+ molecules in unlabeled plasmid DNA, but many in BrdU-labeled samples (Fig. 3D; Supplemental Fig. S8F). Critically, in samples with strand-specific BrdU labeling, BrdU calls were predominantly made on the labeled strand, but not on the unlabeled strand, as indicated by strong segregation strand bias of BrdU in BrdU+ molecules (Fig. 3E; Supplemental Fig. S8G). These results validate our bioinformatic pipeline for BrdU calling and our use of BrdU labeling for distinguishing the parental strand and the daughter strand.
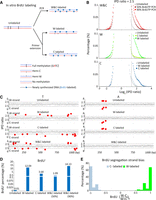
SMRT CCS detection of BrdU incorporation into newly synthesized DNA. (A) In vitro BrdU labeling. Specific labeling of either strand (W-labeled or C-labeled) was achieved by primer extension, whereas labeling of both strands (W&C-labeled) was achieved by PCR. A plasmid fragment containing three fully methylated GATC sites (6mA) was used as the template, as well as the unlabeled control for SMRT CCS. (B) IPDr distributions (log2) of all T sites from: both strands in unlabeled and W&C-labeled DNA (50% or 90% BrdUTP; top); only W in unlabeled, W-labeled, and C-labeled DNA (middle); only C in unlabeled, W-labeled, and C-labeled DNA (bottom). IPDr threshold was set at 2.5 for separating BrdU from T. (C) IPDr for all T (left) or A sites (right) in typical SMRT CCS reads for unlabeled, W-labeled, C-labeled, and W&C-labeled DNA (90% BrdUTP). IPDr thresholds were set at 2.5 for separating BrdU from T, and at 2.7 for separating 6mA from A. (D) Percentage of BrdU+ molecules in unlabeled, W-labeled, C-labeled, and W&C-labeled DNA (50% and 90% BrdUTP, respectively). BrdU+ molecules were defined as DNA molecules with no less than eight BrdU sites on one strand (W||C ≥ 8). (E) Segregation strand biases of BrdU sites in BrdU+ molecules. Segregation strand bias for BrdU was defined as the difference-sum ratio between BrdU sites on W and C: [(W − C)/(W + C)]s.
We performed in vivo BrdU labeling of Tetrahymena cells, sequenced the genomic DNA by SMRT CCS, and called BrdU as well as 6mA (Supplemental Fig. S9A,B). To eliminate interference from 6mA, we masked regions adjacent to 6mApT sites from BrdU calling (Supplemental Fig. S9B). We focused on BrdU+ molecules (Fig. 4E,F). In BrdU-labeled samples, BrdU sites in BrdU+ molecules were mostly segregated to one strand (Fig. 4E). In the unlabeled sample, “BrdU” sites were more evenly distributed across both strands, consistent with miscalls due to random fluctuations in IPDr (Fig. 4E). Our approach was further validated by strong correlations between BrdU labeling and BrdU+ molecules: (1) There were many BrdU+ molecules in BrdU-labeled samples, but few in the unlabeled sample (the percentage was further reduced when focusing on BrdU+ molecules with strong biases in strand segregation) and (2) the percentage of BrdU+ molecules increased progressively with longer labeling time (Fig. 4F). BrdU segregation was often not absolute (Fig. 4E), attributable to false positive BrdU calls (Fig. 4D). Nonetheless, the large number of BrdU sites in BrdU+ molecules allows us to identify the daughter strand with high confidence.
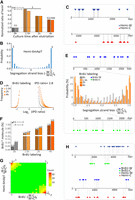
Segregation of hemi-6mApT to the parental strand after DNA replication. (A) Hemi+ molecules are enriched in S phase. Tetrahymena cells were synchronized at G1 phase by centrifugal elutriation and released for growth in the fresh medium (Liu et al. 2021b). Four time points were taken (0, 1.5, 2, and 4 h after release) for SMRT CCS. Hemi+ molecules were defined as DNA molecules with a total count of no less than 11 hemi sites (W + C ≥ 11) or with no less than 11 hemi sites on one strand (W||C ≥ 11). The count of hemi+ molecules was normalized first against the counts of total DNA molecules and then against the 0 h (G1 phase) value. (B) Hemi-6mApT sites in hemi+ molecules exhibit strong segregation strand bias. Segregation strand bias for hemi-6mApT is defined as the difference-sum ratio between hemi-W and hemi-C: [(W − C)/(W + C)]s. (C) Typical DNA molecules with hemi-6mApT fully segregated to W or C, corresponding to segregation strand bias of +1 and −1, as marked in B. (D) IPDr distributions of T sites in genomic DNA samples of synchronized Tetrahymena cells with BrdU labeling (1.5 h, 2 h, and 4 h) or without (0 h). The IPDr threshold for calling BrdU was set at 2.8. (E) BrdU sites in BrdU+ molecules exhibit strong segregation strand biases. BrdU+ molecules were defined as DNA molecules with a total count of no less than 15 BrdU sites (W + C ≥ 15). Segregation strand bias for BrdU was defined as the difference-sum ratio between BrdU sites on W and C: [(W − C)/(W + C)]s. Also shown are typical BrdU+ molecules with BrdU fully segregated to W or C, corresponding to segregation strand bias of +1 and −1, respectively. (F) Correlation between BrdU labeling and BrdU+ molecules. BrdU+ molecules were alternatively defined as DNA molecules with a total count of no less than 15 BrdU sites (W + C ≥ 15), or with no less than 15 BrdU sites on one strand (W||C ≥ 15). The latter is more selective for DNA molecules with strong strand segregation bias. (G) Hemi-6mApT and BrdU are segregated to opposite strands of the DNA duplex. Distribution of hemi+/BrdU+ molecules (hemi-6mApT: W||C ≥ 11; BrdU: W||C ≥ 15) according to their segregation strand bias for hemi-6mApT and BrdU, respectively. (H) Typical hemi+/BrdU+ molecules with hemi-6mApT and BrdU fully segregated to opposite strands, corresponding to segregation strand bias of (−1, +1) and (+1, −1), as marked in G.
There was a significant overlap between BrdU+ and hemi+ molecules in BrdU-labeled samples (Supplemental Fig. S9C–F). We focused on BrdU+/hemi+ double-positive molecules representing postreplicative DNA (Fig. 4G,H). Critically, BrdU and hemi-6mApT always exhibited the opposite biases for strand segregation in BrdU+/hemi+ molecules (Fig. 4G,H). This result indicates that after DNA replication, hemi-6mApT is essentially excluded from the daughter strand and only associated with the parental strand.
AMT1-dependent maintenance methylation
To complete semiconservative transmission of 6mA, hemi-6mApT needs to be restored to full-6mApT by maintenance methylation
before the next round of DNA replication. We investigated whether maintenance methylation was dependent on AMT1. In ΔAMT1 cells, SMRT CCS showed that 6mA was still predominantly associated with the ApT dinucleotide (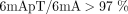 ; Fig. 1D, bottom; Supplemental Fig. S2E, right), in contrast to our previous estimation of a majority of 6mA in non-ApT dinucleotides (53%) based on SMRT CLR (Wang et al. 2019). Although WT cells contained mostly full-6mApT (89%), there were few in ΔAMT1 cells (3%) (Figs. 1E, bottom and 2B,C, right, D, bottom; Table 1). The predominant hemi-6mApT in ΔAMT1 cells is presumably the product of a dedicated de novo MTase. We conclude that AMT1 is required for hemi-to-full conversion,
that is, maintenance methylation.
; Fig. 1D, bottom; Supplemental Fig. S2E, right), in contrast to our previous estimation of a majority of 6mA in non-ApT dinucleotides (53%) based on SMRT CLR (Wang et al. 2019). Although WT cells contained mostly full-6mApT (89%), there were few in ΔAMT1 cells (3%) (Figs. 1E, bottom and 2B,C, right, D, bottom; Table 1). The predominant hemi-6mApT in ΔAMT1 cells is presumably the product of a dedicated de novo MTase. We conclude that AMT1 is required for hemi-to-full conversion,
that is, maintenance methylation.
AMT1 is part of a multisubunit MTase complex (Beh et al. 2019; Chen et al. 2022; Yan et al. 2023). We next reconstituted AMT1 complex comprising bacterially expressed AMT1, AMT7, AMTP1, and AMTP2 (also known as MTA1, MTA9, p1, and p2) (Fig. 5A; Beh et al. 2019). Using a 12 bp DNA substrate with a single centrally located hemi-6mApT, we tested the reconstituted complex for in vitro methylation and evaluated its steady-state kinetics (Km = 0.55 μM, kcat = 0.84 min−1) (Fig. 5B). We also compared methylation rates of two 27 bp DNA substrates with the same primary sequence: the hemi-methylated substrate (with two 6mApT sites segregated to one strand) and the unmodified substrate (Fig. 5C). The hemi-methylated substrate recorded 11.5× higher activity than the unmodified substrate (Fig. 5C), a much bigger advantage than previously reported (Beh et al. 2019). AMT1 complex, therefore, strongly prefers maintenance methylation to de novo methylation.
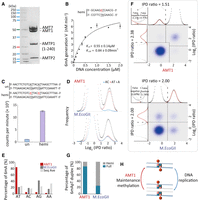
In vitro MTase activity of AMT1 complex. (A) SDS-PAGE of in vitro reconstituted AMT1 complex comprising AMT1, AMT7, AMTP1 (1–240 aa), and AMTP2. (B) The steady-state kinetics of AMT1 complex on a hemi-methylated substrate (hemi), determined by a 3H-SAM-based MTase assay. The substrate contains a single ApT duplex (underlined), which is hemi-methylated (red). (C) Methylation of the unmodified (un) and hemi-methylated (hemi) substrates. Both contain two ApT duplexes (underlined), which are either unmodified or hemi-methylated (red). (D) IPDr distributions for total adenine, adenine at the ApT dinucleotide, and adenine in ApC dinucleotide, after in vitro methylation of human chromatin by either AMT1 complex (top) or M.EcoGII (bottom). (E) 6mA distribution at all four ApN dinucleotides, after in vitro methylation of human chromatin by either AMT1 complex or M.EcoGII. ApN frequencies in SMRT CCS read are also plotted for comparison (Sequence Average). (F) Demarcation of the four methylation states of ApT duplexes by their IPDr on W and C, in human chromatin methylated by AMT1 complex (top) or M.EcoGII (bottom). AMT1 complex methylation pattern is reminiscent of that in WT Tetrahymena cells, with a strong preference for full-6mApT, as indicated by a shift in the IPDr threshold for calling full-6mApT relative to calling bulk 6mA. M.EcoGII methylation pattern is reminiscent of that in ΔAMT1 cells, with no preference for full-6mApT, as indicated by the same IPDr threshold for calling bulk 6mA or full-6mApT. (G) Relative abundance of hemi-6mApT and full-6mApT in human chromatin methylated by either AMT1 complex or M.EcoGII. (H) Model: AMT1-dependent semiconservative transmission of 6mA.
We also performed in vitro methylation of human chromatin using the reconstituted AMT1 complex (Fig. 5D–G; Supplemental Fig. S10); as a control, we used M.EcoGII, a prokaryotic MTase targeting adenine sites in any sequence context (Murray et al. 2018). Due to the scarcity of endogenous 6mA in human genomic DNA (O'Brown et al. 2019; Douvlataniotis et al. 2020; Bochtler and Fernandes 2021; Kong et al. 2022), all 6mA sites revealed by SMRT CCS were essentially attributable to the added MTases. We found that 85% of 6mA sites were at the ApT dinucleotide after AMT1 complex treatment; only 22% of 6mA sites were so after M.EcoGII treatment, close to the ApT frequency in sequenced DNA molecules (Fig. 5D,E; Supplemental Fig. S10C). The substantial 6mA in non-ApT dinucleotides (15%) after AMT1 complex treatment is consistent with previous characterization of 6mA-MTase activity partially purified from Tetrahymena (Bromberg et al. 1982). Therefore, in vitro methylation catalyzed by AMT1 complex occurs preferentially at the ApT dinucleotide, but not exclusively, as in vivo.
Methylation of ApT sites was at similar levels and far from saturation in both AMT1 complex and M.EcoGII-treated samples (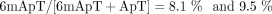 , respectively). However, 85% methylated ApT duplexes were full-6mApT after AMT1 treatment, whereas only 26% were so after
M.EcoGII treatment (Fig. 5F,G). In the case of the AMT1 complex, we found that the IPDr threshold for calling 6mA in ApT duplexes with 6mA on the opposite
strand was substantially lowered, when compared with the IPDr threshold for calling 6mA in bulk ApT duplexes (conditional
probability ≠ unconditional probability) (Fig. 5F, top). Importantly, a very similar shift in the IPDr threshold for calling 6mA in ApT duplexes was observed in WT Tetrahymena cells (Fig. 2C, left). In the case of M.EcoGII, the IPDr threshold for calling 6mA in ApT duplexes stayed the same, regardless of the methylation
state of the opposite strand (conditional probability = unconditional probability) (Fig. 5F, bottom). Therefore, M.EcoGII does not prefer maintenance methylation (hemi-to-full conversion) over de novo methylation
(un-to-hemi conversion); as a corollary, full-6mApT is generated by a random combination of two independent methylation events.
In contrast, AMT1-dependent maintenance methylation is much faster than de novo methylation, leading to the accumulation of
full-6mApT and the depletion of hemi-6mApT under in vitro as well as in vivo conditions. Therefore, preferential targeting
of ApT, especially hemi-6mApT, is an intrinsic and autonomous property of the AMT1 complex. We conclude that 6mA is transmitted
by a semiconservative mechanism in Tetrahymena: full-6mApT is split by DNA replication into hemi-6mApT, which is restored to full-6mApT by AMT1-dependent maintenance methylation
(Fig. 5H).
, respectively). However, 85% methylated ApT duplexes were full-6mApT after AMT1 treatment, whereas only 26% were so after
M.EcoGII treatment (Fig. 5F,G). In the case of the AMT1 complex, we found that the IPDr threshold for calling 6mA in ApT duplexes with 6mA on the opposite
strand was substantially lowered, when compared with the IPDr threshold for calling 6mA in bulk ApT duplexes (conditional
probability ≠ unconditional probability) (Fig. 5F, top). Importantly, a very similar shift in the IPDr threshold for calling 6mA in ApT duplexes was observed in WT Tetrahymena cells (Fig. 2C, left). In the case of M.EcoGII, the IPDr threshold for calling 6mA in ApT duplexes stayed the same, regardless of the methylation
state of the opposite strand (conditional probability = unconditional probability) (Fig. 5F, bottom). Therefore, M.EcoGII does not prefer maintenance methylation (hemi-to-full conversion) over de novo methylation
(un-to-hemi conversion); as a corollary, full-6mApT is generated by a random combination of two independent methylation events.
In contrast, AMT1-dependent maintenance methylation is much faster than de novo methylation, leading to the accumulation of
full-6mApT and the depletion of hemi-6mApT under in vitro as well as in vivo conditions. Therefore, preferential targeting
of ApT, especially hemi-6mApT, is an intrinsic and autonomous property of the AMT1 complex. We conclude that 6mA is transmitted
by a semiconservative mechanism in Tetrahymena: full-6mApT is split by DNA replication into hemi-6mApT, which is restored to full-6mApT by AMT1-dependent maintenance methylation
(Fig. 5H).
Preferential methylation of linker DNA by AMT1 complex
Previous studies have shown that in unicellular eukaryotes, 6mA distribution is connected to nucleosome distribution, suggesting that 6mA transmission relies on the chromatin environment as well as the sequence context (Fu et al. 2015; Wang et al. 2017; Beh et al. 2019). SMRT CCS revealed that on individual DNA molecules from WT Tetrahymena cells, 6mA sites generally distributed in clusters separated by regular intervals (Fig. 6A). Autocorrelation analysis confirmed that 6mA sites were strongly phased at the single-molecule level, oscillating with cycles of ∼200 bp (Fig. 6B). Furthermore, 6mA clusters from different DNA molecules were often coarsely aligned to the same genomic region (Fig. 6A). Indeed, 6mA distribution was also phased at the ensemble level (Fig. 6C, top), just like nucleosome distribution in Tetrahymena (Xiong et al. 2016). Autocorrelation analysis showed that 6mA and nucleosome distributions in Tetrahymena shared the same cycle of ∼200 bp (Fig. 6C, top); cross-correlation analysis showed that 6mA and nucleosome distributions were offset by ∼100 bp and in opposite phases (Fig. 6C, bottom). We also found that 6mA peaks coincided with nucleosome troughs downstream from transcription start sites (Fig. 6A; Supplemental Fig. S10A). Therefore, 6mA is preferentially associated with linker DNA in Tetrahymena.
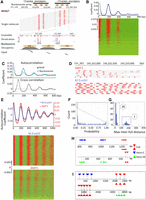
Chromatin-guided 6mA transmission. (A) 6mA and nucleosome distributions in Tetrahymena. A typical genomic region is shown with SMRT CCS reads mapped across it, as well as annotations of genes and canonical nucleosome arrays (Xiong et al. 2016). Note that 6mApT sites (in either full or hemi-methylation, red dot) distributed along individual DNA molecules (gray line) are clustered in linker DNA (LD). LD1 is between the +1 and +2 nucleosome (the first and second nucleosome downstream from TSS); LD2 and beyond are defined iteratively further downstream from the gene body. (B) Periodic 6mA distribution at the single-molecule level in Tetrahymena. Autocorrelation between 6mA sites was calculated for individual DNA molecules, ranked by their median absolute deviations, and plotted as a heat map (bottom) and an aggregated correlogram (top). (C) Autocorrelation of 6mA and nucleosome distributions at the ensemble level in Tetrahymena (top), revealing a ∼200 bp periodicity. Cross-correlation between 6mA and nucleosome distributions (bottom), revealing an ∼100 bp phase difference between them. (D) Typical DNA molecules from human chromatin, after in vitro methylation by AMT1 complex and M.EcoGII, respectively. Note clusters of 6mA sites (red dot) distributed at regular intervals along individual DNA molecules (gray line). The difference in 6mA density is mostly due to the much lower density of the ApT dinucleotide that is preferred by the AMT1 complex, relative to essentially all A sites that can be targeted by M.EcoGII. Additionally, the AMT1 complex may also have reduced chromatin accessibility relative to M.EcoGII, due to its much larger size. (E) Periodic 6mA distributions at the single-molecule level, after in vitro methylation by AMT1 complex and M.EcoGII, respectively. Autocorrelation between 6mA sites was calculated for individual DNA molecules, ranked by their median absolute deviations, and plotted as heat maps (bottom) and aggregated correlograms (top). DNA molecules with regularly spaced 6mA clusters were found across euchromatic and heterochromatic regions. Heterochromatin is known to have low nucleosome positioning, which means at the ensemble level, nucleosomes can occupy alternative genomic positions. However, at the single-molecule level, nucleosomes are still regularly spaced, which is only obvious in long-read, single-molecule sequencing results. (F) Congregation of full-6mApT in DNA molecules undergoing hemi-to-full conversion. Their max inter-full distances were often very small, thus rarely represented (probability ≤0.01) in simulations with permutated full and hemi positions (box); x-axis: the probability for simulated max interfull distances to be no greater than the observed value; y-axis: the count of DNA molecules with the corresponding probability. (G) Distribution of max interfull distances for DNA molecules with strong full-6mApT congregation (probability ≤0.01, Fig. 5F, box). Note the two peaks corresponding to DNA molecules with full-6mApT congregation within an LD (Fig. 5H) and across adjacent LDs (Fig. 5I), respectively. (H) Full-6mApT congregation within an LD. (I) Full-6mApT congregation across adjacent LDs.
We also analyzed human chromatin in vitro methylated by AMT1 complex or M.EcoGII (Fig. 6D,E; Supplemental Fig. S10B–D). We first digested the DNA samples with DpnI (Supplemental Fig. S10A,B), targeting GATC sites with 6mA (Vovis and Lacks 1977). Only a fraction of GATC sites were cleaved, generating a nucleosome ladder strongly suggestive of preferential DNA methylation at linker DNA (Supplemental Fig. S10B). SMRT CCS revealed regularly distributed 6mA clusters on individual DNA molecules from both AMT1 complex and M.EcoGII-treated samples (Fig. 6D; Supplemental Table S3). The difference in 6mA density is mostly due to the much lower density of the ApT dinucleotide that is preferred by AMT1, relative to essentially all A sites that can be targeted by M.EcoGII. Autocorrelation analysis confirmed that 6mA sites were strongly phased, with cycles ranging from 160 to 200 bp (Fig. 6E, bottom). DNA molecules with regularly spaced 6mA clusters were mapped across the entire genome, in euchromatic and heterochromatic regions. Although 6mA density was substantially lower in the sample treated by AMT1 complex (Fig. 6D), the aggregated 6mA distribution correlogram showed the same cycle of ∼190 bp for both samples (Fig. 6E, top), underpinned by the nucleosome distribution pattern in human chromatin.
In contrast to Tetrahymena MAC genomic DNA, 6mA clusters on different DNA molecules from in vitro methylated human chromatin were poorly aligned for most genomic regions (Fig. 6D). Indeed, 6mA distribution autocorrelation was much weaker for in vitro methylated human chromatin at the ensemble level (Supplemental Fig. S11A). In parallel, autocorrelation for nucleosome distribution at the ensemble level was much weaker in human than in Tetrahymena, indicating poor nucleosome positioning overall in human relative to Tetrahymena (Fig. 6C, top; Supplemental Fig. S11B). As an exception that proves the rule, we found that around genomic positions with strong CTCF-binding, which are usually flanked by well-positioned nucleosomes (Fu et al. 2008; Krietenstein et al. 2020), 6mA sites from in vitro methylated human chromatin were strongly aligned, and importantly, 6mA peaks coincided with nucleosome troughs (Supplemental Fig. S11C). We conclude that mutual exclusivity between 6mA and the nucleosome is generally applicable at the single-molecule level, but only manifests at the ensemble level for genomic regions with well-positioned nucleosomes. Our in vitro methylation results also indicate that preferential methylation of linker DNA is an intrinsic property for AMT1 complex, M.EcoGII, and potentially many other MTases.
Processivity of AMT1-dependent methylation
Canonical maintenance MTases (e.g., E. coli Dam DNA MTase) are generally processive rather than distributive (Urig et al. 2002). In other words, upon substrate binding, they tend to catalyze multiple local methylation events before dissociation. To investigate the processivity of AMT1-dependent methylation, we examined DNA molecules undergoing hemi-to-full conversion in WT Tetrahymena cells. We found that hemi-6mApT and full-6mApT distributions were often not random in these molecules (Fig. 6F–I). Many exhibited full-6mApT congregation: The maximum observed distance between adjacent full-6mApT sites (max inter-full distances) was much smaller than expected, and as a result rarely appeared in simulated controls, in which full-6mApT and hemi-6mApT sites were randomly permutated (Fig. 6F). There was a strong tendency for multiple maintenance methylation events to occur in nearby ApT dinucleotides. This tendency was especially prominent for DNA molecules early in the hemi-to-full conversion process, which were more likely to be methylated in a processive run by a single AMT1 complex (Supplemental Fig. S12A).
For DNA molecules with the strong full-6mApT congregation, their max interfull distances were predominantly distributed in two peaks (Fig. 6G): The left peak (max inter-full distances ≤30 bp) corresponds to full-6mApT congregation within the same linker DNA (Fig. 6H), whereas the right peak (130 bp ≤ max inter-full distances ≤ 200 bp) corresponds to congregation across adjacent linker DNA regions (Fig. 6I). Consistent with chromatin-guided 6mA transmission, in some DNA molecules, hemi-to-full conversion was already complete for one linker DNA (or at a higher level, gene), but not even started for its adjacent linker DNA region (or gene) (Fig. 6H,I). More often, full-6mApT were intermixed with hemi-6mApT in one linker DNA region (or gene), whereas its adjacent linker DNA region (or gene) contained only hemi-6mApT (Supplemental Fig. S12B). The processivity of AMT1-dependent maintenance methylation, therefore, manifests as episodes of hemi-to-full conversion events that occur within one linker DNA (or gene), punctuated by transferring of the MTase activity to its adjacent linker DNA (or gene).
AMT1-independent de novo methylation
6mA levels were reduced but not eliminated in ΔAMT1 cells (Table 1). Many ApT positions in the MAC genome were methylated in WT cells but not in ΔAMT1 cells (Supplemental Fig. S13A). For genomic positions methylated in both, methylation penetrance was generally much lower in ΔAMT1 cells (Supplemental Fig. S13B). High penetrance genomic positions were especially depleted in ΔAMT1 cells (Fig. 7A). Assuming exponential decay kinetics, we estimated the apparent half-life values for AMT1-dependent maintenance methylation (0.07× cell cycle) and AMT1-independent de novo methylation (17.6×) (Supplemental Fig. S14). The fast AMT1-dependent maintenance methylation allows effective restoration of full-6mApT within one cell cycle in WT cells, whereas the slow AMT1-independent de novo methylation entails that in ΔAMT1 cells, methylation plateau is only reached after multiple cell cycles. Indeed, in many DNA molecules from ΔAMT1 cells, 6mA counts on W and C were disparate (Figs. 2D, bottom and 7B; Supplemental Fig. S13C,D). The strand with significantly fewer 6mA than expected for random distribution probably corresponds to the daughter strand after DNA replication, which only carries 6mA newly deposited after the last S phase; the strand with significantly more 6mA probably corresponds to the parental strand, which has accumulated 6mA over multiple cell cycles. The difficulty to propagate 6mA across the cell cycle also led to epigenetic instability in ΔAMT1 cells, as different DNA molecules covering the same gene exhibited much higher variability of 6mA counts (Fig. 7C).
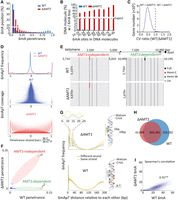
AMT1-independent de novo methylation. (A) Depletion of high penetrance 6mA positions in ΔAMT1 relative to WT cells. (B) Strong 6mA segregation strand biases in ΔAMT1 cells. Chi-squared analysis was performed on DNA molecules with the specified number of total 6mA (full-6mApT counted as
two, hemi-6mApT counted as one; x-axis), the percentage of DNA molecules with a strong bias for 6mA segregation to one strand was indicated (expectance <5%,
assuming random distribution; y-axis). WT cells were also analyzed as a negative control. (C) Increased 6mA variability at the gene level in ΔAMT1 relative to WT cells. For each gene, we calculated the coefficient of variance (CV) of 6mA counts from individual DNA molecules
fully covering the gene, for WT and ΔAMT1 cells, respectively. We then plotted the distribution of the ratio between the two CV values (WT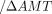 1) across all genes. Note that for most genes, the ratio is <1 (i.e., 6mA variability is higher in ΔAMT1 than WT cells). (D) Penetrance strand bias of 6mA in WT and ΔAMT1 cells. 6mA penetrance strand bias is defined for an ApT position in the genome as the difference-sum ratio between the number
of DNA molecules supporting 6mA on W and C, respectively: [(W − C)/(W + C)]p. We plotted the distribution of ApT genomic positions according to their penetrance strand bias (top). We also plotted their distribution according to both penetrance strand bias and 6mApT coverage (middle: WT; bottom: ΔAMT1). In WT cells, most ApT positions had penetrance strand bias values around 0 (i.e., similar numbers of 6mA on W and C), whereas
few had values at +1 (6mA only on W) or −1 (6mA only on C). The latter most likely corresponds to genomic positions exclusive
for AMT1-independent methylation (Fig. 6F, left panel). The opposite was true for ΔAMT1 cells. (E) Representative genomic positions in Tetrahymena rDNA (top schematic: only the left half of the palindromic dimer, from telomere to dyad, is shown) targeted by AMT1-independent (left) and AMT1-dependent methylation (right). Note that 6mA occurs only on one strand in AMT1-independent methylation, but on both strands in AMT1-dependent methylation.
(F) 6mA penetrance of individual genomic positions in WT and ΔAMT1 cells. Note the two distinct groups corresponding to (1) AMT1-independent and (2) AMT1-dependent methylation. (G) 10 bp cycle of 6mA penetrance strand bias in ΔAMT1 cells (top left), suggesting that the dedicated de novo 6mA-MTase can only approach the DNA substrate from one side (top right). Lack of such a pattern in WT cells (bottom left) supports that the AMT1 complex can approach from different sides (bottom right). (H) Overlap in ApT positions methylated in WT or ΔAMT1 cells (6mA penetrance ≥0.1). (I) 6mA levels of individual genes in WT and ΔAMT1 cells are strongly correlated. Each gene is assigned a coordinate: the sum of 6mA penetrance values for all methylated ApT
positions in the gene body (ΣP) for WT (x-axis) and ΔAMT1 cells (y-axis). The Spearman's rank correlation coefficient is significant: (**) P < 0.01.
1) across all genes. Note that for most genes, the ratio is <1 (i.e., 6mA variability is higher in ΔAMT1 than WT cells). (D) Penetrance strand bias of 6mA in WT and ΔAMT1 cells. 6mA penetrance strand bias is defined for an ApT position in the genome as the difference-sum ratio between the number
of DNA molecules supporting 6mA on W and C, respectively: [(W − C)/(W + C)]p. We plotted the distribution of ApT genomic positions according to their penetrance strand bias (top). We also plotted their distribution according to both penetrance strand bias and 6mApT coverage (middle: WT; bottom: ΔAMT1). In WT cells, most ApT positions had penetrance strand bias values around 0 (i.e., similar numbers of 6mA on W and C), whereas
few had values at +1 (6mA only on W) or −1 (6mA only on C). The latter most likely corresponds to genomic positions exclusive
for AMT1-independent methylation (Fig. 6F, left panel). The opposite was true for ΔAMT1 cells. (E) Representative genomic positions in Tetrahymena rDNA (top schematic: only the left half of the palindromic dimer, from telomere to dyad, is shown) targeted by AMT1-independent (left) and AMT1-dependent methylation (right). Note that 6mA occurs only on one strand in AMT1-independent methylation, but on both strands in AMT1-dependent methylation.
(F) 6mA penetrance of individual genomic positions in WT and ΔAMT1 cells. Note the two distinct groups corresponding to (1) AMT1-independent and (2) AMT1-dependent methylation. (G) 10 bp cycle of 6mA penetrance strand bias in ΔAMT1 cells (top left), suggesting that the dedicated de novo 6mA-MTase can only approach the DNA substrate from one side (top right). Lack of such a pattern in WT cells (bottom left) supports that the AMT1 complex can approach from different sides (bottom right). (H) Overlap in ApT positions methylated in WT or ΔAMT1 cells (6mA penetrance ≥0.1). (I) 6mA levels of individual genes in WT and ΔAMT1 cells are strongly correlated. Each gene is assigned a coordinate: the sum of 6mA penetrance values for all methylated ApT
positions in the gene body (ΣP) for WT (x-axis) and ΔAMT1 cells (y-axis). The Spearman's rank correlation coefficient is significant: (**) P < 0.01.
In ΔAMT1 cells, 6mA was also enriched in linker DNA and toward the 5′ end of Pol II-transcribed genes (Supplemental Fig. S15A,B). 6mApT sites, though present more sparsely, still formed clusters at regular intervals on individual DNA molecules (Fig. 2D, bottom; Supplemental Fig. S15B). Autocorrelation analysis at both the single-molecule level and the ensemble level showed a slight right shift in 6mA peaks, supporting increased linker DNA length (Supplemental Fig. S15C–E). We found many genomic regions that were more variably covered with 6mA in ΔAMT1 than WT cells (Supplemental Fig. S15B). This may reflect reduced nucleosome positioning or increased nucleosome dynamics. In support, 6mA can directly promote nucleosome positioning, as the heavily methylated DNA becomes less bendable and thus prefers to be linker DNA rather than nucleosomal DNA (Fu et al. 2015; Luo et al. 2018; Beh et al. 2019). Nucleosome positioning was indeed weakened in ΔAMT1 relative to WT cells (Supplemental Fig. S15E; Wang et al. 2019). Alternatively, 6mA dispersion in ΔAMT1 cells may be attributed to the slow AMT1-independent de novo methylation, which records nucleosome movement over a much longer period rather than only briefly after DNA replication.
In strong contrast to WT cells, 6mA penetrance for most ApT positions in the MAC genome of ΔAMT1 cells showed strong biases for either W or C, and many were exclusively methylated on one strand (Figs. 2D, bottom and 7D, top); this tendency grew in prominence with increasing sequencing coverage (Fig. 7D, bottom), thus unlikely an artifact of random fluctuations. We also noted a very small minority of ApT positions containing predominantly, if not exclusively, hemi-methylation calls in WT Tetrahymena cells (Supplemental Fig. S6A: hemi-only part of the Venn diagram, 0.36%). These positions, characterized by a strong penetrance strand bias, were most likely methylated by an AMT1-independent de novo methylation pathway (Fig. 7E, left; F, Group 1). In the high copy number Tetrahymena rDNA, we found genomic positions that were exclusively targeted by AMT1-independent de novo methylation (Fig. 7E, left; F, Group 1), as well as those that were AMT1-dependent (Fig. 7E, right; F, Group 2). Genomic positions with strong penetrance bias for W or C exhibited periodic distributions with an ∼10 bp cycle (Fig. 7G, top). This matches the pitch of the DNA double helix, suggesting that the dedicated de novo MTase is constrained to approach the DNA substrate from only one side (Fig. 7G, top right). The strong penetrance bias also precludes this MTase from playing a major role in maintenance methylation.
Despite these distinctions, there were also connections between AMT1-dependent and AMT1-independent methylation. Most ApT positions methylated in ΔAMT1 cells were also methylated in WT cells; the two sets essentially converged at high methylation penetrance (Fig. 7H; Supplemental Fig. S15A). Furthermore, 6mA levels at individual genes and even individual linker DNA regions of a gene showed strong correlations between WT and ΔAMT1 cells (Fig. 7I; Supplemental Fig. S15E). These connections suggest an integrated 6mA transmission pathway: AMT1-independent de novo methylation primes the system by laying down an incipient 6mApT distribution pattern, which is fulfilled and transmitted by AMT1-dependent maintenance methylation.
Discussion
6mA detection by SMRT CCS
In the free 6mA nucleotide, the N6-methyl group minimizes the steric clash by pointing toward the Watson–Crick edge of the purine ring (Bochtler and Fernandes 2021). This is likely also the preferred conformation in single-stranded DNA. However, in double-stranded DNA, the N6-methyl group must adopt the energetically less favorable conformation and point the other way, to allow the N6-lone pair electrons to engage in Watson–Crick base-pairing. This entails a pause in DNA synthesis, as the DNA polymerase waits for 6mA in the template strand to switch conformation. In SMRT sequencing, this is recorded as increased IPD. SMRT CCS allows robust evaluation of IPD at the single site and single-molecule level, as multiple passes by the DNA polymerase overcome random fluctuations. We rationalize that 6mA and unmodified A feature distinct IPDr distributions, which can be deconvoluted effectively. Based on these basic assumptions, we have developed a bioinformatic pipeline to fully exploit the recent progress in SMRT CCS for strand-specific, accurate, and sensitive detection of 6mA on individual DNA molecules multi-kb in length. The result is the first high-quality, single-molecule, genome-wide mapping of endogenous 6mA in eukaryotes.
6mA detection by SMRT CCS is critical for our study in the following aspects. First, SMRT CCS detects 6mA with high accuracy (low false positive rates), which allows us to (1) determine the ApT specificity for AMT1-dependent maintenance methylation and AMT1-independent de novo methylation and (2) distinguish hemi-6mApT from full-6mApT in WT and ΔAMT1 cells. Second, SMRT CCS preserves long-range connectivity information at the single-molecule level, which allows us to (1) identify hemi+/BrdU+ molecules and establish segregation of hemi-6mApT to the parental strand after DNA replication and (2) identify DNA molecules undergoing maintenance methylation and characterize AMT1 processivity. Third, SMRT CCS detects 6mA with high sensitivity (low false negative rates), which, combined with deep sequencing coverage of the Tetrahymena MAC genome, allows us to (1) unambiguously identify rare methylation events and (2) generate absolute and exact quantification of 6mA levels over a genomic region. Furthermore, there is a gross discrepancy between CLR- and CCS-based assessments of many key 6mA parameters in Tetrahymena cells, including the percentage of 6mA in non-ApT context, the percentage of hemi- and full-6mApT, and the percentage of ApT positions with 6mA penetrance bias. In many cases, the misleading CLR results are likely rooted in heterogeneity of 6mA at the single-molecule level, which cannot be readily reduced to a single-value representation at the ensemble level. Lastly, although it has long been implicated and assumed that 6mA is transmitted by a semiconservative mechanism in eukaryotes, our novel application of SMRT CCS in this work represents the first systematic and rigorous proof.
As a gold standard for 6mA detection, SMRT CCS boasts some outstanding features: (1) low background noise, (2) high accuracy, (3) high sensitivity, and (4) long-read length. It is worth noting that these parameters are mutually connected and can be individually optimized according to one's needs. At the cost of CCS read length/DNA insert size (and consequently, sequencing coverage), we chose to increase the number of CCS passes to improve the first three parameters. Shifting the IPDr threshold for 6mA calling affects the accuracy and sensitivity of 6mA detection in the opposite direction. Our deconvolution-based approach automatically sets the threshold to achieve a balanced outcome. SMRT CCS can be used to detect other base modifications, such as BrdU. Although we have limited ourselves to a single readout of the DNA polymerase kinetics (IPD) and a rationally designed algorithm (independent of ground truth training data), there is great potential in incorporating additional readout and implementing neural network-based machine learning algorithms (Tse et al. 2021).
6mA is highly enriched in linker DNA in Tetrahymena. The resulting 6mA clusters, regularly spaced, demarcate individual nucleosomes on a chromatin fiber, providing long-range epigenetic information generally missing from short-read sequencing data. Specific methylation of linker DNA is likely an intrinsic feature of AMT1 complex, M.EcoGII, and many other 6mA-MTases. This property can be exploited to probe chromatin organization via in vitro methylation, analogous to nuclease protection (Abdulhay et al. 2020; Shipony et al. 2020; Stergachis et al. 2020; Altemose et al. 2022). This is an especially powerful approach when combined with 6mA detection by SMRT CCS (Abdulhay et al. 2020; Stergachis et al. 2020).
AMT1-dependent methylation
We have extensively characterized AMT1-dependent 6mA transmission. Our in vivo results demonstrate high specificity for maintenance methylation at the ApT dinucleotide, whereas our in vitro results support substantial de novo methylation activity at ApT sites and, to a lesser degree, non-ApT sites. Note that 6mA at non-ApT sites is necessarily the product of de novo methylation. We emphasize that de novo methylation underpins the biochemical assay by which the Tetrahymena MTase activity and eventually AMT1 complex were identified (Bromberg et al. 1982; Beh et al. 2019). Indeed, DNMT1, the eukaryotic maintenance MTase required for semiconservative transmission of 5-methylcytosine (5mC) in the CpG dinucleotide, also has de novo methylation activity (Bestor 2000; Jeltsch 2006). We argue that AMT1-dependent de novo methylation is amplified under in vitro conditions, while curtailed by various in vivo circumstances. (1) For in vitro methylation of human chromatin, de novo methylation precedes—and is the prerequisite for—maintenance methylation. In contrast, the abundance of hemi-6mApT in Tetrahymena MAC DNA immediately after DNA replication allows maintenance methylation to effectively outcompete de novo methylation in vivo. (2) Processivity of AMT1-dependent methylation may enhance the preference for maintenance methylation in vivo. Multiple hemi-6mApT sites, present in a cluster often fully covering a linker DNA, are readily converted to full-6mApT with little chance of de novo methylation as the side reaction. (3) AMT1-dependent maintenance methylation may be further enhanced by other in vivo factors. In Tetrahymena, 6mA is highly enriched in linker DNA flanked by nucleosomes containing H3K4me3 and H2A.Z (Wang et al. 2017, 2019), which may interact with the AMT1 complex and modulate its substrate specificity.
Comparing 6mA and 5mC pathways in eukaryotes
Our work provides definitive evidence for a eukaryotic 6mA pathway comprising two distinct but linked steps: AMT1-independent de novo methylation and AMT1-dependent maintenance methylation (Fig. 8). Although AMT1-independent de novo methylation is dispensable for maintaining the 6mA pattern in the MAC of asexually propagating Tetrahymena cells (Wang et al. 2019), it is likely to play a critical role during sexual reproduction, as the transcriptionally silent and 6mA-free germline MIC is differentiated into the transcriptionally active and 6mA-rich somatic MAC. This two-step pathway bears resemblance to the eukaryotic 5mC pathway, featuring the DNMT3A/3B-dependent de novo methylation and DNMT1-dependent maintenance methylation for transmission of 5mC at the CpG dinucleotide (Fig. 8; Goll and Bestor 2005; Lister et al. 2009; Sen et al. 2021). As bona fide eukaryotic epigenetic marks, 6mA and 5mC play opposite roles in transcription regulation (Fig. 8). Their transmission pathways are deep-rooted and widespread, but show distinct phylogenetic distributions, with homologs of AMT1 complex components notably missing from land plants, higher fungi, and animals (Supplemental Fig. S16). 6mA and 5mC, therefore, represent a pair of critical switches that can alter the global epigenetic landscape for transcription regulation, and their presence or loss may drive some major branching events in eukaryotic evolution.
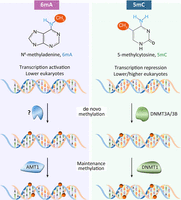
Comparison of 6mA and 5mC pathways in eukaryotes. See text for details.
Methods
Tetrahymena strains
Tetrahymena thermophila WT strain (SB210) was obtained from the Tetrahymena Stock Center. ΔAMT1 was a homozygous homokaryon (MAC and MIC) knockout strain generated in our previous study (Wang et al. 2019). The knockout construct is available at Addgene (https://www.addgene.org/218373/).
In vitro and in vivo BrdU labeling
For in vitro BrdU labeling, a PvuI-digested fragment of the pBluescript II SK(−) plasmid (∼1 kb) was used as the template for primer extension and PCR. For
PCR: Taq DNA polymerase, primers (PSK-Fwd: 5′-CGTTGTCAGAAGTAAGTTGGCCGC-3′; PSK-Rev: 5′-CGCCCTTCCCAACAGTTGCGC-3′), and dNTP
mix 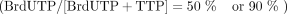 . For primer extension: Taq DNA polymerase, either PSK-Fwd or PSK-Rev, and dNTP mix
. For primer extension: Taq DNA polymerase, either PSK-Fwd or PSK-Rev, and dNTP mix 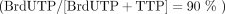 . SMRT sequencing was performed on five samples: unlabeled plasmid, PSK-Fwd primer extension, PSK-Rev primer extension, 50%
BrdUTP PCR, and 90% BrdUTP PCR.s
. SMRT sequencing was performed on five samples: unlabeled plasmid, PSK-Fwd primer extension, PSK-Rev primer extension, 50%
BrdUTP PCR, and 90% BrdUTP PCR.s
For in vivo BrdU labeling, Tetrahymena cells were synchronized at the G1 phase by centrifugal elutriation (Liu et al. 2021b), released into the fresh medium with 0.4 mM BrdU, and collected after 0 h, 1.5 h, 2 h, or 4 h for genomic DNA extraction and SMRT sequencing.
DNA methyltransferase assay of reconstituted AMT1 complex
Synthesized 12-mer DNA oligos containing one central 6mA-modified ApT site were annealed to generate the substrate (upper strand: 5′-GCAAG (6mA) TCA ACG-3′, lower stand: 5′-CGTTGATCT TGC-3′). For the steady-state kinetic assay, a 20 μL reaction mixture contained the hemi-methylated substrate at various concentrations (0, 0.04, 0.1, 0.16, 0.24, 0.36, 0.5, 0.75, 1, 1.5, 2 μM), 0.01 μM AMT1 complex, 0.55 μM S-adenosyl-l-[methyl-3H] methionine (specific activity 18Ci/mmol, PerkinElmer), 1.9 μM AdoMet in 50 mM Tris-HCl, pH 8.0, 0.05% β-mercaptoethanol, 5% glycerol, and 200 μg/mL BSA. For substrate specificity assay, a 15 μL reaction mixture contained 2 μM unmodified or hemi-methylated substrates, 0.1 μM AMT1 complex, 3 μM S-adenosyl-l-[methyl-3H] methionine (specific activity 18Ci/mmol, PerkinElmer). For unmodified DNA duplex, upper strand: 5′-AACTTCTGTCATTACATTAAGCTTTAA-3′, lower stand: 5′-TTAAAGCTTAATGTAATGACAGAAGTT-3′. For hemi-methylated DNA duplex, upper strand: 5′-AACTTCTGTC (6mA) TTAC (6mA) TTAAGCTTTAA-3′, lower stand: 5′-TTAAAGCTTAATGTAATGACAGAAGTT-3′. The assays were performed in triplicate at room temperature for 30 min.
In vitro methylation of human chromatin
1.7 × 105 OCI-AML3 cells were lysed in 0.5 mL of nuclei extraction buffer (20 mM HEPES pH 7.9, 10 mM KCl, 0.1% Triton X-100, 20% glycerol, 0.5 mM spermidine, 1× Protease Inhibitor Cocktail) for 8 min on ice. Purified nuclei were methylated in a 30 μL of 50 mM Tris-HCl pH 8.0, 2 mM EDTA, 0.5 mM EDGA, 160 μM SAM, 1× Protease Inhibitor Cocktail, and 38 μM AMT1 complex or M.EcoGII or no enzyme control for 1 h at 37°C. Genomic DNA was extracted with Monarch HMW DNA Extraction Kit for Cells & Blood (New England Biolabs), digested with DpnI overnight at 37°C, and resolved on 1% agarose gel. DNA fragments 3–5 kb in length were gel purified for SMRT sequencing.
SMRT CCS detection of 6mA
Native genomic DNA was extracted from Tetrahymena WT (SB210, with or without BrdU labeling) and ΔAMT1 cells using Wizard Genomic DNA Purification Kit (Promega). REPLI-g Single Cell Kit (Qiagen) was used for WGA (negative control). DNA samples were sheared to 3–5 kb in length with Megaruptor (Diagenode Diagnostics) and prepared for sequencing by the Sequel II System.
Single-molecule SAM files were extracted from the SMRT sequencing data using a custom Perl script and transformed into single-molecule BAM files by SAMtools (Li et al. 2009). CCS was calculated for each DNA molecule using the CCS module (SMRT Link v10.2, Pacific Biosciences). Only DNA molecules with high subread coverage (≥30×) were retained. Single-molecule-aligned BAM files were generated using BLASR (Chaisson and Tesler 2012), which in turn served as the input for the ipdSummary module to calculate IPDr. Self-referencing not only allows 6mA calling at the single-molecule level, but also greatly speeds up computation. We aligned CCS reads to the latest Tetrahymena genome references for the MAC (Sheng et al. 2020), MIC (Supplemental File_Tet MIC, updating the published MIC reference [Hamilton et al. 2016]), and mitochondrion (Brunk et al. 2003). Additional details are available in Supplemental Methods “SMRT CCS data analysis,” “Mapping CCS reads back to genome references,” and “CCS versus CLR.”
SMRT CCS detection of BrdU
For analyzing in vitro BrdU-labeled DNA, SMRT CCS reads with high passes (≥60×) were aligned to the reference sequence using BLASTN, and only molecules satisfying a strict criterion of 0 mismatches, 0 gaps, and an alignment length exceeding 1000 bp were analyzed. Furthermore, strand-specific IPDr standard deviations (SDs) of guanine sites (presumably all unmodified) were calculated and molecules with SD ≤ 0.35 for both strands were retained. The threshold for calling BrdU was set at an IPDr value of 2.5 or 2.8. BrdU+ molecules were defined as DNA molecules with no less than eight BrdU sites on one strand (W||C ≥ 8).
For analyzing in vivo BrdU-labeled Tetrahymena genomic DNA, regions adjacent to 6mApT sites (both strands: −10 to +10 bp) were masked from BrdU calling to avoid interference between 6mA and BrdU. IPDr 2.8 was set as the threshold for calling BrdU. Note that BrdU+ molecules, defined as DNA molecules with no less than 11 BrdU sites in total (W + C ≥ 11) or on one strand (W||C ≥ 11), represent a small fraction of BrdU-labeled DNA molecules (only ∼10% of SMRT CCS reads were BrdU+ in G2 cells, in which nearly all DNA should be labeled by BrdU), due to the high false negative rate of BrdU calls and the high threshold for BrdU+ molecules.
Penetrance strand bias and segregation strand bias
6mA penetrance strand bias, 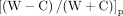 , is defined for an ApT position in the genome as the difference-sum ratio between the number of DNA molecules supporting
6mA on W and C, respectively (W + C ≥ 10). The values range between −1 (6mA only on C) and 1 (6mA only on W). 6mA penetrance
strand bias was calculated for both WT and ΔAMT1 cells. We identified ApT positions with penetrance strand bias of +1 or −1 in ΔAMT1 cells and generated phasogram (defined as histogram of distances between specified positions) to reveal their periodic distribution
relative to each other.
, is defined for an ApT position in the genome as the difference-sum ratio between the number of DNA molecules supporting
6mA on W and C, respectively (W + C ≥ 10). The values range between −1 (6mA only on C) and 1 (6mA only on W). 6mA penetrance
strand bias was calculated for both WT and ΔAMT1 cells. We identified ApT positions with penetrance strand bias of +1 or −1 in ΔAMT1 cells and generated phasogram (defined as histogram of distances between specified positions) to reveal their periodic distribution
relative to each other.
6mA segregation strand bias, 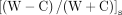 , is defined for a hemi+ molecule (W + C ≥ 11 or W||C ≥ 11) as the difference-sum ratio between the count of hemi-6mApT on W and C, respectively.
The values range between −1 (only hemi-C) and 1 (only hemi-W). Note that 6mA in full-6mApT is not included in this calculation.
Segregation strand bias is also calculated for BrdU sites in BrdU+ molecules (W + C ≥ 15 or W||C ≥ 15).
, is defined for a hemi+ molecule (W + C ≥ 11 or W||C ≥ 11) as the difference-sum ratio between the count of hemi-6mApT on W and C, respectively.
The values range between −1 (only hemi-C) and 1 (only hemi-W). Note that 6mA in full-6mApT is not included in this calculation.
Segregation strand bias is also calculated for BrdU sites in BrdU+ molecules (W + C ≥ 15 or W||C ≥ 15).
Autocorrelation and cross-correlation analysis
A vector consisting of a series of 0's (no 6mA) and 1's (6mA at the position on either strand) was encoded for each DNA molecule with 6mA sites (≥2). This vector was the input for the acf function in the statsmodel Python package (Seabold and Perktold 2010) for computing 6mA autocorrelation coefficients at the single-molecule level. For correlation analysis at the ensemble level, the MAC reference genome was divided into 5 kb regions. We then used the BEDTools coverage subcommand (Quinlan and Hall 2010) to count 6mA or nucleosome dyad across all genomic positions, generating two encoding vectors for each such genomic region (focusing on genomic positions with 6mApT coverage ≥2; 6mApT genomic positions supported by only one 6mA call are excluded to reduce background noise). This pair of vectors were the input for the acf function for computing the autocorrelation coefficients of 6mA and nucleosome distributions, respectively; they were also the input for the ccf function for computing the cross-correlation coefficients between 6mA and nucleosome distributions.
Full-6mApT congregation
For each DNA molecule undergoing hemi-to-full conversion (full-6mApT ≥ 3, hemi-6mApT ≥ 9), we first calculated the observed maximum value of distances between adjacent full-6mApT duplexes (Dobs). We then calculated the equivalent values for 1000 simulations, in which the full-6mApT and hemi-6mApT positions in the same DNA molecule were randomly permutated (Dsim). This allowed us to estimate the probability for the observed full-6mApT congregation (Dsim ≤ Dobs), assuming that maintenance methylation is random.
Data access
The SMRT CCS data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA932808. All custom scripts and code are available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Tetrahymena WT strains were obtained from the Tetrahymena Stock Center (https://tetrahymena.vet.cornell.edu). SMRT sequencing was performed at the Genomics Core Facility, Icahn School of Medicine at Mount Sinai, and Novogene Co., Ltd. (Beijing, China). High-performance computing resources for data processing were provided by the Institute of Evolution and Marine Biodiversity at the Ocean University of China, the Center for High Performance Computing and System Simulation at Laoshan Laboratory, the Marine Big Data Center of the Institute for Advanced Ocean Study at OUC, and the Center for Advanced Research Computing (CARC) at the University of Southern California. This study was supported by the National Natural Science Foundation of China 32125006, 32070437 (S.G.), the National Natural Science Foundation of China 32200336 (Y.S.), the National Natural Science Foundation of China 32200399 (Y.W.), and the Department of Biochemistry and Molecular Medicine at the University of Southern California (Yi.L.).
Author contributions: Yi.L. and S.G. conceived the project, supervised most of the experiments and data analyses, and prepared the manuscript; Y.S. and W.Y. analyzed SMRT CCS data; Y.W. set up an in vitro and in vivo BrdU labeling system and performed the Tetrahymena experiments; J.L. (supervised by J.S.) and B.N. (supervised by S.G.) reconstituted and characterized the AMT1 complex, which was used by X.Q.W. (supervised by Y.D.) to perform in vitro methylation of the human chromatin; Yo.L. provided the synchronized Tetrahymena cells; F.Y. calculated the conversion rate of WT and ΔAMT1 cells; B.P. analyzed Illumina sequencing data and phylogenetic distribution of DNA MTases; C.L. performed statistical analyses.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277843.123.
- Received March 3, 2023.
- Accepted April 15, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








