
A universal framework for detecting cis-regulatory diversity in DNA regions
Abstract
High-throughput sequencing-based assays measure different biochemical activities pertaining to gene regulation, genome-wide. These activities include transcription factor (TF)–DNA binding, enhancer activity, open chromatin, and more. A major goal is to understand underlying sequence components, or motifs, that can explain the measured activity. It is usually not one motif but a combination of motifs bound by cooperatively acting proteins that confers activity to such regions. Furthermore, regions can be diverse, governed by different combinations of TFs/motifs. Current approaches do not take into account this issue of combinatorial diversity. We present a new statistical framework, cisDIVERSITY, which models regions as diverse modules characterized by combinations of motifs while simultaneously learning the motifs themselves. Because cisDIVERSITY does not rely on knowledge of motifs, modules, cell type, or organism, it is general enough to be applied to regions reported by most high-throughput assays. For example, in enhancer predictions resulting from different assays—GRO-cap, STARR-seq, and those measuring chromatin structure—cisDIVERSITY discovers distinct modules and combinations of TF binding sites, some specific to the assay. From protein–DNA binding data, cisDIVERSITY identifies potential cofactors of the profiled TF, whereas from ATAC-seq data, it identifies tissue-specific regulatory modules. Finally, analysis of single-cell ATAC-seq data suggests that regions open in one cell-state encode information about future states, with certain modules staying open and others closing down in the next time point.
High-throughput sequencing technologies are routinely used to map multiple types of biochemical activities occurring across the genome. Examples include protein–DNA binding events (Johnson et al. 2007; Vogel et al. 2007), open chromatin regions (Giresi et al. 2007; Boyle et al. 2008; Buenrostro et al. 2013), interacting chromatin domains (Fullwood et al. 2009; Lieberman-Aiden et al. 2009), active transcription start sites (TSSs) (Shiraki et al. 2003), and many more (Davis et al. 2018). A major goal of these efforts is to understand what part of the underlying sequence might be driving that particular activity. Now, although the measured activity might be of a specific nature, the same sequence signature may not be responsible for it at all locations. Consider, for example, an assay such as transposase-accessible chromatin with sequencing (ATAC-seq) or DNase I hypersensitive sites sequencing (DNase-seq), which identifies open chromatin regions. The reason behind the accessibility of a region may be one of several: It may be an active promoter, an enhancer, an insulator, or even a matrix-attachment region. Naturally, then, the pertinent sequence components in those regions will also be different. In some cases, the heterogeneity is less obvious, but present, all the same. For instance, although the primary objective of a high-throughput chromatin immunoprecipitation assay (ChIP-seq) is to identify regions bound by a specific transcription factor (TF), in reality the experiment reports a miscellaneous set of genomic regions: those making direct contact with the TF, those indirectly bound to the TF via an intermediate, those where the TF binds along with a cofactor, and perhaps, those that are simply proximal to the TF in 3D space (Farnham 2009).
Although the existence of such an assortment of regions is well accepted in most high-throughput experiments, methods used to learn the regulatory architecture at these regions do not effectively account for it. Reported regions are generally analyzed by identifying individual overrepresented motifs by sequentially searching for motifs one after the other, either from a known database or de novo. This strategy can fail in certain situations. Motifs may be missed because they are present only in a small set of regions and therefore are not statistically overrepresented in the entire set. A few recent methods do account for this by posing this as a mixture problem with each component of the mixture being enriched with a potentially different motif (Eggeling et al. 2017; Agrawal et al. 2018; Mitra et al. 2018). But these approaches do not take into account combinations of motifs, which may be critical to drive the biochemical activity at the region. Additionally, there may be multiple distinct motif combinations across the reported regions, with each combination explaining a fraction of the regions.
Here we propose a new method called cisDIVERSITY, which attempts to explain the whole set of the reported regions in terms of motifs and their combinations, all computed de novo. We take inspiration from topic modeling in computer science, in which the goal is to cluster documents (here DNA regions) into different topics (here functions/modules) based on word frequencies (here DNA motifs). In contrast to typical topic modeling in which the words are established and parsed a priori, here the motifs are also unknown and learned along with the modules.
Results
A sequence model for representing cis-regulatory diversity
A high-throughput sequencing experiment typically identifies a set X of genomic regions that are enriched with a specific biochemical activity. We assume that r different regulatory modules may be responsible for the activity and that each module is defined by the presence or absence of m motifs. More specifically, each module is modeled as a product of m Bernoulli distributions corresponding to the presence or absence of each motif. Different modules will have different Bernoulli distributions over the same motifs. A motif is modeled with the standard position weight matrix (PWM) (Staden 1984). Figure 1A shows an instance of a simulated data set of 1000 sequences, in which five motifs were planted from the JASPAR database (Khan et al. 2018), with specific Bernoulli distributions across three modules. For example, motif 1 is present in all sequences of module 2, in a fifth of sequences of module 1, and never in module 3. On the other hand, motif 5 is present in all sequences of module 3, but because the module consists of only about 40 sequences, overall, motif 5 occurs less frequently in the data. The aim of cisDIVERSITY is to learn the m motif parameters, the r × m Bernoulli distributions, and the sequences that belong to each of the r modules.
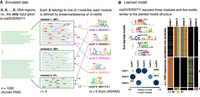
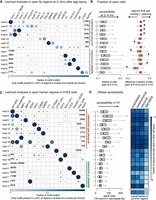
cisDIVERSITY. (A) DNA regions reported by the experiment are given as input to cisDIVERSITY. In this simulation, the n = 1000 regions are a mixture of three kinds of regions: Each region resembles one of r = 3 regulatory modules. Each module can be represented in terms of the probability of occurrence of m = 5 motifs. For example, motif 1 is present in all sequences of module 2, 20% of sequences in module 1, but not at all in module 3. In contrast, motif 4 is present only in module 2 and that, too, only in 70% of the its sequences. (B) cisDIVERSITY is run with upper bounds of r ≤ 10 and m ≤ 20. cisDIVERSITY learns the planted structure in the data set. The output has three components. First is the set of motifs that are learned, second (below) is r × m Bernoulli distributions describing the learned modules, and the third is an image matrix of the data, where each DNA sequence is a row and the sites corresponding to each motif are represented in the column. If a site is absent, those cells in the column are shown in black. cisDIVERSITY recovers the five motifs (motifs 1 and 3 are the reverse complements of the planted motifs) and the three modules to a great extent. The slight variability in the number of sites and sequences in each module is expected owing to the stochastic nature of both, the PWMs as well as the learning algorithm.
Gibbs sampling is used to iteratively sample each of these unknown values, with the aim of finding the set that maximizes the posterior distribution (Methods). cisDIVERSITY reports the output in three parts (Fig. 1B). The first is the set of de novo motifs ordered according to the number of sites that contribute to each motif. The second is the overall structure of the modules describing the contribution of each motif in every module. The color and the size of the circles denote the proportion of sites of the corresponding motif in each module. The last part of the output is the sequences clustered together as per the identified modules, displaying the sites that contribute to the PWMs in four colors for the four nucleotides. If a site is absent, those nucleotides are shown in black. The modules are ordered according to their size, the largest one shown on top. cisDIVERSITY finds all the motifs and recovers the general module structure in this case.
Performance on simulated data sets
cisDIVERSITY can be thought of as a joint clustering and de novo motif discovery method. To systematically assess how well cisDIVERSITY is able to retrieve modules and motifs, we simulated more such data sets. To better emulate reality, we used random nonrepetitive regions from the human genome as the data set X. The number of planted motifs m was from the set {5, 10}, and for each m, the number of modules was varied between the set {1, 2, 3, 5}. Now the performance of any clustering approach will depend on how separable the clusters are, whereas that of a motif discovery method will depend on how informative the motifs are. For the latter, we simply use real motifs from the JASPAR database (Methods). The former is decided by the Bernoulli parameters (probability of presence of a motif) in each regulatory module. The more extreme (close to zero or one) this probability, the more informative the motif is in describing the module. A value closer to 0.5 for all r × m Bernoulli parameters will cause the modules to be less separable from each other. This variation was included in the simulated data sets by sampling the Bernoulli parameters from a beta distribution with a symmetric hyperparameter β taken from the set {0.01, 0.1, 1, 10}. A value much smaller than one results in extreme values of Bernoulli probabilities, a value of one is akin to uniform sampling between zero and one, whereas a value of 10 results in probabilities closer to 0.5 and hence more “confused” modules. Ten sets were generated for each combination of parameters (m, r, β), resulting in 320 simulated sets, and cisDIVERSITY was run on each. Module recovery was measured by the adjusted Rand index (ARI), popularly used to compare the similarity of two clusterings of the same data. Two identical clusterings get an ARI value of one, whereas two random clusterings get a value of zero. Predictably, the ability of cisDIVERSITY to identify the module structure goes down as the beta hyperparameter goes up (Fig. 2A). It is reassuring to see the modules are picked up, although the m and r used in model learning (20 and 10, respectively) are larger than the true number of planted motifs and modules.

Performance on 320 simulated data sets. (A) Recovery of modules. Low values of beta result into modules with more extreme (zero or one) probability distributions of motifs. This is where cisDIVERSITY does better in recovering the planted modules. For beta = 10, the performance with respect to recovery of modules is similar to what a random clustering approach would do. (B) Recovery of motifs. Precision, recall, and F-score of recovered motifs across the 320 data sets for three different programs. (C) Time taken. All programs were run on a single core (Methods).
Although plenty of methods solve the problem of de novo motif discovery, we are not aware of any other method that solves this problem of simultaneously identifying multiple modules. However, to evaluate how cisDIVERSITY compares with state-of-the-art de novo motif discovery methods, we ran two commonly used programs—MEME (Bailey and Elkan 1994) and HOMER (Heinz et al. 2010)—using default parameters on these data sets. The recovery of a planted motif was evaluated using TOMTOM (Gupta et al. 2007), also from the MEME suite (Methods). Figure 2B shows that cisDIVERSITY performs better in terms of both precision and recall. Overall, of the total 2400 planted motifs across the 320 sets, cisDIVERSITY missed ∼12.5%, MEME missed 15.2%, and HOMER missed 15.1%. The difference is more stark in the specificity: Both MEME and HOMER falsely identified 23.5% and 27.8% additional incorrect motifs compared with the 4.5% additional motifs identified by cisDIVERSITY. We admit that this is not a fair comparison, because the data contain modules and no motif discovery method accounts for this fact. That said, when diverse modules are not a feature of the data, that is, all data sets where there is a single planted module (r = 1) or when the modules are close to being indistinguishable (β = 10), cisDIVERSITY still is highly competitive (Supplemental Fig. S1A), with similar precision and recall rates. This shows that even if the data do not contain distinguishable modules, cisDIVERSITY is capable of finding motifs at an accuracy comparable to state-of-the-art methods. Similarly, we also tested the false-positive rate of the methods in randomly chosen 1000 nonrepetitive regions of the human genome, containing no planted module or motif. cisDIVERSITY again identifies fewer motifs, that is, has a lower false-positive rate than either of the two motif discovery methods (Supplemental Fig. S1B). All programs were run in their serial mode, using a single core on an Intel Xeon CPU E5-2630 v3 machine. HOMER is by far the fastest, with there being not much difference between MEME and cisDIVERSITY (Fig. 2C).
In the next sections, we apply cisDIVERSITY to data sets arising from a range of different types of high-throughput assays (Methods). We start with modules discovered in core promoters and then investigate various ChIP-seq data sets, followed by diverse assays targeting enhancers and accessible regions.
Core promoter architectures retrieved from genome-wide TSS maps
cisDIVERSITY was first run on core promoters to see if it can recover established promoter architectures and elements. A 200-bp neighborhood around 4159 TSSs identified using paired-end analysis in fly embryos (Ni et al. 2010) was used as input. Motif discovery was restricted to the given strand (Methods), because TSS data are inherently strand oriented. A total of eight modules and 24 motifs were identified (Supplemental Fig. S2). The nine motifs that occur in at least a fifth of some module are displayed here (Fig. 3A,B). Most of these motifs, their combinations in terms of modules, and their positional preferences with respect to the TSS (Fig. 3D) have been identified before (Ohler et al. 2002): TATA box is present ≈30 bases upstream, the initiator (INR) is at the TSS, and the downstream promoter element (DPE) is 20 bases downstream. Ni et al. (2010) classified TSSs as narrow (reads map within a small window of 25 bases with a clear peak), broad (reads map to a larger window but still have a peak), or weak (all other promoters) and analyzed each set separately for differential enrichment of established fly promoter motifs. Here we have treated all regions together and note that the detected modules encode information about both motif enrichment and transcriptional activity. Modules characterized by the TATA box, INR, and DPE are significantly enriched with narrow promoters, whereas modules 1, 3, 5, and 7 are significantly depleted of them (P < 10−10). Motif 5, which has a weak AT-rich sequence, is more prominent in these modules and has not been categorized as a promoter element earlier. It is, however, visible in the representative modules 1 and 3 shown in Figure 3C, specifically downstream from the TSS. Module 6 is characterized largely by the INR-DPE motif but has a small number of promoters with a TATA box as well. The TATA box is enriched in promoters in which the INR-DPE is shifted downstream from the TSS. We are unsure of the significance of this observation.
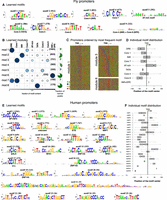
Promoter modules in fly and human. (A) cisDIVERSITY identifies 24 motifs in fly promoter data, but only the nine that contribute to at least 20% of some module are shown here. Core motifs are numbered according to the method of Ohler et al. (2002). (B) Eight modules are learned. The fraction of sequences in each module that are focused promoters, namely, have a narrow peak are shown in green. (C) Three representative modules are shown as sequence heat maps sorted based on the position of the most frequently occurring motif. Module 1 contains DRE, which is visible owing to the manner in which the sequences were ordered, but Core 7, which is present in >40% of the sequences, is not visible because it has no clear positional relationship with DRE or the TSS. The weak presence of the AT-rich motif downstream from the TSS is, however, visible. In contrast, module 3 displays a clear relationship between Core 1 and Core 6: Core 6 is present about 20 bases upstream of Core 1 and is especially prominent when Core 1 is close to the TSS. Module 6 is largely composed of INR + DPE but also contains the TATA box specifically when the INR + DPE is a few bases downstream from the TSS. (D) Each motif has a distinct distribution about the TSS. (E) cisDIVERSITY identifies 25 motifs in pooled human promoter data, with motifs 21–25 contributing on one module with TSSs of 37 zinc finger genes. All the other motifs are strand invariant, except for motif 2 (TATA) and motif 6 (unknown). (F) Each motif here too has a distinct distribution about the TSS.
Module 4 is not significantly enriched or depleted of narrow promoters. It is also the only module with a significantly lower tag count, implying that it comprises primarily of weak promoters. We suspect that is why cisDIVERSITY finds no motifs in these regions.
We next ran cisDIVERSITY on 14,408 nonoverlapping human core promoters determined by pooling CAGE tags across tissues (Frith and The FANTOM Consortium 2014) in the same strand-specific manner. A total of 25 motifs and 13 modules are detected (Fig. 3E; Supplemental Fig. S3). The last five motifs (21–25) are part of the smallest module comprising TSSs of a family of zinc finger proteins with near-identical promoters. We omit these special motifs and module from the subsequent analysis.
In contrast to the fly motifs, almost all motifs appear in both orientations or the motif itself is palindromic. Motifs 8, 10, 13, 14, and 16 are examples of the latter. On the other hand, motifs 1 & 4, motifs 3 & 17, motifs 5 & 7, motifs 9 & 20, motifs 11 & 12, and motifs 15, 18, & 19 are copies of the same motif in both orientations. Their position with respect to the TSS does not differ (Fig. 3F), suggesting these motifs function in a strand-invariant way, although two motifs (3 & 9) do occur more frequently than their reverse complements (17 & 20). The TATA box, present 29–35 bp upstream, and motif 6, present downstream, are the only motifs discovered in a single orientation. Whereas the TATA box appears in a single module enriched with narrow promoters, motif 6 is scattered across modules (Supplemental Fig. S3). It does not resemble the fly DPE, nor could we find evidence in literature supporting its existence as a human promoter element. The closest match is to the human donor splice site consensus sequence, but we do not see how that might play a role here.
Similar to fly promoters, the detected modules have distinct characteristics in terms of enrichment or depletion of narrow promoters (Supplemental Fig. S3). Here, too, we note that modules with few binding sites have a significantly lower tag density.
CTCF-bound regions display remarkable sequence-level diversity
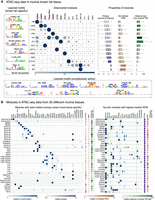
We next investigated ChIP-seq data sets. CTCF is a highly conserved TF, known to play different critical roles in regulation from binding insulators to forming chromatin loops in different contexts (Phillips and Corces 2009; Matthews and White 2019). Here we apply cisDIVERSITY to see if these roles can be characterized in terms of modules from ChIP-seq data targeting CTCF. We used data from Drosophilia melanogaster in the white prepupa developmental stage (Ni et al. 2012). We also looked at the ChIP signal and the distance from the closest TSS at the sequences to see if the identified modules had specific properties/activities. Ni et al. (2012) had identified a 9-bp motif, AGSKGGCGC, using MEME in the set, which resembles the canonical fly CTCF motif (Khan et al. 2018). This motif was present in approximately half of the regions. cisDIVERSITY reports 17 motifs spread across 13 modules (Fig. 4A–C). The top module—with the most sequences—is dominated by the first motif, which matches the reported CTCF motif. This module also has a significantly higher (P < 10−10) ChIP signal, which is expected if we assume this is where the binding of CTCF is strongest. Motif 1 contributes partially (27%) to module 4, where it co-occurs with motif 5. Motif 5 resembles the motif of another insulator binding protein suppressor of hairy wing, Su(Hw) (Khan et al. 2018). This module also has the maximum overlap with the ChIP-seq regions bound by Su(Hw) in the same developmental stage (Nègre et al. 2011), suggesting that sequences in this module may be bound by Su(Hw) directly.
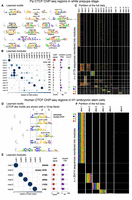
CTCF displays contrasting diversity in fly and human. (A–C) cisDIVERSITY identifies 17 motifs and 13 modules in the fly CTCF data. Motif 1 shown in a red box is the canonical fly CTCF motif. (D–F) cisDIVERSITY identifies only six motifs and six modules in the human CTCF data. Again, motif 1 in the red box matches the canonical vertebrate CTCF motif. Motifs 2–5 resemble the vertebrate CTCF but differ at one of three parts denoted with dotted lines. These motifs are shown with 10-bp flanks to ensure that they are genuine variants of the motif.
Multiple studies (Smith et al. 2009; Ni et al. 2012) have shown that in fly, the highest proportions of CTCF binding regions are in promoters. Indeed, if we look at this data set as a whole, over half of the bound regions are within 1 kb of some TSS. However, when we look at the modules individually, we note that the two modules described above, which contain the CTCF motif, in fact have fewer promoters (P < 10−5 for both). In contrast, modules 5–8 largely overlap promoters (<100 bp from a TSS) and, except for module 8, do not contain CTCF motifs. All of them are enriched with motif 3, that is, promoter element Core 3 (Ohler et al. 2002), but with diverse cofactors. These are in fact different core-promoter architectures, which we have seen in the earlier section. Taken with the fact that ChIP signal is also low at these modules, CTCF probably makes indirect contacts here. Module 2 has no motif and has the lowest ChIP signal, which is also significantly lower than the other modules (P < 10−5), again suggesting nonspecific or weak binding of CTCF.
Motifs 2 and 4 are highly similar and resemble the motif of the zinc finger TF Pita (Maksimenko et al. 2015). Both occur in modules 3 and 7. This implies that sequences in these modules have two copies of the Pita motif, and therefore, both are required to describe the data set. Now cisDIVERSITY does not take into account the spatial distribution of the motifs in its model. However, in over two-thirds of the sequences, the distance between the two copies is <50 bp, suggesting cooperative binding.
Unlike vertebrates, there is no evidence to support cohesin-CTCF-mediated chromatin loop formations in the fly (Matthews and White 2019). Instead, it is believed that looping may be mediated by interactions of CTCF with other insulator binding proteins like Su(Hw), BEAF-32, Ibf1/2, and Trl (Cuartero et al. 2014), all of which are identified by cisDIVERSITY (Fig. 4A, motifs 5, 6, 9, and 13). We were unable to find strong matches for the other discovered motifs to any known TF motif in literature. However, sites at many of these motifs are conserved across flies (Supplemental Fig. S4), which suggests they may play a role in CTCF-related regulation.
These results are much in contrast with those obtained on human CTCF. cisDIVERSITY was run on approximately 35,000 ENCODE CTCF ChIP-seq regions from human H1 embryonic stem cells (ESCs). Although the number of bound sequences is far greater than in the fly, cisDIVERSITY reports only six modules and six motifs (Fig. 4D–F). The top motif is the canonical CTCF motif, and four are variants with some parts of the motif missing or displaying nucleotide dependencies. These variants possibly correspond to the variable usage of CTCF zinc fingers (Phillips and Corces 2009); however, although nucleotide dependencies within vertebrate CTCF have been well documented (Narlikar 2013; Eggeling et al. 2014), we have not previously seen variants that are missing parts of the motif. Furthermore, these five motifs almost always occur in separate modules and together explain >85% of the sequences. The only non-CTCF motif matches that of ZNF143, a transcriptional activator that binds at promoters, associated with CTCF (Heidari et al. 2014), and in loop formations (Ye et al. 2020). Of the sequences that contain ZNF143, ∼90% also contain a CTCF variant, implying that interaction at these sequences with CTCF is not necessarily indirect/via ZNF143. Compared with fly CTCF, there is far more variability in the human CTCF motif itself, and it does not appear to make as many indirect or nonspecific interactions with DNA. However, the ChIP signal is significantly higher at the module with the canonical motif, suggesting that the modules with the variants are less strongly occupied by CTCF. CTCF is known to form loops and topological domains in mammals (Phillips and Corces 2009). It is possible that the various zinc fingers of CTCF interact at different chromosomal locations, thereby facilitating loops between them, but more experiments would be required to definitively establish this.
GR binds to diverse regions after activation
The human glucocorticoid receptor (GR; encoded by NR3C1) binds to thousands of sites in response to exposure to the glucocorticoid (GC) hormone cortisol. GR is understood to bind primarily to DNase I hypersensitive sites (DHSs) (John et al. 2011) and often with other pioneer factors (Biddie et al. 2011; Grøntved et al. 2013). McDowell et al. (2018) have probed binding of multiple factors, including GR, before and after treating A549 cells with the synthetic GC dexamethasone (dex).
Figure 5A shows the results of applying cisDIVERSITY on 6694 GR-bound 200-bp regions identified 1 h after dex treatment. Six modules and five motifs are identified. The largest module contains motif 1 that matches the GR motif. Modules 2–5 are dominated by the other four motifs, which match motifs of TFs that are known to play a role in recruiting GR to its binding sites (Biddie et al. 2011; Grøntved et al. 2013).
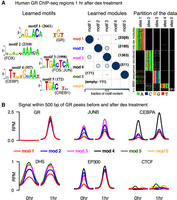
Diverse signals discovered in GR-bound ChIP-seq regions. (A) cisDIVERSITY identifies six modes and five motifs in the GR ChIP-seq regions. (B) The average DNase hypersensitivity signal and input-subtracted ChIP-seq signal in reads per million (RPM) of five TFs—GR, JUNB, CEBPA, EP300, and CTCF—before and after treatment at the GR-bound regions are shown for each module.
We looked at the ChIP-seq signal of profiled TFs at these modules both before and 1 h after dex treatment. As expected, GR signal is nonexistent before dex treatment at any module (no GR-bound regions were reported in this stage) and goes up at all modules after treatment (Fig. 5B) but significantly more at module 1 (P < 10−10). ChIP-seq signal of JUNB and CEBPA (previously known as CEBP) is also higher at modules with their cognate sites. However, they appear to occupy those modules even before dex treatment, albeit with lower intensity. Their signal at all modules, including module 1, goes up after dex treatment, suggesting GR contributes to some sort of cooperative binding at all modules.
As has been noted before (John et al. 2011), all modules have some DNase hypersensitivity signal before treatment, but the difference before and after treatment is the most at the module with the GR motif. This difference is even more pronounced when we look at the enhancer mark: EP300 binding signal. McDowell et al. (2018) used the GR motif to scan the GR-bound regions and observed that regions with initial EP300 binding (before treatment) had a weaker median GR motif strength than did those without initial EP300 binding. Our results are consistent with this, but in addition, they show that not only is the GR motif absent in regions with initial EP300 binding but also those regions can be explained by the presence of other specific motifs. When cisDIVERSITY is run on the EP300 ChIP-seq regions separately, it identifies near identical motifs, but with different module distributions; the module with GR motif is the fourth largest (Supplemental Fig. S5). CTCF ChIP-seq signal is plotted as a negative control: There is no difference before and after dex treatment of CTCF occupancy at these modules.
The total number of detected motifs and the number contributing to each module are both lower than the fly CTCF data examined earlier. To investigate whether more signal of cooperativity exists in a larger window around the summit, we reran cisDIVERSITY on a twice-as-long, 400-bp neighborhood (Supplemental Fig. S6). Indeed, more than twice as many motifs (13) and nine modules are detected. The GR motif continues to be the most abundant. Motifs resembling FOX, JUN, CEBP, and CREB1 are also discovered but with an additional copy of CEBP: Both copies co-occur. Together these motifs continue to explain most of the regions. Three of the remaining seven motifs resemble TFs active in the lung: HNF4A, HNF1A, and MAF::NFE2. However, they are dispersed across multiple modules, not contributing in a significant manner to any specific module. The remaining four motifs are low complexity di- or trinucleotide repeats and unlike the other motifs, which are concentrated near the summit, occur evenly across the 400-bp regions. These are likely structural features of the DNA (Yanez-Cuna et al. 2014). Increasing the region length further to 1000 bp, yields 21 motifs. Although all the earlier motifs continue to be identified, GR is no longer the most abundant motif. Instead, several more FOX and JUN motifs along with lung-specific TF binding sites and multiple low-complexity DNA motifs are discovered (Supplemental Fig. S7), suggesting the diminishing influence of GR motifs over the regions away from the ChIP summit.
Enhancers have a different structure based on the detection assay
Enhancers play critical roles in activating gene transcription while often being distant from the target gene. Although certain TF binding sites are enriched at enhancers, there is no consensus sequence-based rule that can explain or characterize these regions. Over the last few years, several new high-throughput assays have been developed that measure a biochemical activity that is indicative of enhancers, either directly or indirectly. For example, short, bidirectional, and largely unstable transcripts have been shown to originate at active enhancers (Andersson et al. 2014). The function(s) of these enhancer RNAs (eRNAs) are as yet not completely understood, but active TF binding at enhancers has been shown to increase corresponding eRNA levels (Danko et al. 2015). We applied cisDIVERSITY to 14,300 distal eRNAs (Azofeifa et al. 2018) detected in the GRO-cap data set from the K562 erythroid cell line (Core et al. 2014). Figure 6A shows nine motifs and eight modules learned in this data. There are actually five more motifs, which together explain the ninth module of 32 sequences (Supplemental Fig. S8). The UCSC Genome Browser marks these sequences as segmental duplications (Haeussler et al. 2019). Indeed, clear conserved structures were identified by the multiple sequence alignment tool CLUSTAL (Madeira et al. 2019) when module 9 was given as input (Supplemental Fig. S9). For clarity, we have removed this module and the corresponding motifs here.

cisDIVERSITY run on putative enhancers in K562. (A) On distant eRNAs, overlap with ChIP-seq data is significant (hypergeometric P < 10−4; shown in bold) in modules that contain the matching TF motif. Note that in some cases the overlap looks large but does not show up as significant, because the hypergeometric test corrects for the sizes of the overlaps and the modules. (B) Similar motifs are found in EP300 ChIP-seq data. (C) In STARR-seq peaks, YY1 is additionally discovered. CTCF and NFYA/NFYB are not enriched. (D,E) Distant (D) and proximal enhancer-like sequences (E) deduced from chromatin signatures have fewer motif-like signatures. cisDIVERSITY run on putative enhancers in K562.
In contrast to the models obtained from ChIP-seq regions, here the largest module, composed of over half the data set, is largely empty, with no significant occurrences of any motifs. All other modules have one dominant motif, which matches a motif of a TF active in K562. We therefore assessed overlaps between modules and ChIP-seq regions of all these TFs. As expected, the overlap is significant (hypergeometric P < 10−4) for the corresponding TF, although the overall overlap with the complete set is, for most TFs, small. Overlap with the enhancer mark EP300 and active chromatin marks is higher (Supplemental Fig. S10), but there too, we see differences in the degree of overlap across the modules.
The largely empty module in the distal eRNAs has no significant overlap with any of the external data sets. In fact, the overlaps are significantly less than that expected by chance (hypergeometric P < 10−4) with EP300 and other active chromatin marks. This, taken together with the fact that no significant sequence motif is detected in this module, suggests that perhaps these regions do not conform to the standard definition of enhancers, that is, enriched with TF binding and active chromatin marks.
Azofeifa et al. (2018) had removed bidirectional transcripts that overlapped with annotated promoters to get the distal set of eRNAs. We applied cisDIVERSITY to these 8324 excluded proximal transcripts and retrieved a starkly different model with 10 modules and a much larger set of 25 motifs (Supplemental Fig. S11). Some motifs such as those matching SP1, NFYA/B, and ZNF143 are common between both sets, but proximal eRNAs are enriched with many other promoter motifs such as YY1, RFX1, CREB1, and NRF1, etc. Contrary to the distal modules, there are more motifs per module, implying more cooperative binding at these regions, which is a hallmark of promoters (Maston et al. 2006). The enrichment of active chromatin marks is also different across these modules. These regions are more often accessible and have more overlaps with all active marks, except for H3K4me1, which is associated with enhancers (Heintzman et al. 2009) and less so with promoters (Cheng et al. 2014). This is consistent with the data set being separated based on overlaps with annotated promoters. However, even within the modules in both data sets, there are differences across the marks, implying that not all eRNAs have the same chromatin signatures, even after separating them as distal and proximal. But more importantly, these differences can be partially explained with modules and motifs.
We next ran cisDIVERSITY on four other data sets, which also report enhancer activity in K562. The first is the EP300-bound sequences in K562, where we again get nine motifs, almost identical to ones in distal eRNAs but present in different fractions of the sequences (Fig. 6B). In contrast to the distal eRNA set but similar to the other TF ChIP-seq data sets, a large majority of sequences contain some motif in the EP300 set. We next looked at STARR-seq data. In a STARR-seq assay, random genomic fragments are placed in the 3′ UTR of a reporter gene with a minimal promoter, and the resulting plasmids are transfected into the cells of interest, K562 here. The enhancer activity of these fragments is then measured by sequencing the 3′ UTR of the reporter gene transcripts. Unlike the other methods, this assay considers regions outside of their chromatin context so it can report regions that are inaccessible but have a potential for enhancer activity (Liu et al. 2017). We used the peaks reported by Lee et al. (2020), using their STARRPeaker method, which resulted in a little over 9000 sequences (Fig. 6C). In spite of very few sequences overlapping between the STARR-seq regions and the eRNAs or EP300-bound regions (Supplemental Fig. S12), many motifs are common between the three sets. Only YY1 is discovered additionally in the STARR-seq regions (module 4), whereas NFYA/NFYB and CTCF are absent. Because STARR-seq assays consider regions outside of chromatin context, we looked at whether the modules had accessibility profiles distinct from the first two data sets. Indeed, modules 2 (devoid of motifs) and 4 (dominated by YY1) are significantly (P < 10−5) depleted of DHSs. This suggests that these modules are suppressed by endogenous chromatin in K562. On the other hand, modules 1 (AP1-dominated) and 6 (GATA-dominated) are significantly enriched with DHSs: These structures were also identified in the eRNA and EP300 assays.
The last two data sets are from ENCODE phase III, where The ENCODE Project Consortium et al. (2020) have published a registry of candidate cis-regulatory elements based on results from multiple high-throughput experiments. They report two disjoint subsets, which they propose have distal enhancer-like signatures (dELSs) and proximal enhancer-like signatures (pELSs), respectively. The criteria for a sequence to be included in either of these sets is that it should have high DNase and H3K27ac signals. pELSs are within 2000 bp of an annotated TSS, whereas dELSs are away. pELSs additionally must have low relative H3K4me3 signal to ensure they are not active promoters. Other than an SP1/SP2 motif, there are no common motifs in the two sets (Fig. 6D,E). CTCF is found in the dELS and ZNF143 in pELS, which is expected, because these TFs’ binding sites are enriched in regions distal and proximal to TSSs, respectively (Kim et al. 2007; Bailey et al. 2015). In contrast to the first three enhancer data sets, cisDIVERSITY finds no motifs matching SPI1, CEBP, or NFYA/B. No GATA motif is found in the pELS set, possibly because GATA proteins primarily bind to distal enhancers (Romano and Miccio 2020). Overall, fewer signatures that look like TF-motifs are identified in these enhancer-like regions from ENCODE III.
Modules in open regions have differing future fates
We next looked at data from ATAC-seq and DNase-seq, two different technologies that measure chromatin accessibility. Cusanovich et al. (2018) used single-cell ATAC-seq on Drosophila embryos at three different stages after egg laying. Here we report cisDIVERSITY results on the earliest stage: 2–4 h after egg laying. We considered regions that were open in at least 10% of the cells assayed at that stage. This resulted in 9963 unique peaks. cisDIVERSITY finds 28 motifs and 13 modules (Supplemental Fig. S13). For clarity, Figure 7A shows only those motifs that contribute to at least a quarter of the sequences in some module. Without any additional information, cisDIVERSITY largely partitions the data into modules that are significantly enriched with promoters (within 500 bp of an annotated gene) and those that are significantly depleted (P < 10−4). Most of the motifs that contribute to the promoter-enriched modules are well-established core-promoter motifs. The two Pita motifs from fly CTCF data (Fig. 4A,B) are recovered here as well, in module 10. Modules distant from TSSs are potentially enhancers and/or insulators. Indeed, enrichment of CTCF and Su(Hw) in module 11 suggests this module harbors insulators. Similarly, dinucleotide repeats of GA (bound by GAGA binding factor Trl) and CA are known to be features of fly enhancers (Yanez-Cuna et al. 2014). One of the promoter modules is comprised of tRNAs, and cisDIVERSITY captures the highly conserved hair-loop structure of tRNAs as a “motif” (module 12).
cisDIVERSITY run on open regions. (A) Thirteen modules and 28 motifs (Supplemental Fig. S13) are learned on ATAC-seq regions, which are open in at least 10% of the cells probed 2–4 h after egg laying. Only the 19 motifs that contribute to at least a quarter of the sequences in some module are shown here for clarity. Modules are reordered: The red and green modules are significantly (hypergeometric P < 10−4) enriched with promoters and depleted of them, respectively. (B) Gray indicates there are only a few differences in the fraction of cells open within each module. Orange indicates modules 2, 3, 7, 8, 4, and 6 are significantly more open in the cells 6–8 h after egg-laying, whereas modules 12, 1, and 9 are closing at that time point. (C) Eighteen modules and 21 motifs (Supplemental Fig. S14) are learned on DNase-seq regions in H1 ESCs. Again, only the motifs appearing in a quarter of sequences of some module are shown here. Red and green modules are as in A. (D) Gray indicates promoter modules have a higher DNase signal in general, but there are variations among them. Blue indicates the fraction of each module (and total below) that is also open in trophoblast, mesenchymal, and neuronal stem cells (all derived from H1 ESCs), and GM12878 shows considerable variation across modules and cell types.
There is little difference in the fraction of cells open at the regions across the modules: Only modules 4 and 6 (distal regions with dinucleotide repeats) are significantly more open. However, when we compare the fraction of cells open at these regions with the fraction open 4 h later (6–8 h after egg laying), we see several modules significantly changing their accessibility (Fig. 7B). Almost all promoter modules are opening up, except for the tRNAs. We found no evidence of tRNA gene expression going down during these stages of development. However, tRNA genes are known to play a role in remodeling chromatin and act as insulators or harbor origins of replication in other organisms (Su et al. 2020; Sreekumar et al. 2021). Modules 1 and 9 are the only other modules that are significantly closing. Module 1 has very few predicted binding sites, with no motif that occurs in even 10% of the sequences. This is in concordance with recent results that suggest two classes of enhancers are active during early Drosophila embryogenesis, one of which has significantly lower TF occupancy, in general (Arbel et al. 2019). All sequences in module 9, however, contain a motif matching that of the TF zelda (zld). zld is a pioneer TF that is critical for maternal to zygotic transition, which happens during this stage (Hamm and Harrison 2018). cisDIVERSITY results suggest that these zld-enriched regions are less accessible in the subsequent stages.
cisDIVERSITY was next run on 47,279 DNase hypersensitive regions of H1 ESCs (Fig. 7C; Supplemental Fig. S14). The original option of r = 15 resulted in 15 modules, so cisDIVERSITY was rerun with r = 20, reporting a total of 18 modules. As in fly data, here too, the modules are significantly enriched with promoters or depleted of them. All promoter modules have high DNase accessibility, but there are significant differences between them. For example, although modules 3 and 6 are both enriched with promoters and with the SP1/SP2 motif, module 6, which also contains an NFYA/NFYB motif, has a significantly (P < 10−10) higher DNase hypersensitivity signal. Module 16 also contains SP1/SP2 and NFYA/NFYB but comprises primarily of core-promoters of a KRAB family of zinc fingers. This similarity is captured as a distinct long motif, resulting in a module distinct from module 6. Modules depleted of promoters are characterized with motifs of H1 ESC-specific TFs (SOX2 and POU5F1::SOX2), along with motifs of other pioneer factors (AP1, REST, and CTCF).
We looked at the DNase accessibility at these regions in stem cells derived from H1 ESCs available in ENCODE: trophoblast, mesenchymal, and neuronal stem cells (Fig. 7D). Although the overall fraction of regions overlapping with DHSs in these stem cells is similar (0.76 for trophoblast, 0.71 for mesenchymal, and neuronal), the overlap fraction within modules ranges from 0.42 to 1.0. Indeed, >90% of sequences of modules 8, 12, 14, 16, and 17 are accessible in these three stem cells. In contrast, modules dominated by motifs of pioneering ES TFs (SOX2, POU5F1, and KLF4) are less accessible, which is expected. Module 11, containing the REST motif, has a high accessibility in all the stem cells. The canonical CTCF motif (module 2) is significantly more often accessible than the variant (module 4) in the other cells, although in H1 ESCs, the variant has a higher accessibility signal (P < 10−5). DHSs in the lymphoblastoid GM12878 cell-line (derived from blood) are used as control. Overall, the overlap is lower for all modules, but we see the same trend of higher overlaps at promoters and lesser ones at distal modules.
Tissue-specific modules discovered in open regions
We then analyzed ATAC-seq data in 20 different murine tissues (Liu et al. 2019). The investigators report regions with normalized read counts in each tissue. They also compute an entropy value for each region, based on these read counts: a lower entropy value implies that the regulatory region is open in a tissue-specific manner. We considered all those regions with at least 10 normalized reads and took a longer, 400-bp region around the center to account for the fact that these regions were merged across experiments. cisDIVERSITY was run on every tissue separately. Figure 8A shows the motifs and modules obtained in the brown fat tissue, ordered according to the median entropy value. The first six modules have a median entropy lower than 4.0; that is, they are specifically reported in the brown fat tissue. These modules are characterized by motifs that match those of TFs known to be expressed in the fat tissue (Pradhan et al. 2017; Lee et al. 2019). These modules also have lower reads on average, implying that although they are tissue specific, they are also relatively less accessible than the other modules. They are also more likely to be distal elements. In contrast, the modules characterized by constitutively active TFs such as ZFP263, SP1, etc., indeed have a higher entropy, are promoter proximal, and are more open than the other modules. This trend largely persists across the other 19 tissues as well (Fig. 8B). Modules that have a significantly lower entropy value, that is, are specifically reported in the respective tissues, are characterized with motifs of corresponding tissue-specific TFs and have lower accessibility in general. They are also likely to be distal from any TSS. In contrast, modules with the highest accessibility are promoter proximal, are accessible across tissues, and, barring a few exceptions, are largely characterized by motifs of TFs that are constitutively active (Baldarelli et al. 2021). As expected, a common set of TFs is present in these modules. MYF6 and HNF4a are additionally present in liver modules and GABPA in the lung; these TFs are known to be overexpressed in the respective tissues (Baldarelli et al. 2021). We stress that cisDIVERSITY cannot determine identity of the TFs, because it only detects sequence signatures associated with each module. These results are therefore based on the matches to TF motifs as determined by TOMTOM.
cisDIVERSITY run on accessible regions in mouse tissues. (A) Thirteen modules are learned in ATAC-seq regions from the brown fat tissue, sorted according to median entropy (most brown fat–specific on top). The 15 discovered motifs can be split into two sets: those that are enriched in tissue-specific regions, that is, those with median entropy value less than 4.0, and those that are constitutively active. Boxplots on the right indicate the relationship between tissue specificity, accessibility, and proximity to TSSs. (B) Left panel shows modules with the lowest median entropy for each tissue. Many tissues have multiple modules with lower than 4.0 median values: All such modules are shown. Motifs that are present in at least 10% of the sequences in any module are shown. The right panel shows the top two modules based on highest median RPM values per tissue. Median entropy, RPM, and distance to closest TSS are displayed on the right for each module. TOMTOM matches were used to assign putative TF identities to motifs and combined across the cisDIVERSITY individual runs.
We note that ATAC-seq data are usually not analyzed in this manner. Especially if multiple tissues have been profiled, as in this case, motif discovery is typically targeted in regions preidentified to have high tissue specificity or be distal to any TSS. Indeed, Liu et al. (2019) use HOMER to detect motifs in tissue-specific accessible regions. cisDIVERSITY finds those motifs in spite of not being supplied information about tissue specificity: Instead, it automatically identifies modules that are tissue specific.
Discussion
The importance of combinatorial binding of TFs in gene regulation is well established (Reiter et al. 2017) as is the fact that most high-throughput experiments report regions with diverse sequence characteristics. Several attempts have been made to detect regulatory modules from high-throughput data. One of the earliest motif-module detection programs was CisModule (Zhou and Wong 2004), where the goal was to learn the location of a module in each sequence along with motifs. The module, however, was of a single kind. Self-organizing maps (Xie et al. 2013), nonnegative matrix factorization (Giannopoulou and Elemento 2013), and topic models (Guo and Gifford 2017) have been subsequently used to cluster regions based on multiple ChIP-seq data sets, with the goal of identifying different modules. These methods have been used to predict complexes forming along the chromatin and colocalization of TFs. However, they explicitly require regulatory information in terms of other high-throughput experiments for each region, which act as features for clustering. They do not incorporate sequence information or motifs as part of the model. cisDIVERSITY is the first attempt to cluster regions based on motifs that are themselves learned during the process, requiring no additional experimental or TF binding information. The discovered modules indeed correlate with experimental binding data of TFs with matching motifs, showing that regions contain sequence information that can characterize functional modules. On simulated sets, as a motif discovery method, it performs better than standard approaches, especially in terms of precision or false-positive predictions. We believe this is because of its model-based approach, in which the goal is to learn a model that explains the full data set and not to learn motifs that individually explain a portion of the sequences.
Regulatory modules are typically studied in accessible regions or putative enhancers, assuming cooperativity of TFs there. However, cisDIVERSITY gives new insights even in the ChIP-seq data sets investigated here. Take, for example, the heavily studied CTCF protein. The original study also mentioned that many sequences did not contribute to the overrepresented CTCF motif in the fly data, but all subsequent location and evolution analysis was performed considering all ChIP regions equally. However, cisDIVERSITY clearly shows that some sequences are core-promoters with no CTCF motif, whereas some are enriched with other insulator-binding TF motifs. The evolutionary profile of the motifs is also diverse in the data set. On the other hand, the human CTCF behaves differently, with ZNF143 being the only non-CTCF motif that is discovered. Instead, several individual variants of the CTCF motif are enriched in different modules, suggesting a possibility of differential usage of its zinc fingers while binding DNA.
Putative enhancers have been detected using multiple high-throughput assays in the same cell type or context. The data sets arising from these assays differ in terms of their cardinality, length distributions, and evolutionary features (Benton et al. 2019). cisDIVERSITY shows the differences in terms of motifs and modules. No motif is common across all the enhancer-detection strategies, at least in the cell type assessed here. It is important to note that each data set used here comes from an assay that measures a different biochemical activity or combination of activities. cisDIVERSITY can be used to further tease out the differences between the sequences reported by these strategies in terms of motifs and their combinations.
The strength of cisDIVERSITY is in its lack of reliance on known PWMs or modules, making it general enough to be used on any set of DNA sequences. The only assumption it makes is that the sequences arise from one cellular context, implying that (1) a finite set of TFs is active during the experiment, and (2) binding sites for those TFs are the underlying cause for the regions to be reported. It does not need to be given a focused set such as distal regions or preprocessed tissue-specific regions. Instead, it recognizes that the data—even when it is from a single high-throughput experiment—can be a diverse set. cisDIVERSITY automatically clusters accessible regions into promoters and TSS-distal regions. This is not surprising because the sequence architecture of promoters is different. However, even within promoters and putative insulators/enhancers, it captures considerable sequence-level diversity as well as tissue specificity. Although the modules in Figure 7 are reordered based on their propensity to have more or less promoters, no module is completely composed or devoid of promoters. Distal sequences in a promoter-enriched module should be further studied for potential promoter activity and vice versa.
cisDIVERSITY does not explicitly model multiplicity of motifs or distance between them. These aspects can be studied from the learned parameters. For example, cisDIVERSITY identifies homotypic binding if it appears in a significant fraction of sequences, by learning more than one copy of the same motif. See, for example, the pair of Pita motifs in the fly (Figs. 4A and 7A) and the multiplicity of CEBP motifs in mammals (Fig. 8A; Supplemental Fig. S6). The distance between motifs and relative orientation can be assessed as well. Similarly, the distance of each motif from the summit of the peak can give insights into the likelihood of direct binding of the profiled TF in case of ChIP experiments (Bailey and Machanick 2012). cisDIVERSITY can be restricted to look for motifs on a given strand especially when dealing with data sets like 5′ CAGE TSSs or UTRs, which contain inherent directionality.
In all results described here, we have reported the top-scoring model, treating it as a hard clustering method. However, we note that cisDIVERSITY learns a probability distribution over the modules for each sequence, and multiple module usage can also be explored (Guo and Gifford 2017). Currently cisDIVERSITY reports no significance value or false-discovery rate for a learned model. However, it does report the posterior probability associated with the learned model. These values are far lower in data sets with random regions with no planted modules/modules (Supplemental Fig. S15). Such distributions can be learned to report an associated empirical “P-value.” In terms of run-time, cisDIVERSITY is comparable to MEME but is much slower than HOMER. In principle, we can initialize the model with at least some user-defined motifs, which could potentially speed up the model discovery.
Across the data sets investigated here, far fewer TF-binding sites characterize modules in mammalian regions compared with the fly. Cooperativity between TFs can arise from multiple mechanisms. The simplest situation is of DNA mediating it by harboring respective binding sites within a short linear region. This most likely explains cases in which multiple motifs characterize discovered modules. However, we see increasing evidence of interplay between 3D genome architecture and TF activity, which affects TF cooperativity, especially in higher eukaryotes (Ma et al. 2018). TF-driven loops and condensates are mechanisms of cooperativity, which do not require the presence of multiple binding sites in a linear genomic neighborhood (Kim and Shendure 2019). Similarly, the presence of clusters of low-affinity binding sites—which may not be captured by cisDIVERSITY’s motif models—has also been suggested to influence TF cooperativity (Malin et al. 2015). The extent to which these mechanisms play roles in control of gene expression is still unclear. More experiments and analyses will help determine whether the sparse modules identified by cisDIVERSITY are indicative of these mechanisms.
Methods
Model framework
The goal of most high-throughput experiments is to identify all regulatory regions with a certain biochemical property in a specific context. These regions might display that owing to r different mechanisms, and we assume one of these mechanisms is encoded in the DNA sequence of each region. A “mechanism” is represented as what is commonly known as a regulatory module. Each of the r modules is characterized by the presence or absence of m motifs. Each level of the model M, characterized by r and m, is described below in a bottom-up manner:
-
Motif level. We assume at most m motifs are present and contribute to modules, across the complete set of reported regions. Motif k is modeled as a PWM with width wk; that is, it is represented as a product of wk categorical distributions over the four bases. ϕk is used to denote these distributions, where
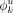 (B) is the probability of finding base B at position u within motif k,B ∈ {A, C, G, T}. Given any wk length DNA sequence
(B) is the probability of finding base B at position u within motif k,B ∈ {A, C, G, T}. Given any wk length DNA sequence 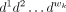 , the likelihood of it being an instance of motif k is
, the likelihood of it being an instance of motif k is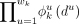 .
.
-
Module level. We assume at most r modules are present across the complete set of reported regions. Each module is modeled as a product of Bernoulli distributions over the m motifs: Module j has a probability, fjk, of containing motif k and a probability (1 − fjk) of not containing it.
-
Region level. The experiment reports a total of n genomic regions X1, X2…, Xn. Region Xi is a DNA sequence of length li:
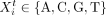 , where 1 ≤ t ≤ li. Parts of each sequence are instances of a subset of the m motifs; other parts, namely, the “background,” are modeled using a second-order Markov chain over the nucleotides, whose
parameters are denoted by ϕ0. Zik denotes the position within Xi, where motif k is present; namely, 1 ≤ Zik ≤ li − wk + 1. Zik = −1 in cases in which there is no motif k in sequence Xi. Motif occurrences are not allowed to overlap, and each motif can have at most one occurrence in each sequence. Sequence
Xi has a module identity Ii that is modeled with a categorical distribution γ over the r modules: γj is the probability of a sequence having a module identity j.
, where 1 ≤ t ≤ li. Parts of each sequence are instances of a subset of the m motifs; other parts, namely, the “background,” are modeled using a second-order Markov chain over the nucleotides, whose
parameters are denoted by ϕ0. Zik denotes the position within Xi, where motif k is present; namely, 1 ≤ Zik ≤ li − wk + 1. Zik = −1 in cases in which there is no motif k in sequence Xi. Motif occurrences are not allowed to overlap, and each motif can have at most one occurrence in each sequence. Sequence
Xi has a module identity Ii that is modeled with a categorical distribution γ over the r modules: γj is the probability of a sequence having a module identity j.
The set of regions X is all we are given, which we use first to compute the background Markov model as well as the lengths l. We are also told whether motifs can occur on either strand or on the given strand only. The default is the former, in which
case the sequences are appended to their reverse complements and their lengths are effectively doubled. In case of core promoters,
we use the strand-specific option, that is, searching only on the given strand. We assume we know the structure of the model;
that is, an upper bound on the r and m is given. The unknown parameters Θ are therefore ϕ1…m, w, Z, f, γ, and I. The likelihood of region Xi, based on these assumptions can be computed as
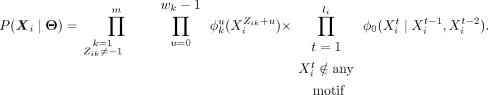 (1)
(1)
The first term denotes the probability associated with all the motifs that are present in Xi, whereas the other explains the sequence not overlapping with any of the motifs, using the background Markov model. cisDIVERSITY uses the second-order Markov model as default, where each sequence has a background model built only from its 3-mers, to accommodate the vast heterogeneity in eukaryotic sequences (Narlikar 2013). But this can be changed by the user if required.
The complete likelihood is simply
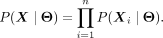 (2)
Given a set X, the goal is to find the Θ that maximizes the posterior distribution, which can be computed as
(2)
Given a set X, the goal is to find the Θ that maximizes the posterior distribution, which can be computed as
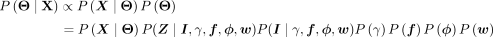 (3)
(3)
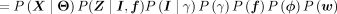 (4)
(4)
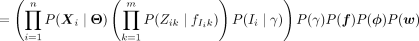 (5)
(5)
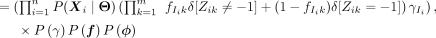 (6)
where δ[condition] = 1 only when condition is satisfied and zero otherwise. The simplification in Equation 4 arises from the structure of the model: The site positions Z are independent of γ, ϕ, w when conditional on the module identity I and its associated probabilities f; I is independent of f, ϕ, w, when its categorical distribution γ is known. The second term in Equation 5 is the product of Bernoulli probabilities, which are assumed to be independent. The third term is simply the categorical
probability of the module identity. The rest are independent priors over γ, f, and ϕ, which are discussed in the next section. The prior over the widths is considered uniform.
(6)
where δ[condition] = 1 only when condition is satisfied and zero otherwise. The simplification in Equation 4 arises from the structure of the model: The site positions Z are independent of γ, ϕ, w when conditional on the module identity I and its associated probabilities f; I is independent of f, ϕ, w, when its categorical distribution γ is known. The second term in Equation 5 is the product of Bernoulli probabilities, which are assumed to be independent. The third term is simply the categorical
probability of the module identity. The rest are independent priors over γ, f, and ϕ, which are discussed in the next section. The prior over the widths is considered uniform.
Model learning
To find the value of Θ that maximizes Equation 6, we need to design appropriate priors over the parameters. We assume conjugate symmetric Dirichlet priors over all categorical
parameters: (1) prior over γ, which characterizes the distribution of module identity I, has r equal hyperparameters defined by αmodule (1 by default); (2) prior over each fjk, which characterizes the distribution of presence or absence of motif k in module j, has hyperparameters, defined by αYES and αNO (both set to 0.1 by default); and (3) prior over each 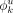 , which characterizes the distribution over the four nucleotides at position u in motif k, has four equal hyperparameters defined by αPWM for all u and k (all set to 0.1 by default). Hyperparameters less than one assume extreme final distributions: We expect motifs to be informative;
namely, nucleotide probabilities will be close to zero or one. Similarly, we expect modules to be specifically described with
the presence or absence of motifs. However, we do not know the number of nonempty modules, in other words, the inherent diversity
in the data. Therefore, we set αmodule to one by default, which assumes all distributions are equally likely a priori. However, all these values can be changed
by the user.
, which characterizes the distribution over the four nucleotides at position u in motif k, has four equal hyperparameters defined by αPWM for all u and k (all set to 0.1 by default). Hyperparameters less than one assume extreme final distributions: We expect motifs to be informative;
namely, nucleotide probabilities will be close to zero or one. Similarly, we expect modules to be specifically described with
the presence or absence of motifs. However, we do not know the number of nonempty modules, in other words, the inherent diversity
in the data. Therefore, we set αmodule to one by default, which assumes all distributions are equally likely a priori. However, all these values can be changed
by the user.
We use Gibbs sampling to draw samples from the posterior distribution with the aim of learning the parameter values that maximize
it. To reduce the search space and speed up the sampling, we use collapsed Gibbs sampling (Liu 1994), marginalizing over f, ϕ, and γ while sampling only the other unknowns. The objective function then reduces to
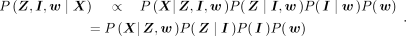 (7)
(7)
We therefore need to iteratively sample Zik, Ii, wk for each i and k. If we assume a binding site is equally likely to be anywhere within a sequence, after dividing the posterior distribution
by the background for each sequence, we can compute the sampling expressions for Zik as
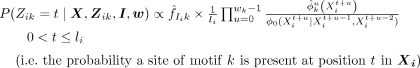 (8)
(8)
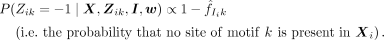 (9)
(9)
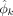 and
and 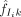 are the posterior probabilities after marginalizing, namely, integrating out ϕk and
are the posterior probabilities after marginalizing, namely, integrating out ϕk and 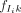 from Equation 7 when sampling
from Equation 7 when sampling 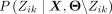 , assuming Dirichlet priors over both:
, assuming Dirichlet priors over both:
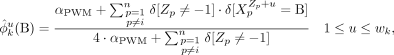 and similarly,
and similarly,
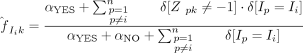 We note that the first term of Equation 8 is similar to standard collapsed Gibbs sampling applied to motif discovery, except that the counts are computed only from
those Xp that contain a motif k at the current iteration, whereas the second term arises from the contribution motif k makes to the current module Ii.
We note that the first term of Equation 8 is similar to standard collapsed Gibbs sampling applied to motif discovery, except that the counts are computed only from
those Xp that contain a motif k at the current iteration, whereas the second term arises from the contribution motif k makes to the current module Ii.
The sampling expression for Ii can be similarly derived by collapsing γ and f from Equation 7:
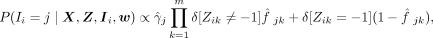 (10)
where
(10)
where 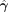 is the posterior probability distribution after integrating out γ
is the posterior probability distribution after integrating out γ
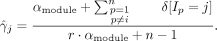
The width wk of motif k is sampled a little differently. Instead of looking at all possible widths at one time, the width is allowed to increase or decrease by one on either side or stay the same as performed before (Mitra et al. 2018).
Each of Zik, Ii, and wk are sampled iteratively using the expressions as described. The full posterior probability (Equation 7) is computed after each sampling step and stored if found to be the best thus far. If the probability does not increase for a predetermined number of iterations (500 by default), the sampling stops and a hill climbing approach (Mitra et al. 2018) starting from the sample with highest probability is taken until the probability stops increasing. The final model is cleaned up: All motifs that are composed of fewer than a minimum number of sites (20 by default, can be changed by the user) are emptied out, and all modules with fewer than minimum number of sequences (20 by default, can also be changed by the user) are combined into one. By default, cisDIVERSITY starts from 10 different initializations and reports the model that scores the best across the 10 trials.
Simulated data sets
A total of 320 data sets were simulated for testing the efficacy of cisDIVERSITY. The JASPAR2018 CORE vertebrate motifs were used for this purpose, regardless of their information content. However, many of these motifs are similar to each other. To create a nonredundant set, going serially, we removed all motifs that had a match with any earlier one according to the motif comparison tool TOMTOM (Gupta et al. 2007). This resulted in a total of 189 JASPAR motifs. Each simulated data set had 1000 DNA sequences of length 200 bp each, sampled randomly from the nonrepetitive part of the human genome. Each data set was constructed as an instance of the model described earlier. At the motif level, m motifs were drawn at random from the set of nonredundant motifs. At the module level, the Bernoulli parameters fjk were sampled from beta distributions with two symmetric (equal) parameters (αYES, αNO). At the sequence level, the categorical distribution γ was first sampled using the uniform parameter value of αmodule = 1 and set for that model instance. For each sequence, this γ was used to first sample the module j. Then, the fjk was used to sample whether motif k was to be included in the sequence or not. If yes, a site was sampled from the PWM k and randomly planted in the sequence, on either strand with equal chance, ensuring no overlap with any other planted site. Multiple data sets were created: m was one of {5,10}, r was one of {1,2,3,5}, and αYES was one of {0.01,0.1,1.0,10.0}. There were 10 data set instances of each parameter set of {m, r, αYES}, resulting in a total of 320 different data sets on which cisDIVERSITY and other programs were tested.
We note that two modules can have very similar Bernoulli distributions, making them less separable, but the models were not screened for this possibility because (1) that would be rare with m ≥ 5 and (2) this would only mean we are underestimating cisDIVERSITY’s performance.
cisDIVERSITY was run with m = 20 and r = 10. All other parameters were set to default values. MEME (Bailey and Elkan 1994) was run on these data sets using the command line options: -nmotifs 20 -maxsize 100000000 -revcomp -dna -evt 10. MEME was run with background Markov models of orders zero through five separately, learned on the nonrepetitive human DNA sequences used to create the data sets. Markov order of three gave the best F-scores; those results are reported here. HOMER (Heinz et al. 2010) was run using the command line options: -noknown -nogo -basic. HOMER was also given the background file of the nonrepetitive human DNA sequences. TOMTOM was used to compare the learned motifs with the JASPAR planted motifs. An E-value of 0.01 was considered a match.
Biological data sets
All data sets were processed in the following manner. A 200-bp (or 400-bp, where stated) neighborhood of the summits (if reported, else the midpoint) of the regions was extracted. If two regions overlapped, only the first one in the list was kept. This was to ensure that the model would not identify the same site in the genome twice. Only those regions with at least 100 bp of nonrepetitive bases in them were retained and used as input to cisDIVERSITY. All repetitive bases were replaced with Ns; cisDIVERSITY skips over all such bases. All data sets with their accession numbers are listed in Supplemental Table S1. cisDIVERSITY was run on each data set with the default values of m = 30 and r = 15. Only in the case of CAGE data, it was run by using the option that searches for motifs only on the given strand, because the data are strand specific and the background Markov order was set to one. Reported modules were assessed for enrichment with promoters, assay signal, etc., with either a Wilcoxon test or a hypergeometric test. All P-values in the text have been reported after Bonferroni multiple hypothesis correction. Sequence conservation plots were computed from phastCons scores (Haeussler et al. 2019).
Software availability
The code is freely available at GitHub (https://github.com/NarlikarLab/cisDIVERSITY), and as Supplemental Code S1.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Rahul Siddharthan and Uwe Ohler for useful suggestions and discussions. This study was supported by grants from Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India BT/PR16240/BID/7/575/2016 and BT/IN/BMBF-BioHr/32/LN/2018-19.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.274563.120.
- Received November 22, 2020.
- Accepted July 9, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








