
Novel features of telomere biology revealed by the absence of telomeric DNA methylation
- Alejandro Vega-Vaquero1,4,
- Giancarlo Bonora2,4,
- Marco Morselli2,4,
- María I. Vaquero-Sedas3,
- Liudmilla Rubbi2,
- Matteo Pellegrini2 and
- Miguel A. Vega-Palas3
- 1Technical Superior School of Informatics Engineering, University of Seville, 41080 Seville, Spain;
- 2Department of Molecular, Cell and Developmental Biology, University of California, Los Angeles, California 90095, USA;
- 3Institute of Vegetal Biochemistry and Photosynthesis, CSIC-University of Seville, IBVF (CSIC-US), 41092 Seville, Spain
- Corresponding authors: vega-palas{at}ibvf.csic.es, matteop{at}mcdb.ucla.edu
-
↵4 These authors should be considered equal first authors.
Abstract
Cytosine methylation regulates the length and stability of telomeres, which can affect a wide variety of biological features, including cell differentiation, development, or illness. Although it is well established that subtelomeric regions are methylated, the presence of methylated cytosines at telomeres has remained controversial. Here, we have analyzed multiple bisulfite sequencing studies to address the methylation status of Arabidopsis thaliana telomeres. We found that the levels of estimated telomeric DNA methylation varied among studies. Interestingly, we estimated higher levels of telomeric DNA methylation in studies that produced C-rich telomeric strands with lower efficiency. However, these high methylation estimates arose due to experimental limitations of the bisulfite technique. We found a similar phenomenon for mitochondrial DNA: The levels of mitochondrial DNA methylation detected were higher in experiments with lower mitochondrial read production efficiencies. Based on experiments with high telomeric C-rich strand production efficiencies, we concluded that Arabidopsis telomeres are not methylated, which was confirmed by methylation-dependent restriction enzyme analyses. Thus, our studies indicate that telomeres are refractory to de novo DNA methylation by the RNA-directed DNA methylation machinery. This result, together with previously reported data, reveals that subtelomeric DNA methylation controls the homeostasis of telomere length.
Telomeres guarantee the complete replication of chromosomal termini, prevent genome instability, and influence relevant systemic processes like aging, cancer, or illness (Blackburn 2010). The length of telomeres and the chromatin organization of telomeric regions influence telomere functions. Hence, the epigenetic marks that label telomeric regions, which include telomeres and subtelomeres, play important roles in telomere biology (Blasco 2007; Galati et al. 2013; Giraud-Panis et al. 2013).
One of the major epigenetic signatures found in eukaryotes is cytosine methylation. This DNA modification regulates multiple processes in plants and animals, including the homeostasis of telomere length (Blasco 2007; Suzuki and Bird 2008; Ooi et al. 2009; Law and Jacobsen 2010; Castel and Martienssen 2013; Ogrocká et al. 2014; Vaquero-Sedas and Vega-Palas 2014). Mammalian DNA methylation is primarily found in the CG context (Ramsahoye et al. 2000; Lister et al. 2009). In contrast, plants have significant levels of DNA methylation in all sequence contexts (CG, CHG, and CHH, where H can be A, C, or T) (Law and Jacobsen 2010).
Although subtelomeric DNA methylation has been reported in animals and plants, the presence of DNA methylation at telomeres remains an open question in both kingdoms (Blasco 2007; Vrbsky et al. 2010; Vaquero-Sedas et al. 2011; Ogrocká et al. 2014). The methylation status of mammalian telomeres has not been investigated because, as mentioned above, mammals have low levels of non-CG methylation, which is the type of DNA methylation that should be associated with telomeric sequences (CCCTAA in mammals and CCCTAAA in plants). In turn, although the methylation levels of plant telomeres have been studied by different groups, they remain controversial (Vrbsky et al. 2010; Majerová et al. 2011a,b; Vaquero-Sedas and Vega-Palas 2011a,b; Vaquero-Sedas et al. 2011, 2012; Ogrocká et al. 2014). Therefore, it is important to settle the methylation status of telomeres.
The experimental analysis of the epigenetic marks that label telomeres is complicated by the influence of subtelomeres and/or the Interstitial Telomeric Sequences (ITSs), which are usually present at pericentromeric regions and subtelomeres (Vaquero-Sedas and Vega-Palas 2011b). On the one hand, telomeres and subtelomeres cannot be differentiated by microscopy techniques. On the other hand, ITSs can interfere with the analyses of telomeric chromatin structure by chromatin immunoprecipitation followed by hybridization with a telomeric probe. Moreover, ITSs might be identified as telomeres in massively parallel DNA sequencing studies (Vaquero-Sedas et al. 2012; Vega-Palas and Vaquero-Sedas 2013). Hence, the analysis of the epigenetic modifications present at telomeres should be carefully designed.
The study of telomeres independently of ITSs may be facilitated by the fact that they usually have different sequence organizations. Although telomeres are essentially composed of tandem arrays of perfect telomeric repeats, ITSs usually contain perfect telomeric repeats interspersed with degenerate repeats. In fact, it is uncommon for ITSs to contain long stretches of perfect tandem telomeric repeats (Lin and Yan 2008; Gámez-Arjona et al. 2010).
Here, we have addressed the methylation status of Arabidopsis thaliana telomeres by analyzing data produced by genome-wide bisulfite sequencing studies and by performing methylation-dependent restriction analyses. These studies revealed that Arabidopsis telomeres are not methylated.
Results
In silico analysis of telomeric DNA methylation
To gain insight into the methylation status of Arabidopsis telomeres, we estimated their methylation levels from different genome-wide bisulfite sequencing studies (Supplemental Table S1). These studies had been performed in different laboratories and involved the treatment of DNA with sodium bisulfite, the PCR amplification of the resulting DNA samples, and the sequencing of the bisulfite modified DNA strand. Since bisulfite deaminates unmethylated cytosines generating uracil, unmethylated cytosines are detected as thymines after PCR amplification. In contrast, methylated cytosines are not modified by bisulfite and remain as cytosines after amplification (Frommer et al. 1992; Clark et al. 1994).
The reads representing telomeres in the bisulfite sequencing studies should follow a perfect tandem telomeric repeat pattern, represented as (YYYTAAA)n, in which Y is C or T depending on whether the telomeric cytosines are converted or not. We estimated that reads containing about seven perfect tandem telomeric repeats should essentially represent telomeres in the bisulfite experiments (Methods). Only 20 genomic (YYYTAAA)7 sequences can be found in the Arabidopsis genome, with one of them being 300 bp in length (Fig. 1).

Major Arabidopsis ITSs containing perfect telomeric repeats. Genomic DNA sequences containing (YYYTAAA)7 strings and several nucleotides upstream and downstream were aligned using the computer program, CLUSTALW. These sequences were identified using the PatMatch tool and the TAIR10 Whole Genome database (BAC clones) in The Arabidopsis Information Resource (TAIR; http://www.Arabidopsis.org). The name of the clones corresponding to each sequence is indicated as well as the hit corresponding to each clone. The right border of the 300-bp ITS is included in the alignment. Red letters label DNA sequences that continuously follow the (YYYTAAA)n pattern. Asterisks mark conserved residues.
Since the length of the reads in many of the studies selected was 50 bp, we considered telomeric reads as those in which the first 50 bp follow the (YYYTAAA)n pattern, even if they were longer. We denoted these reads or fragment of reads as 50-bp Telomeric Reads (50TRs).
Variable levels of telomeric DNA methylation are estimated from different bisulfite sequencing studies
Before studying the methylation status of Arabidopsis telomeres, we decided to evaluate the efficiency of telomeric C-rich strand generation after bisulfite treatment. We found that the production efficiency of the C-rich strand varied among the different bisulfite experiments. These efficiencies were calculated as the number of 50TRs divided by the total number of mapped reads and were expressed as percentages (Supplemental Table S1).
We next decided to determine two parameters to estimate the degree of cytosine methylation at telomeres: first, the percentage of unconverted cytosines within 50TRs, and second, the percentage of 50TRs that contained at least one unconverted cytosine. Our analyses revealed that the percentages of unconverted cytosines within 50TRs derived from wild-type Arabidopsis plants varied depending on the study analyzed (Fig. 2). These percentages ranged from 1.5% to 40%. Interestingly, the comparison of the different studies indicated that the percentage of unconverted cytosines varied with the telomeric C-rich strand production efficiency. High percentages of unconverted cytosines, from 5% to 40%, were detected when the telomeric C-rich strand production was < 8% × 10−4. In turn, the percentages of unconverted cytosines were between 1.5% and 5% when the telomeric C-rich strand production was > 8% × 10−4 (Fig. 2). In agreement with this observation, we noticed that the percentage of 50TRs containing cytosines also decreased when the production efficiency of the C-rich strand increased (Fig. 2). Hence, the levels of cytosine methylation estimated at telomeres decreased with the production efficiency of the telomeric C-rich strand.
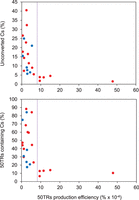
Telomeric DNA methylation estimates vary with the telomeric reads' production efficiency. Data from different genome-wide bisulfite sequencing experiments were analyzed. The upper panel shows the scatter plot representation of the percentage of unconverted cytosines (Cs) detected within 50TRs (estimated methylation levels) versus the telomeric C-rich strand production efficiency. Production efficiencies were calculated as the number of 50TRs divided by the total number of mapped reads and expressed as percentages. The lower panel shows the scatter plot representation of the percentage of 50TRs containing cytosines versus the telomeric C-rich strand production efficiency. Data obtained from wild-type experiments are represented as red circles and data obtained from DNA methylation mutants are represented as blue circles. These mutants include single (met1-3, cmt2, ddm1-2, drd1-7), double (met1-3 and cmt3-11, ddm1-2 and drd1-7), and triple (drm1-2, drm2-2 and cmt3-11) mutants.
Experimental limitations of the bisulfite technique lead to overestimation of telomeric DNA methylation
To determine whether the anti-correlation detected between production efficiency and cytosine unconversion was also observable at other genomic loci, we assessed both variables in mitochondrial DNA. We detected higher levels of unconverted mitochondrial cytosines in experiments with lower mitochondrial read production efficiencies (Fig. 3A). In addition, we also observed higher levels of unconversion in cytosines with lower read coverage in all the experiments analyzed (e.g., Fig. 3B). These results lead us to conclude that the methylation levels of telomeres should be better estimated from the experiments with the high telomeric C-rich strand production efficiencies.
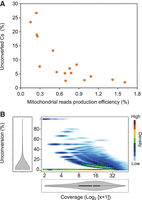
Mitochondrial DNA methylation estimates vary with the mitochondrial reads' production efficiency. Data from different genome-wide bisulfite sequencing experiments were analyzed. (A) Scatter plot representation of the percentage of unconverted cytosines (Cs) within mitochondrial reads versus the mitochondrial read production efficiency in wild-type plants. The production efficiencies were calculated as the number of mitochondrial reads divided by the total number of mapped reads. They are expressed as percentages. (B) Density plot representation of unconversion versus coverage for all the mitochondrial cytosines analyzed in experiment SRR578938 (Zemach et al. 2013). The distribution of unconversion and coverage is represented in both axes.
Before further analyzing the methylation status of Arabidopsis telomeres, we decided to investigate telomeric production efficiency and cytosine unconversion in DNA methylation mutants. We analyzed several mutants affected in CG, CHG, and CHH methylation. These mutants were altered in DNA methyltransferases or chromatin remodeling proteins. Among the DNA methyltransferase mutants were met1-3, cmt2, a double met1-3, cmt3-11 mutant, and a triple drm1-2, drm2-2, cmt3-11 mutant. Among the chromatin remodeling mutants were ddm1-2, drd1-7, and a double ddm1-2, drd1-7 mutant. Previous results have shown that DNA methylation is affected at a genome-wide level in all these mutants. Whereas CG methylation is catalyzed by MET1, non-CG methylation is catalyzed by CMT3 (CHG) and DRM1, DRM2 or CMT2 (CHH and also CHG). These DNA methyltransferases access DNA with the help of chromatin remodeling proteins like DDM1 and DRD1 (Vongs et al. 1993; Finnegan et al. 1996; Ronemus et al. 1996; Jeddeloh et al. 1999; Bartee et al. 2001; Lindroth et al. 2001; Cao and Jacobsen 2002a,b; Law and Jacobsen 2010; Stroud et al. 2013; Zemach et al. 2013).
Not surprisingly, we found that the percentage of unconverted cytosines in 50TRs and the percentage of 50TRs containing cytosines detected in all the methylation mutants varied with the telomeric C-rich strand production efficiency (Fig. 2). Hence, these results confirmed that the methylation status of telomeres is confounded by experimental limitations of the bisulfite technique.
The double met1-3, cmt3-11, and drm1-2, drd1-7 mutants have been shown to lack significant levels of CHH methylation. However, we detected telomeric DNA methylation in these mutants, which revealed that their telomeric DNA was not fully converted by bisulfite. To further explore conversion efficiencies, we performed bisulfite sequencing experiments using a synthetic nonmethylated telomeric DNA template. We used two different temperatures (54°C and 40°C) during the bisulfite treatment step. The percentage of unconverted cytosines detected in these experiments was ∼1% at 54°C and 2.3% at 40°C, revealing that bisulfite conversion was more effective at higher temperature (Fig. 4A). These data demonstrate that the experimental conditions influence the degree of telomeric cytosine conversion.
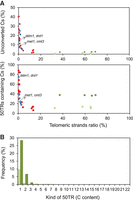
A synthetic nonmethylated telomeric DNA template is not fully converted during bisulfite treatment. (A) Scatter plot representations of the percentages of unconverted cytosines (Cs) within 50TRs and of 50TRs containing cytosines versus the telomeric strands ratio. Ratio values were calculated as the number of 50TRs divided by the number of reads that follow the TTTAGGG pattern along the first 50 bp. They were expressed as percentages. Whole-genome bisulfite sequencing studies performed with the Arabidopsis wild-type strain (red dots) and with DNA methylation mutants (blue dots) were analyzed. Similarly, bisulfite sequencing experiments performed with a 105-bp-long telomeric synthetic fragment at 54°C (light green dots) or at 40°C (dark green dots) were also analyzed. Data corresponding to the met1-3, cmt3-11 and ddm1-2, drd1-7 double mutants are indicated. (B) Bar plot representation showing the distribution of cytosines within 50TRs derived from the synthetic telomeric fragment treated with bisulfite at 54°C (light green bars) or at 40°C (dark green bars). The percentages of 50TRs containing different numbers of cytosines are represented. Average values corresponding to four independent experiments are shown for each temperature.
We next wanted to address why high levels of cytosine methylation were estimated from some of the Arabidopsis bisulfite sequencing studies. Two different kinds of 50TRs could be detected in all the Arabidopsis studies according to their cytosine content: those with a low content of cytosines, and those with a high content (Fig. 5A). This kind of distribution has been previously observed after bisulfite treatment of nonmethylated phage DNA (Guo et al. 2013). Although phage reads with few cytosines were proposed to originate from random bisulfite conversion failure or sequencing errors, phage reads with high content of cytosines were thought to arise from secondary structure formation and overall failure of bisulfite conversion (Guo et al. 2013).
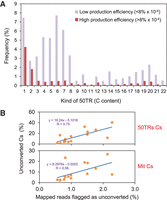
Low levels of converted reads underlie the overestimation of cytosine methylation. (A) Bar plot representation showing the distribution of cytosines within 50TRs. The percentages of wild-type 50TRs containing different numbers of cytosines are represented. Low ( < 8% × 10−4) and high ( > 8% × 10−4) telomeric C-rich strand production efficiency experiments were grouped and their average percentages were independently represented. (B) Scatter plot representation of the percentage of unconverted cytosines detected within 50TRs or mitochondrial reads versus the percentage of mapped reads flagged as potentially unconverted. Potential unconverted reads were flagged by BS-Seeker2 as follows: #(mCH sites) > 5 and [#(mCH sites)/#(all CH sites)] > 0.5. P-values for 50TRs and mitochondrial reads are 0.0008 and 0.0241, respectively.
We observed that the low telomeric C-rich strand production experiments generated higher percentages of 50TRs with a high content of cytosines than the high production experiments (Fig. 5A). These results support the hypothesis that the low production efficiency experiments might be related, at least in part, to secondary structure formation and overall failure of bisulfite conversion of the Arabidopsis telomeric DNA. Indeed, we found a strong positive correlation between the percentage of unconverted cytosines detected within 50TRs or mitochondrial reads and the percentage of unconverted reads using BS-Seeker2 (Fig. 5B; Guo et al. 2013).
We next decided to analyze the production efficiency of the telomeric template DNA strands. We found that the telomeric C-rich strand was produced with lower efficiency than the G-rich strand (Fig. 4A). In addition, we also found this telomeric strand imbalance in the experiments performed with Arabidopsis genomic DNA, although it was more pronounced in this latter case. Although the levels of unconverted cytosines at Arabidopsis telomeres were lower in experiments with lower strand imbalance, the levels of unconverted cytosines detected for the synthetic template did not decrease with the ratio of the telomeric strands (Fig. 4A). These results could be explained if we assume that the telomeric template had a lower tendency to remain double stranded or to form secondary structure than the Arabidopsis telomeric fragments during bisulfite experiments. This assumption is supported by the fact that unconverted cytosines do not tend to accumulate within the same telomeric template reads and by the virtual absence of telomeric template reads with a high density of cytosines (Fig. 4A,B). The nature of the synthetic telomeric template could have influenced its capability to remain double stranded or to form secondary structures as it was synthetized by annealing two telomeric oligos and, most importantly, was shorter (105 bp) than the Arabidopsis DNA fragments analyzed in the whole-genome bisulfite sequencing studies (typically several hundred bp).
We found that cytosines within mitochondrial DNA regions with high CG density had a higher tendency to be unconverted and, to a lesser extent, uncovered than cytosines within low CG density regions (e.g., Fig. 6A). This tendency was particularly evident in experiments with high levels of total mitochondrial unconverted cytosines (Fig. 6B). It will be interesting to ascertain whether high CG density regions have a high propensity to remain as double-stranded DNA or to form noncanonical secondary structures during bisulfite experiments.
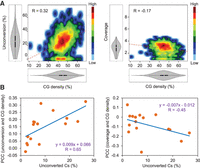
High CG density challenges cytosine conversion and coverage of mitochondrial DNA. Data from different genome-wide bisulfite sequencing experiments were analyzed. (A) Density plot representations of cytosine unconversion or coverage versus the percentage of CG residues within 100-bp tiles (CG density). All the mitochondrial cytosines from experiment SRR492932 (Dowen et al. 2012) were grouped in 100-bp tiles and analyzed. The distributions of unconversion, coverage, and CG densities are represented in both axes. Pearson correlation coefficients (R) are also indicated. (B) Scatter plot representations of Pearson correlation coefficients between cytosine unconversion or coverage and CG density per 100-bp tiles (PCC values) versus the mean mitochondrial methylation estimated in every experiment (percentage of unconverted Cs). P-values for cytosine unconversion and coverage are 0.0064 and 0.0802, respectively.
High-confidence data reveal the absence of DNA methylation in Arabidopsis telomeres
As mentioned above, the experiments with high telomeric C-rich strand production efficiencies should better represent the methylation status of Arabidopsis telomeres. Indeed, these experiments revealed that Arabidopsis telomeres are not methylated or undergo very low levels of DNA methylation (Fig. 2). We focused on the only high C-rich strand production efficiency study analyzed here that generated reads longer than 50 bp (Supplemental Table S1). The Sequence Read Archive accession number of this experiment is SRR578938 (Zemach et al. 2013). The percentages of unconverted cytosines within 50TRs and of 50TRs containing cytosines detected in SRR578938 were 1.5% and 10%, respectively (Fig. 2). Hence, the levels of telomeric cytosine methylation estimated from SRR578938 are within the technical background levels achieved for the whole genome in bisulfite experiments (Lister et al. 2008).
Since the reads from SRR578938 were 100 bp in size, we decided to use the complete sequence of all the 50TRs from this experiment to map their genomic origin. We found that all the 50TRs mapped to telomeres, to ITSs, or to the left telomere-subtelomere junction of Chromosome I (Fig. 7; Supplemental Fig. S1). Most of the telomeric reads detected lacked cytosines along their entire length (Fig. 7; Supplemental Fig. S1). In contrast, the reads corresponding to ITSs or to the telomere-subtelomere junction of Chromosome I exhibited high levels of cytosines, which is in agreement with the high levels of DNA methylation previously reported for these loci (Fig. 7; Cokus et al. 2008; Vrbsky et al. 2010; Vaquero-Sedas et al. 2011, 2012; Ogrocká et al. 2014). Thus, the analysis of the complete 100-bp reads from SRR578938 confirms the virtual absence of DNA methylation in Arabidopsis telomeres.

A high-confidence experiment reveals the absence of telomeric DNA methylation. The complete 100-bp sequences of all the 50TRs from SRR578938 (Zemach et al. 2013) containing one or more cytosines are displayed, together with the complete sequences of some of the 50TRs lacking cytosines. The number of cytosines within 50TRs and the genomic origin estimated for all the reads are indicated in blue. Among the 222 50TRs identified in SRR578938, 199 did not contain cytosines (see Supplemental Fig. S1), 19 contained one or two cytosines, and four contained five cytosines or more. The parts of the reads that follow a continuous perfect telomeric (YYYTAAA)n pattern are shown in black with the exception of cytosines that are labeled red. The remaining bases are shown in green and have been used for mapping purposes. All the reads could be mapped to the TAIR10 database except one, which mapped to the left telomere-subtelomere junction of Chromosome I (Kuo et al. 2006). Note that all the 50TRs lacking cytosines (Supplemental Fig. S1) and those containing one or two cytosines follow a quasi-perfect telomeric (YYYTAAA)n pattern along their entire 100-bp length. Most of these reads (218 of 222) should originate from telomeres. The 50TR containing 19 cytosines also follows the perfect telomeric pattern along its entire 100-bp length and should be telomeric and originate from experimental limitations of the bisulfite technique or originate from the 300-bp ITS. The 50TRs that contain more than five cytosines originate from ITSs or from the left telomere–subtelomere junction of Chromosome I (IL). Since the frequency of these 50TRs (3 of 222; 1.4%) is expected according to the distribution of ITSs and telomere–subtelomere junctions in the Arabidopsis genome, their cytosines might reflect real methylation events.
Restriction enzyme sensitivity analyses confirm the absence of DNA methylation in Arabidopsis telomeres
To verify our in silico analyses, we decided to study DNA methylation at Arabidopsis telomeres using an alternative technique. We performed Southern blot hybridization experiments using three restriction enzymes with different DNA methylation dependence/sensitivity. One of the enzymes (HpaII) recognizes and cuts the sequence CCGG only when both cytosines are unmethylated. The other two enzymes (McrBC and FspEI) recognize methylated cytosines and cut the surrounding nucleotides. McrBC recognizes the sequence RmC(N40-3000)RmC and should target the first cytosine of the telomeric repeat unit (CCCTAAA) if it were methylated. FspEI recognizes the sequence CmC and should target the second and third cytosines of the telomeric repeat unit if they were methylated (Fig. 8A).
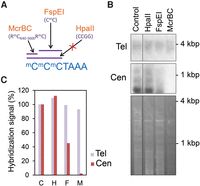
Methylation-dependent restriction enzyme analyses confirm the absence of telomeric DNA methylation. (A) Cartoon representing the sequence specificity of the restriction enzymes used to digest Arabidopsis genomic DNA. (B) Southern blot hybridizations of Arabidopsis genomic DNA digested with the restriction enzymes indicated in A. The upper panel shows equal amounts of undigested (Control) or digested (HpaII, FspEI, or McrBC) DNA samples hybridized with a telomeric probe. The four DNA samples were also digested with Tru9I prior to hybridization. The middle panel shows the hybridization of the same samples with a 180-bp centromeric repeat probe. The ethidium bromide staining of the samples is shown in the lower panel. The control DNA sample was run in the same gel as the rest of the samples, so that their corresponding hybridization signals were processed equally. (C) Bar plot representation of the telomeric and centromeric bottom band hybridization signals expressed as percentages of the control. C, H, F, and M represent Control, HpaII, FspEI, and McrBC, respectively.
We digested equal amounts of Arabidopsis genomic DNA with the restriction enzyme Tru9I or with Tru9I plus one of the enzymes mentioned above. We then ran the resulting DNA samples on an agarose gel and hybridized them with a telomeric probe or with a centromeric probe. Tru9I recognizes and cuts the sequence TTAA, which localizes in subtelomeric regions at ∼200 bp from the perfect telomeric repeats, on average. Therefore, this enzyme cuts the Arabidopsis genome frequently, but leaves telomeres essentially uncut. We observed a 2- to 5-kbp fuzzy telomeric band after digesting Arabidopsis genomic DNA with Tru9I, as previously reported (Richards and Ausubel 1988; Fitzgerald et al. 1999; Vaquero-Sedas et al. 2011). This telomeric band remained unaffected after digestion with HpaII, FspEI, or McrBC (Fig. 8B,C). In contrast, three centromeric DNA bands generated after Tru9I digestion were readily digested with FspEI and McrBC, as would be expected due to the high levels of DNA methylation previously observed for the centromeric repeats. As expected, these bands remained unaffected in the presence of HpaII (Vongs et al. 1993; Lindroth et al. 2001). Although these results cannot completely rule out the existence of a very minor portion of methylated telomeres, we should consider that a single cut within a telomeric fragment should shorten this fragment. In addition, the very low proportion of 50TRs containing one or two unconverted cytosines that we have observed in the wild-type high production efficiency experiments should be better explained by experimental limitations of the bisulfite technique, as should be the 50TRs containing unconverted cytosines observed in the experiments performed with the DNA methylation mutants lacking CHH methylation. Thus, taken together our results support that DNA methylation is essentially absent from Arabidopsis telomeres.
Discussion
Limitations of the bisulfite sequencing technique
Although bisulfite sequencing is probably one of the most accurate techniques to analyze cytosine methylation, some problems can arise during the bisulfite treatment. We believe that these problems may explain the very high levels of cytosine detection that we found at Arabidopsis telomeres in experiments with low telomeric C-rich strand production efficiencies. If some unmethylated cytosines do not react with bisulfite, they will be detected as methylated cytosines leading to overestimation of the methylation events. Thus, overestimation of telomeric methylation should be influenced by the degree of telomeric DNA denaturation or the formation of telomeric C-rich strand secondary structures like the i-motif (Leroy et al. 1994; Fernández et al. 2011; Guo et al. 2013). In addition, since bisulfite treatment modifies DNA and even degrades it (Frommer et al. 1992; Grunau et al. 2001), some unmethylated cytosines might partially or improperly react with bisulfite reagents leading to the generation of aberrant cytosines. These aberrant cytosines might still be recognized as cytosines but undergo amplification with less efficiency than uracil during the first cycle of PCR. Alternatively, these aberrant cytosines might be amplified efficiently during the first cycle of PCR but incorrectly copied. Such phenomena might explain the anti-correlation observed for the telomeric C-rich strand production efficiency and the unconversion of cytosines at Arabidopsis telomeres.
The third cytosine of the Arabidopsis telomeric repeat unit has been found to be unconverted more frequently than the first and second cytosines after bisulfite treatment (Cokus et al. 2008). This cytosine is unconverted more frequently than the first and the second in the 50TRs originating from the double met1-3, cmt3-11 and ddm1-2, drd1-7 mutants, which lack significant levels of asymmetric DNA methylation (Supplemental Fig. S2; Stroud et al. 2013; Zemach et al. 2013). Hence, the lower conversion rate of this cytosine should result from experimental limitations like those mentioned above. The third cytosine of the Arabidopsis telomeric repeat unit is also unconverted more frequently than the first and second cytosines in subtelomeric regions and ITSs of wild-type plants (Fig. 7; Cokus et al. 2008; Vrbsky et al. 2010; Ogrocká et al. 2014). However, these cytosines reflect real methylation events. Thus, taken together, our results suggest a special DNA conformation of the telomeric repeat arrays.
We hypothesize that incomplete telomere denaturation and/or telomeric secondary structure could prevent bisulfite conversion, enhance bisulfite driven DNA breakage, and/or alter the efficiency/accuracy of the first cycle of PCR amplification after bisulfite treatment. On the other hand, the efficient denaturation of telomeres and the absence of telomeric secondary structures could favor cytosine conversion, although the conversion efficiencies achieved depend on different experimental parameters, such as the temperature of incubation with bisulfite.
Telomeres are refractory to de novo DNA methylation
Our results indicate that DNA methylation is absent from Arabidopsis telomeres. However, subtelomeric regions display a high level of cytosine methylation, which is in agreement with previously published results (Fig. 9A; Vrbsky et al. 2010; Vaquero-Sedas et al. 2011; Ogrocká et al. 2014).
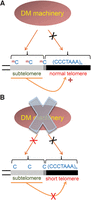
Model representing the influence of DNA methylation on the homeostasis of telomere length. (A) The DNA methylation machinery of wild-type plants can target subtelomeric regions (gray bar) and telomere-subtelome junctions (dark gray bar) but not telomeres (black bar). In this scenario, subtelomeric DNA methylation contributes to positively regulate telomere length. (B) The homeostasis of telomere length is lost when subtelomeric DNA methylation cannot be achieved due to the impairment of the DNA methylation machinery.
Small interfering RNAs (siRNAs) can target DNA methylation in all sequence contexts through the RNA-directed DNA Methylation pathway (RdDM). This pathway involves multiple proteins including DNA and histone methyltransferases, chromatin remodeling proteins, RNases, and DNA or RNA polymerases (Law and Jacobsen 2010). Plants have two specific DNA polymerases involved in RdDM: pol IV and pol V. Pol IV together with the RNA dependent RNA polymerase 2 (RDR2) and the Dicer-like RNase 3 (DCL3) participates in the generation of 24-bp siRNAs. In turn, pol V, the siRNA single strand binding protein Argonaute 4 (AGO4) and the non-CG Domains Rearranged DNA methyltransferase 2 (DRM2) are required for siRNA-dependent DNA methylation. The RdDM pathway is involved in de novo DNA methylation. Through this process, siRNAs containing a specific nucleotide sequence are believed to drive DRM2-dependent DNA methylation of the same DNA sequence at different genomic loci. For example, 24-bp siRNAs containing perfect telomeric repeats have been described in Arabidopsis (Gustafson et al. 2005; Vrbsky et al. 2010). These siRNAs drive RDR2, DCL3, AGO4, and DRM2-dependent DNA methylation of telomeric repeats at ITSs and subtelomeres (Cokus et al. 2008; Vrbsky et al. 2010; Stroud et al. 2013). However, we have shown here that the RdDM pathway cannot target DNA methylation at telomeres, which probably reflects their special architecture (Fig. 9A).
Subtelomeric DNA methylation controls telomere length homeostasis
We have previously reported that Arabidopsis telomeres have lower levels of DNA methylation than ITSs or subtelomeres (Vaquero-Sedas et al. 2011). Here, we have described that DNA methylation is indeed absent from Arabidopsis telomeres. Notably, Arabidopsis mutants affecting CG and non-CG DNA methylation have shorter telomeres than wild-type plants (Ogrocká et al. 2014; Vaquero-Sedas and Vega-Palas 2014). Therefore, DNA methylation must be involved in the control of telomere length homeostasis in Arabidopsis. Since the expression of proteins that regulate telomere length have not been found to be altered in the aforementioned mutants (Zhang et al. 2006; Hudson et al. 2011), we conclude that subtelomeric DNA methylation positively controls the homeostasis of telomere length in Arabidopsis (Fig. 9). In contrast, DNA methylation controls telomere length negatively in mammals (Gonzalo et al. 2006). Thus, long telomeres are observed in mammalian DNA methylation mutants. However, these telomeres might arise after telomeric shortening, as previously reported for the Saccharomyces cerevisiae est1 (ever shorter telomeres 1) survivors (Lundblad and Blackburn 1993). In addition, it is unknown whether the control of telomere length exerted by DNA methylation in mammals occurs at telomeres or subtelomeres (Blasco 2007).
Cytosine methylation has been related to a wide variety of biological features from the maintenance of genome stability to cell differentiation, development, or illness. Many of these features are also affected by telomere length (Osborne and Boubriak 2002; Ooi et al. 2009; Law and Jacobsen 2010; Guy et al. 2011; Bodelon et al. 2014; Heard and Martienssen 2014; Plasschaert and Bartolomei 2014). Hence, it will be interesting to ascertain the relevance of subtelomeric DNA methylation in all these processes in mammals and plants.
Methods
In silico analysis of telomeric DNA methylation
Data from different genome-wide bisulfite sequencing studies were analyzed (Schmitz et al. 2011; Ausin et al. 2012; Dowen et al. 2012; Moissiard et al. 2012; Stroud et al. 2012, 2013, 2014; Zhong et al. 2012; Greenberg et al. 2013; Law et al. 2013; Zemach et al. 2013). Since the original Watson and Crick strands, but not their reverse complements, were sequenced in these studies, only the telomeric C-rich strand was monitored to estimate methylation levels. Therefore, the telomeric G-rich strand did not influence the methylation analyses. The Gene Expression Omnibus and the Sequence Read Archive accession numbers of the studies are indicated in Supplemental Table S1, together with the codes of the specific runs analyzed.
Considering that the length of the reads in many of the studies selected was 50 bp, we decided to start by studying only the first 50 bp of the reads in all the experiments even if they were longer. In principle, we considered telomeric reads as those that followed the (YYYTAAA)n pattern, where Y is C or T, along the 50 bp. These sequences could start at any nucleotide (C, T, or A) and corresponded to about seven methylated/unmethylated tandem telomeric repeats. We denoted these reads/fragment of reads as 50-bp Telomeric Reads (50TRs).
To verify that the 50TRs were generated essentially from telomeres, we determined the number of (YYYTAAA)7 nonoverlapping sequences in the Arabidopsis thaliana Col-0 genome, including ITSs and telomeres. First, we determined the number of these sequences at ITSs. We used the PatMatch tool in The Arabidopsis Information Resource (TAIR; http://www.Arabidopsis.org) and searched for the sequence (YYYTAAA)7 in the TAIR10 Whole-Genome (BAC clones) database, which contained 135,670,227 bytes. This search was performed using the default options that included searching in both strands with no mismatches allowed. The search resulted in 41 hits, from which only 31 were not repeated in different BAC clones. Six of the unique hits corresponded to sequences that were organized in tandem at the pericentromeric region of Chromosome III generating a 300-bp ITS composed of perfect tandem telomeric repeats. In addition, six of the hits were telomeric (five from pAtT51 and one from TEL3N). An alignment of the remaining 19 and the right border of the 300-bp ITS is shown in Figure 1. This alignment reveals that none of the 19 (YYYTAAA)7 aforementioned sequences are included in a DNA string containing more than 11 repeats.
Once we determined the number of ITSs containing the (YYYTAAA)7 sequence, we estimated the number of times that this sequence is found at telomeres. We estimated that telomeres contained about 765 nonoverlapping (YYYTAAA)7 sequences (3750 bp of average telomere length divided by 7 bp of the telomeric repeat unit and multiplied by 10 telomeres). Therefore, only ∼3% of the (YYYTAAA)7 sequences are present at ITSs (25 divided by 765 and multiplied by 100). However, the percentage of 50TRs corresponding to ITSs in the bisulfite studies should be lower than 3% as the size of the DNA inserts used to construct the DNA libraries are typically 150–300 bp in size. Similarly, a low percentage of the 50TRs should correspond to the telomere–subtelomere junctions, which usually contain perfect telomeric repeats interspersed with degenerate repeats. In summary, if the bisulfite experiments work properly, we expect that at least 97% of the 50TRs correspond to telomeres.
To perform the analyses of cytosine methylation at telomeres we used an in-house computer program that allowed the identification of 50TRs, the determination of the number of unconverted cytosines within 50TRs, and the number of reads that followed the (TTTAGGG)n pattern along the first 50 bp (Supplemental Table S1, Supplemental Telomeric analyses source code).
In silico analysis of mitochondrial DNA methylation
Reads for the aforementioned Arabidopsis data sets were downloaded from the GEO repository and were mapped to the entire TAIR10 reference genome without allowing mismatches using BS-Seeker v2.0.32 (Guo et al. 2013), which calls Bowtie v0.12.9 (Langmead et al. 2009) in turn. Reads were not filtered for poor conversion prior to base calling. Counts of C and T base calls per mitochondrial cytosine for both strands were extracted from the resulting CG map files. Subsequent statistical analysis and plots were generated using a custom R environment (R Core Team 2008).
Telomeric template synthesis, bisulfite treatment, library preparation, and next generation sequencing
Equal amounts of oligos containing the sequences (TTTAGGG)15 and (CCCTAAA)15 were mixed, heated for 10 min to 98°C and allowed to cool down for 15 min at room temperature. Then, the annealed DNA fragments were gel purified and their quality assessed using the D1000 assay on the TapeStation 2200 (Agilent Technologies). End repair, A-tailing, and adapter ligation was performed on the annealed oligos according to the Illumina TruSeq Nano kit protocol (catalog #15041759, Illumina). Adapter-ligated oligos were denatured in the presence of bisulfite (Zymo Lightning MagPrep, catalog #D5046, Zymo Research) for 8 min at 98°C, and two temperatures were used for the conversion step: 54°C (manufacturer's suggested temperature) and 40°C. Desulfonation was performed according to the manufacturer's instructions. Converted DNA was amplified using MyTaq Mix (Bioline, Taunton) and Illumina TruSeq PCR Primer Cocktail according to the following protocol: initial denaturation for 30 sec at 98°C; 12 cycles of 15 sec at 98°C, for 30 sec at 60°C, for 30 sec at 72°C; final extension for 5 min at 72°C. AMPure XP beads were used to clean up the final PCR reactions before sequencing. Libraries were sequenced with an Illumina HiSeq 2500 system using 100-bp single-end reads.
Methylation-dependent restriction enzyme analyses of telomeric DNA methylation
Equal amounts of genomic DNA (500 ng) were undigested or digested with the restriction enzymes HpaII, FspEI, or McrBC following the supplier's instructions (New England Biolabs). The resulting DNA samples were purified, further digested with Tru9I, resolved on an agarose gel, transferred to a Hybond-N+ membrane, and hybridized with a telomeric probe, as previously described (Gámez-Arjona et al. 2010). Then, the same membrane was stripped and hybridized again with a 180-bp centromeric repeat probe. The 180-bp centromeric template was synthetized as previously reported (Nagaki et al. 2003).
Data access
Illumina bisulfite sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE83510.
Acknowledgments
We thank Suhua Feng for critical reading of the manuscript and NCBI and TAIR for sharing their resources.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.202465.115.
- Received November 26, 2015.
- Accepted June 20, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








