
Genome-Wide Epistatic Interaction Analysis Reveals Complex Genetic Determinants of Circadian Behavior in Mice
Abstract
Genetic heterogeneity underlies many phenotypic variations observed in circadian rhythmicity. Continuous distributions in measures of circadian behavior observed among multiple inbred strains of mice suggest that the inherent contributions to variability are polygenic in nature. To identify genetic loci that underlie this complex behavior, we have carried out a genome-wide complex trait analysis in 196 (C57BL/6J X BALB/cJ)F2 hybrid mice. We have characterized variation in this panel of F2 mice among five circadian phenotypes: free-running circadian period, phase angle of entrainment, amplitude of the circadian rhythm, circadian activity level, and dissociation of rhythmicity. Our genetic analyses of these phenotypes have led to the identification of 14 loci having significant effects on this behavior, including significant main effect loci that contribute to three of these phenotypic measures: period, phase, and amplitude. We describe an additional locus detection method, genome-wide genetic interaction analysis, developed to identify locus pairs that may interact epistatically to significantly affect phenotype. Using this analysis, we identified two additional pairs of loci that have significant effects on dissociation and activity level; we also detected interaction effects in loci contributing to differences of period, phase, and amplitude. Although single gene mutations can affect circadian rhythms, the analysis of interstrain variants demonstrates that significant genetic complexity underlies this behavior. Importantly, most of the loci that we have detected by these methods map to locations that differ from the nine known clock genes, indicating the presence of additional clock-relevant genes in the mammalian circadian system. These data demonstrate the analytical value of both genome-wide complex trait and epistatic interaction analyses in further understanding complex phenotypes, and point to promising approaches for genetic analysis of such phenotypes in other mammals, including humans.
Circadian rhythms represent a complex set of phenotypes for genetic analysis. Such rhythms, present in a broad array of biological processes and taxa, are endogenously generated. These cycles persist in the absence of exogenous time cues with a period close, but rarely equal to, 24 h. In the presence of a daily light-dark cycle, these circadian rhythms are synchronized (entrained) to a 24-h period. The precise nature of the time-keeping mechanism itself remains unclear, but defining work from the modern field's inception during the mid-twentieth century demonstrated a number of unique characteristics of circadian clocks, including their self-sustaining nature, their ability to be precisely synchronized by the environmental light-dark cycle, and the persistence of similar formal properties among diverse organisms. These initial studies also demonstrated that the clock's action is genetic in origin, not relying on the rhythmic nature of the environment for its daily oscillation.
Molecular genetic analysis of circadian rhythms, beginning with the identification of the period (per) gene inDrosophila melanogaster (Konopka and Benzer 1971; Bargiello et al. 1984; Zehring et al. 1984), has met with undeniable success in identifying genes in this timekeeping mechanism. Analysis of theper gene's temporal expression profile led researchers to propose a framework model for circadian timekeeping, based on a negative feedback loop of per transcription and translation (Siwicki et al. 1988; Hardin et al. 1990, 1992). This general model has proven to be quite broad in its applicability, with evidence for transcription/translation feedback loop clocks extending from cyanobacteria to mammals.
A mammalian feedback loop model has been developed recently, concurrent with the rapid identification of at least nine genes proposed to be involved in mammalian circadian rhythms (Bunger et al. 2000; King and Takahashi 2000; Lowrey et al. 2000). These genes have been identified in a multitude of ways—by deliberate or serendipitous mutations (the mouse Clock and hamster tau [CKIɛ] genes, respectively), by molecular interactions with other clock genes (the Bmal1 gene), and by homology to clock or photoreceptor genes from other organisms (the mouse Period, Timeless, and Cryptochrome homologs, mPer1, mPer2, mPer3, mTim, mCry1, andmCry2). Four of these genes, mPer2, mCry1, mCry2, andBmal1, have been linked to circadian function through analysis of gene-targeting mutations.
As more circadian genes have been characterized, and as the model of the mammalian circadian clock has been elaborated, it has become evident that multiple functional interactions between molecules are necessary for the generation and regulation of mammalian circadian rhythms. Among these include the dimerization and transactivational capabilities of CLOCK and BMAL1 (a positive aspect of the feedback loop) (Gekakis et al. 1998), and the PER–TIM and PER–CRY interactions and their inhibition of the CLOCK–BMAL1 transactivation (negative aspects of the feedback loop) (Sangoram et al. 1998; Kume et al. 1999). In addition, CKIɛ phosphorylates the PER proteins, leading to their destabilization within the cell (Keesler et al. 2000; Lowrey et al. 2000). Evidence for interaction is also apparent at a phenotypic level; for example loss-of-function mutants of the two Cryptochrome genes have opposite effects on circadian period, but in combination produce a novel, striking phenotype: arrhythmicity (van der Horst et al. 1999; Vitaterna et al. 1999). The emerging picture of the mammalian circadian clock mechanism is clearly one of multiple and complex genetic interactions.
Such complexity might have been predicted in hindsight. Although a transcriptional feedback loop with a limited number of components may be sufficient to generate a rudimentary circadian rhythm (Leloup and Goldbeter 1998; Scheper et al. 1999), a variety of other proteins are likely to be involved in refining and amplifying this basic circadian oscillation. This could be achieved, for example, by proteins regulating the stability of RNA (So and Rosbash 1997) or restricting nuclear entry of clock proteins to specific times of the circadian day (Curtin et al. 1995). The complex nature of circadian behavior is reflected in the phenotypic variability observed in mammalian species; this has been described particularly well in mice. Significant differences in circadian behavior have been demonstrated when comparing multiple inbred strains of mice (Ebihara et al. 1978; Possidente and Hegmann 1982; Schwartz and Zimmerman 1990). In particular, when endogenous period has been measured, differences of nearly a full hour have been observed among the most divergent strains. For example, the BALB/c strain has a short period, <23 h, whereas the C57BL/6 strain has a period close to 24 h. Strain differences are also present in other aspects of circadian behavior, including entrainment to light-dark cycles and the robustness of the rhythm. Crosses among such inbred strains have indicated a polygenic basis for these differences in circadian behavior.
Furthermore, no comprehensive attempts have yet been made to identify all (or even most) mammalian circadian clock genes (e.g., by a recessive saturation mutagenesis screen). Thus, it is likely that many genes that underlie circadian rhythms remain to be discovered. Unlike the present understanding of the Drosophila circadian clock, in which all of the proposed genetic components have been characterized by mutations bearing effects on circadian behavior, many of the proposed components of the mammalian clock do not have demonstrated effects on circadian behavior. Identifying the nature or source of the genetic variability present within strains of mice provides an alternative means to illuminating underlying genetic contributions to this behavior.
In light of the evident polygenic underpinning to mammalian circadian behavior, and the uncertainty of the contributions of many of the proposed clock genes to this phenotype, we undertook a genome-wide quantitative trait mapping strategy using an F2 hybrid intercross between BALB/cJ and C57BL/6J mice to characterize loci in the mouse genome that contribute to variability of circadian behavior. A similar intercross strategy, limited to an analysis of circadian period, has led to the identification of loci significantly affecting this circadian behavior in the flowering plant, Arabidopsis thaliana (Swarup et al. 1999), demonstrating the usefulness of this technique. From our analysis of these hybrid mice, we have characterized several loci in the mouse genome having significant main effects on five specific aspects of circadian behavior.
Because of the genetic complexity of the circadian pacemaker, in particular the multiple molecular interactions required for its proper function, any analysis limited to treating each locus independently is not sufficient. In a complex system like this, for which the underlying genetic contributions are also likely to be complex, it is important to establish whether and how the loci contributing to phenotype interact. There are likely to be interactions among specific alleles at different loci that also contribute to the circadian phenotypes. Such epistatic interactions have been reported to contribute to the variability of other complex phenotypes, including diabetes and cancer susceptibility (Fijneman et al. 1996; Frankel and Schork 1996;Reifsnyder et al. 2000). We have investigated the role of gene interactions in circadian behavior by performing genome-wide pairwise interaction analyses. We have found that epistatic interactions have a pronounced, significant effect on this behavior. From our complete analysis (including both main effects and interactions), we have identified 14 loci that make significant contributions to multiple aspects of circadian behavior. To assess the novelty of these loci, we have also compared these map positions to the genetic locations of the nine proposed genes of the mammalian circadian clock, five of which are mapped here for the first time.
RESULTS
Inheritance of Circadian Rhythms in an Intercross between the C57BL/6J and BALB/cJ Inbred Mouse Strains
To collect our phenotypic data, we began by examining the distribution of circadian activity among mice from a two-generation intercross arranged between the C57BL/6J (B6) and BALB/cJ (BALB) strains. We produced a total of 196 F2 progeny for phenotypic and genetic analysis. We used a wheel-running assay of circadian behavior to determine the activity phenotypes of mice from this intercross. Mice exhibit a robust circadian rhythm of wheel running that is under direct control of the circadian pacemaker (Schwartz and Zimmerman 1990). From an analysis of this behavior we are able to extract multiple measures of circadian behavior. We focused specifically on five circadian traits which had been shown or were likely to differ significantly between the B6 and BALB parental strains: free-running period (period), phase angle of entrainment (phase), amplitude of circadian rhythmicity (amplitude), daily activity level (activity), and dissociation of the activity component (dissociation). The first four traits are quantitative, whereas dissociation is a qualitative trait. Previous studies of the BALB strain have described its characteristically short circadian period and the lability of its activity rhythm (Possidente and Stephan 1988). In contrast, the B6 strain is characterized by its longer circadian period, and the stability of its free-running rhythms. Figure1 shows typical activity records for the B6 (A), BALB (B) parental mice as well as for the F1(C) and F2 (D) generations. Table 1summarizes the mean phenotypic values and variance for mice from all generations of the intercross. In general, for each trait analyzed we observed significant differences between the parental strains, and these did not differ significantly from values published previously. In the F1 generation the distribution of phenotypic values indicated that all circadian traits were genetically determined, but also indicated that B6 haplotypes act dominantly over BALB to determine overt phenotypes in this hybrid background (Table 1, Figs.2, 3). As expected, the F2 generation showed greater variability in all observed phenotypes and it is consistent with the explanation of genetic variance. For all traits we also observed continuous distributions in the F2 generation (Figs. 2, 3) suggesting that the regulation of each is polygenic.
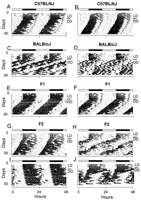
Representative wheel-running activity records of C57BL/6J, BALB/cJ, (BALB/cJ X C57BL6J)F1, and (BALB/cJ X C57BL6J)F2mice. Each record is for an individual mouse, double-plotted by convention so that a continuous 48 h are presented on each line, and each day's data is presented both beneath and to the right of that of the previous day. Times of activity are indicated by the vertical black marks. All animals were maintained on a 12 h light:12 h dark (LD) cycle for the first seven days shown, indicated by the bar above the record, followed by a transfer to continuous darkness (DD) begun at the usual lights-off time (17:00 Central standard time). (A) Female C57BL/6J; (B) male C57Bl/6J; (C) female BALB/cJ; (D) male BALB/cJ; (E) female F1; (F) male F1; (G) C57BL/6J-like F2; (H) BALB/cJ-like F2; (I,J) F2 mice with circadian behavior unlike C57BL/6J or BALB/cJ.
The Summary of the Mean Phenotypic Values for Mice from all Generations of the Intercross
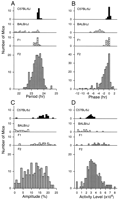
Frequency histogram of phenotypic measures for each generation. For each panel, the distribution histograms are presented in the following order from top to bottom: C57BL/6J, BALB/cJ, (BALB/cJ X C57BL/6J)F1, (BALB/cJ X C57BL/6J)F2.(A) The distribution of free-running period in constant darkness, as determined by χ2periodogram analysis of 20 days. Data is presented in 1/10 h bins. (B) The phase angle of entrainment, relative to the time of lights off. Values are plotted in 1/2 h bins, with positive values indicating activity onset preceding lights-off, and negative values indicate activity onset following lights-off. (C) The distributions of amplitude of circadian rhythmicity in constant darkness. Amplitude is estimated from the Fast Fourier Transform of 15 days' data in constant darkness. The total power from 0 to 1 cycles/hour is normalized to a value of 1.0, and the resultant relative power of the peak in the circadian range (18–30 h period) is taken as a measure of circadian amplitude. (D) The distributions of mean total daily activity levels. Values are individual mean numbers of wheel revolutions/24 h for 15 d in constant darkness.

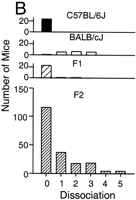
Phenotype distribution of dissociation trait. Dissociation of individual activity records were assigned to one of six classes of dissociation scores by two observers, with zero representing the least and five representing the most dissociated activity pattern. (A) Representative activity records of each dissociation score are presented. The plotting format is identical to Figure 1. (B) Distribution histograms of dissociation scores are presented in the following order from top tobottom: C57BL/6J, BALB/cJ, (BALB/cJ X C57BL/6J)F1, (BALB/cJ X C57BL/6J)F2.
Mapping Loci Underlying Complex Phenotypes in (C57BL/6J X BALB/cJ) F2 Hybrid Mice
In recent years, mapping complex phenotypes by quantitative trait locus (QTL) analysis has become an increasingly efficient technique for the genetic dissection of complex traits. Although a number of variants on the basic protocol that can increase power and reduce material costs have been proposed and successfully applied, including selective genotyping strategies (Lander and Botstein 1989) and preliminary analysis of pooled DNA samples (Michelmore et al. 1991; Asada et al. 1994), a more efficient strategy for our screen involved scanning the genomes of the entire (B6 X BALB) F2 population of 196 mice owing to our interest in mapping multiple phenotypes, each of which may have different segregation patterns. We did this with an array of 89 simple sequence length polymorphism (SSLP) markers spaced at intervals of ∼20 cM throughout the genome (Table2). In our initial analysis of linkage, we then used the program MAPMAKER/QTL to calculate the probability of linkage of each of these markers to differences in the five circadian phenotypes (Lander and Botstein 1989). For loci confirmed statistically with suggestive levels of significance (LOD≥2.8) (Lander and Kruglyak 1995), we then identified and genotyped multiple flanking markers within a 30 cM interval to search for stronger linkages. (This occasionally led to the association of multiple SSLPs with a single QTL.) With the combined data, we performed genome-wide single marker and pairwise scans using our own analysis tools (based on permutation testing; see Methods) (Churchill and Doerge 1994).
List of Markers Initially Used for Genome-Wide Scan
Free-Running Period
The period of oscillation is a key factor in describing an observed rhythm. The activity-rest behaviors of mice housed under a 24 h light-dark cycle (LD) entrain to the 24 h period of the lighting regime to which they are exposed. Once released into constant darkness, free of entrainment cues, each mouse will begin to free run with a period that soon reflects not the cycle to which it was formerly entrained, but the innate cycle of its own circadian pacemaker. Previous studies have indicated that B6 mice express circadian periods of activity that can range from 23.2 to 24.1 h (Schwartz and Zimmerman 1990). The BALB/c mouse, on the other hand, is notable for an extremely short period around a mean of 22.6 h (Possidente and Hegmann 1982). As anticipated from these previous reports, the free-running periods of the BALB parental mice observed in our study (22.88 h ± 0.602) were significantly shorter than those of the B6 mice (23.61 h ± 0.068,P<10−6). Figure 2A shows a histogram of this trait in each generation. Although the B6 population exhibited a distribution with little variance, we observed considerable variation among BALB mice. The initial outcross between B6 and BALB mice yielded F1 progeny with a distribution of periods that closely resembles that of the B6 parental strain, ranging from 23.2 to 23.7 h, indicating the presence of B6 alleles that regulate this trait in a dominant manner. Consistent with the divergent phenotypes of its founder strains, the F2 generation from this cross expressed a much wider period distribution, ranging from 22.5 to 24.7 h. We also observed significant differences (P<0.05) between male and female mice in the F2 generation (Table 1).
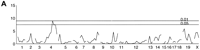
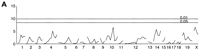
To detect loci with significant main effects, we computed one-way ANOVA F-statistics (with 2 and n-3 degrees of freedom [df]) at each marker in a genome-wide scan. Significance thresholds were estimated by the analysis of 1000 permutations of the original data. We recorded the maximal F value for each permuted data set and found the critical values to be 7.80 and 10.52 (for alpha = 0.05 and 0.01, respectively). This genome scan (Fig. 4A) identified significant associations with markers on chromosomes 4 and 5. Peaks occur at markers D4Mit178 (F = 13.46) and D5Mit30 (F = 9.80). We note that the chromosome 4 peak is sharp whereas the chromosome 5 peak extends over much of the chromosome. We named these loci Free-running period (Frp)-1 andFrp-2. The average free-running period for mice homozygous at the Frp-1 locus for the B6 allele (B/B) was 23.32 h, versus 23.49 h for heterozygotes (B/C) and 23.68 h for BALB homozygotes (C/C). Interestingly, the B6 allele at this locus is associated with shorter period lengths, whereas the BALB allele is associated with longer period lengths, which is in direct contrast to the period lengths observed in the parental strains. Effects of Frp-1 appear to be additive, such that each BALB allele adds about 10 min to the free-running period. The average free-running period for mice homozygous for the B6 allele (B/B) at Frp-2 was 23.39 h, versus 23.44 h for heterozygotes (B/C) and 23.68 h for BALB homozygotes (C/C), again contrary to strain behavior. The B6 allele appears to be dominant for period at the Frp-2 locus. Additional peaks are noted on chromosomes 12 and 18 in this genome scan.

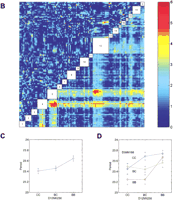
Genome-wide scan for free-running period phenotype. (A) Single marker regression genome scan for free-running period. Thevertical axis is the regression F-statistic (with 2 and n-3 degrees of freedom). Threshold values estimated from 1000 permutations of the trait data are indicated as horizontal lines at genome-wide significance levels of 0.05 and 0.01. The horizontal axis is indexed by the marker loci. Locations of markers are indicated but the relative spacing of the markers is not conserved in the graph. (B) Pairwise marker regression genome scans. The results of the pairwise search for loci associated with free-running period is shown. The x and y axes of the figure are genetic markers arranged in order along the length of the 20 mouse chromosomes. Joint analysis of markers on the same chromosome was not carried out because of numerical problems. Chromosomal locations of the markers are indicated in the empty boxes along the main diagonal of the figure. The color scale indicates the −log10 of the nominalP-value of the F-statistics. The primary scan F-statistic (with 8 and n-9 degree of freedom) is shown below the main diagonal and the secondary F-statistic for interaction (with 4 and n-9 d.f.) is shown above the diagonal. Significant locus pairs identified by the genome scans are listed in Table 3. (C) Main effect ofFrp-3 on free-running period. X-axis represents genotypes at Frp-3 locus. C represents BALB/cJ and B represents C57BL/6J allele. BC represents heterozygote at this locus.Y-axis is free-running period. (D) Pairwise interaction of Frp-2 and Frp-3 on free-running period. X-axis is for genotypes at Frp-2 locus.Y-axis is free-running period. Blue line is CC, green line for BC, and red line for BB at Frp-2 locus.
To detect epistatic interactions, we examined all pairwise combinations of marker loci for association with the trait in a two-dimensional genome scan (see Methods). We computed the two-way ANOVA F-statistic (F-all, with 8 and n-9 df) for every marker pair. The F-all statistic is our primary screen for identifying interesting locus pairs. Significance thresholds of 5.10 and 6.03 (for alpha = 0.05 and 0.01, respectively) were determined by analysis of 500 permuted data sets. These critical values account for the large number of pairs tested in the genome-wide search. To determine if a locus pair detected by the F-all statistics is either (1) two interacting QTL, (2) two additive main effects, or (3) an artifact resulting from the strength of one of the loci that makes the pair significant, we carried out additional secondary tests as described in Methods. In Figure 4B, the primary test statistic F-all is shown below the main diagonal and the test statistic for interaction F-int is shown above the diagonal. Three clusters of locus pairs passed the genome-wide criteria for this trait (Table3). The first cluster has a peak (F-all = 6.57) at the pair D4Mit178 (corresponding to Frp-1) and D5Mit188 (corresponding to Frp-2, as with D5Mit30; see above). There is no evidence for interaction between these loci. The second cluster has a peak (F-all = 5.54) at markers D4Mit178 and D12Mit236; again there is no evidence for interaction. The F-statistic for an additive effect of the chromosome 12 locus after taking account of the chromosome 4 effect is F-add = 5.12,P = 0.007. The third cluster has a peak (F-all = 5.73) at loci D5Mit98 and D12Mit236. There is moderate evidence for interaction between these loci (F-int = 3.06,P = 0.011) and the F-statistic for the additive effect of the chromosome 12 locus after accounting for the chromosome 5 locus is F-add = 7.00, P = 0.0012. This locus on chromosome 12 we named Frp-3. The moderate effects of Frp-3 and its interaction effects with Frp-2 are shown in Figure 4C,D, respectively. A multiple regression model that includes each of the main effects and the Frp-2 by Frp-3 interaction explains 30% of the total variance of the period phenotype (Table4).
Summary of Significant Marker Pair from Genome-Wide Interaction Analysis
Multiple Regression Model of Circadian Phenotypes
Phase Angle of Entrainment
The phase angle of entrainment is defined as the phase difference between the onset of the activity rhythm and the light cycle to which the animal is entrained. By convention, the phase angle has a negative value when activity leads the entrainment cue, and a positive value when it lags. In the case of nocturnal animals, onset of activity is compared relative to the onset of darkness. To entrain to a 24 h cycle, mice, whose rhythms are typically shorter than 24 h, need to see light at dusk, which produces phase delays. Measuring this pre-dark arousal is complicated however, because in mice light exposure tends to suppress locomotor activity, thus masking the true timing of circadian arousal. The most reliable way to measure this trait is therefore to transfer an entrained animal into constant darkness and then record the onset of activity (in darkness) and compare this to the expected time of the entrainment cue (DeCoursey 1960). Figure 2B shows phenotype distribution of this trait in each generation.
We observed significant (P<10−9) differences between B6 and BALB mice in the phase angle of entrainment. B6 mice became active an average of 12 min after the scheduled time of lights off, whereas activity of BALB mice preceded lights off by an average of 3 h and 36 min. BALB mice show significantly greater variance than B6 mice. The phase angle distributions of both the F1 and F2 generations were generally continuous but deviated significantly from normal, skewing towards the B6 parent — once again reflecting the dominance of B6 alleles. We observed no significant differences between male and female cohorts in each generation. The single marker genome scan for phase (Fig.5A) shows two distinct peaks on chromosome 12 at D12Mit81 (F = 9.22) and at D12Mit7 (F = 10.31) both of which exceed the alpha = 0.01 permutation critical value of 8.62. These loci are named Angle-1 and Angle-2, respectively. The average phase angle for mice homozygous at Angle-1 locus for the B6 allele (B/B) was −1.08 h, versus −0.79 h for heterozygotes (B/C) and −2.10 h for BALB homozygotes (C/C). The average phase angle for mice homozygous at Angle-2 locus for the B6 allele (B/B) was −0.27 h, versus −1.48 h for heterozygotes (B/C) and −1.77 h for BALB homozygotes (C/C). These peaks appear to be distinct from the chromosome 12 locus Frp-3. Additional peaks are noted on chromosomes 7 and 18 in the main effects genome scan. The pairwise genetic interaction genome scan (Fig. 5B, Table 3) did not detect any marker pairs that exceeded the critical values 6.48 and 7.24 (for alpha = 0.05 and 0.01). This trait has a very skewed distribution (Fig. 2B), and thus the thresholds were much higher than for other traits. When we first consider loci that meet a less stringent genome-wide significance level (alpha = 0.1) for which the threshold value is 5.31, a consistent picture emerges. Three clusters of marker pairs with peaks at D7Mit30 and D12Mit81 (corresponding toAngle-1) (F-all = 5.77), D12Mit81 and D18Mit17 (F-all = 6.21), and D12Mit195 (corresponding to Angle-2, as with D12Mit7; see above) and D18Mit17 (F-all = 5.47) meet this criterion. (An additional pair, D7Mit30 and D12Mit195, falls just below the threshold; F-all = 5.18). We have named these loci on chromosomes 18 and 7 Angle-3 and Angle-4, respectively (Table 3). There is weak evidence for interaction between Angle-1 andAngle-3 (F-int = 2.65, P = 0.016). An additional cluster with peak (F-all = 5.23) at D8Mit13 and D12Mit263 approaches the significance threshold. There is significant evidence for interaction between these loci(F-int = 5.89, P = 0.00053). The chromosome 12 locus D12Mit263 is within the region spanned byAngle-2. The chromosome 8 locus is named Angle-5. A multiple regression model (Table 4) with all five phase angle loci (Angle-1, Angle-2, Angle-3, Angle-4, and Angle-5) and two interaction effects explains 37% of the total variance in the phase angle phenotype. The multiple regression without Angle-5 and its interaction withAngle-2 (data not shown) explains only 29% of the variance in phase angle. Graphical summaries of the two interaction effects are shown in Figure 5C,D.

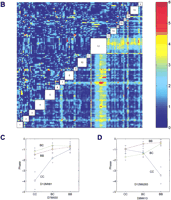
Genome-wide scan for phase angle of entrainment phenotype. (See Fig. 4for plotting details.) (A) Single marker regression genome scans for phase angle of entrainment. (B) Pairwise marker regression genome scans. (C) Pairwise interaction ofAngle-1 and Angle-4. (D) Pairwise interaction of Angle-2 and Angle-5.
Amplitude of Circadian Rhythm
The amplitude of a circadian rhythm reflects the strength or robustness of the rhythm, i.e., the extent to which the circadian behavior is separated into distinct active and resting periods within each cycle. In this study, we have estimated the amplitude of the circadian rhythm from the power spectral density of the circadian peak obtained by Fourier analysis (Bracewell 1986). This measure is scored based on the percentage of activity falling within the circadian peak, with higher percentage scores reflecting higher amplitude rhythms. Figure 2C shows the distribution of this trait in each generation. We observed that amplitudes of circadian activity were significantly greater (P<10−9) in B6 mice than in BALB mice, but that distributions in both strains were not continuous, possibly reflecting environmental effects and the low numbers of animals assayed. We observed a significant sex difference in F1(P<0.05) and F2 (P<0.001) generation, where amplitude was significantly lower in male than female mice; no such sex differences were detected in B6 and BALB mice. Again in the F1 generation, we observed a discontinuous distribution that skewed towards the B6 parentals. Measure of amplitude in the F2 generation, however, were distributed both normally and continuously.
The single marker genome scan (Fig. 6A) shows a significant peak at marker D4Mit27 (F = 8.86). We identified genome-wide thresholds based on 1000 permutation test of 7.50 and 8.90 (for alpha = 0.05 and 0.01, respectively). We named this locusAmplitude (Amp)-1. The average amplitude score for mice homozygous at the Amp-1 locus for the B6 allele (B/B) was 13.99%, versus 12.67% for heterozygotes (B/C) and 9.74% for BALB homozygotes (C/C). This suggests that the B6 Amp-1allele acts semidominantly over the BALB allele. The pairwise genome scan (Fig. 6B, Table 3) shows a single significant pair at D1Mit33 and D4Mit27 (corresponding toAmp-1) (F = 6.46). The genome-wide permutation critical values are 5.15 and 5.63 (for alpha = 0.05 and 0.01). This marker pair shows a significant interaction (F = 6.88, P = 0.000036) which is summarized in Figure 6D. The locus on chromosome 1 we named Amp-2. High values of the amplitude score were associated with homozygous B6 atAmp-1 in combination with homozygous BALB at Amp-2(Fig. 6D). A multiple regression model was fit to amplitude including both markers and the interaction. Because gender has a significant effect on amplitude, we included a term for gender in the joint model; there was, however, no evidence for interactions between gender and the QTLs. The regression model explains 26% of the total variance of this trait in the F2 population (Table 4). Of this, 20% is attributable to the genetic effects and 5% is attributable toAmp-1 alone, thus the Amp-1–Amp-2interaction is the major contributor to genetic variance in this trait.
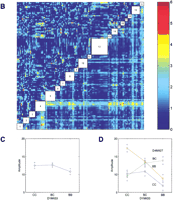
Genome-wide scan for amplitude phenotype. (See Fig. 4 for plotting details.) (A) Single marker regression genome scans for circadian amplitude. (B) Pairwise marker regression genome scans. (C) Main effect of Amp-2. (D) Pairwise interaction of Amp-1 and Amp-2.
Activity Level
Although the relationship between the level of wheel running activity and circadian rhythmicity is poorly defined, an animal's activity level may reflect broad parameters of the circadian axis, such as the amplitude of the pacemaker's oscillator, the strength of the oscillator's coupling to its output, or the amplitude of the behavioral output. In addition several studies have demonstrated that activity itself can act as an entrainment cue, feeding back to the circadian pacemaker (Edgar and Dement 1991; Edgar et al. 1991a,b;Mrosovsky 1995).
We determined the level of each animal's daily activity by averaging the number of daily wheel revolutions over 15 consecutive cycles in constant darkness. Figure 2D shows phenotype distribution of this trait in each generation. We observed significantly lower (P<10−3) daily activity levels in BALB mice than in B6 parentals. Once again we observed significant sex difference in F1 (P<0.001) and F2(P<10−8) mice, with the activity levels of male mice significantly lower than female mice.
The single marker genome scan (Fig. 7A) for this trait shows no significant peaks. However the pairwise genome scan (Fig. 7B, Table 2) identifies the marker pair D16Mit106 andDXMit27 (F-all = 5.04), whose significance falls at the alpha = 0.05 permutation threshold. There is a significant interaction effect (F = 7.23, P<10−4) between these loci. We named the locus on chromosome 16, Activity(Act)-1 and the locus on the X chromosome,Act-2. It is important to note that X chromosome loci in males in this F2 population are hemizygous (B or C). In females, the X chromosome genotypes are either BALB homozygote (C/C) or B6/BALB heterozygote (B/C) because the F1 generation was produced using BALB female by B6 male matings, thus females in the F2generation will have at least one nonrecombinant BALB chromosome X. Therefore we carried out separate analyses of male and female F2 progeny. There was no evidence for a significant effect ofAct-1, Act-2, or the interaction in males. However there is a significant effect in females as indicated by the multiple regression model shown in Table 3. There are no significant main effects at Act-1 or Act-2 but their interaction is significant. The interaction is summarized graphically in Figure 7C,D. Heterozygosity at Act-2 increases activity in females that are homozygous for the B6 allele (B/B) atAct-1, decreases activity in females that are heterozygous atAct-1 and has no effect in females that are homozygous for the BALB allele (C/C) at Act-1. These loci explain 19% of the variation in activity among F2 females (Table 4).
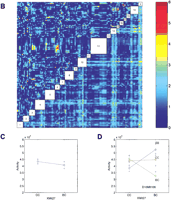
Genome-wide scan for daily activity level phenotype. (See Fig. 4 for plotting details.) (A) Single marker regression genome scans for activity level. (B) Pairwise marker regression genome scans.(C) Main effect of Act-2. (D) Pairwise interaction of Act-1 and Act-2.
Dissociation
A clear difference between the locomotor activity of BALB and B6 mice is that the BALB activity rhythm is much more fragmented (dissociated) than B6. B6 mice usually have a major activity onset component followed by a very small offset component. In contrast, BALB mice typically have multiple activity components, particularly in constant darkness (DD). In the F1 generation the pattern of activity is similar to B6 in this respect. Some F1 mice showed no visible activity offset components. The period of each component was similar in this generation and the parentals. In the F2 generation, this phenotype was quite variable. Some mice had activity profiles similar to B6 or BALB. Interestingly, some mice exhibited patterns that had not been observed in the parental population. For example, in some mice, two major activity components are present (onset and offset), which free-run with different periods and later consolidate. In others, a major activity component is present with multiple fragmented components, as though discrete aspects of both the B6 and BALB phenotypes were being expressed simultaneously. This kind of dissociation of activity has been observed in mice and other rodents. At an extreme, it can lead to a split of the locomotor activity into two components that drift apart and then synchronize at phases that are 12 h (180°) apart. Such splitting of the locomotor activity in the golden hamster is well documented (Pittendrigh and Daan 1976; Pickard and Turek 1982). This behavioral splitting is reflected in physiological and molecular measures of the suprachiasmatic nuclei (SCN) as well. The circadian rhythm of both electrophysiological activity and expression of clock-related genes (Per1 andPer2) can also split into two components (de la Iglesia et al. 2000; Jagota et al. 2000). Such dissociated or split rhythms have also been observed in mice (Abe et al. 1999). In this case the effect of light on the phase of the split rhythm was also affected, again indicating that the central pacemaker is affected, not merely the behavioral output.
We developed a scoring method to characterize this dissociation phenotype in our F2 panel. This is a qualitative trait with rankings from 0 to 5 by the degree of dissociation of activity fragments. We defined d = 0 as an activity rhythm which has a large activity onset component followed by a very small or undetectable activity offset component, both of which have the same or very similar endogenous period. The subsequent scores, d = 1 through d = 5, correspond to activity rhythms that are increasingly disrupted. Figure3A shows a typical example of each ranked mouse activity record in the F2 population. All B6 mice analyzed were categorized as d = 0. For the BALB strain, only 1 of 17 mice were categorized as d = 0, whereas the rest showed some degree of dissociation. In the F1 generation, 22 of 24 mice were categorized as d = 0, whereas the remaining two were scored as d = 1 and d = 2. The phenotype of the F1 population was close to the B6 parent, once again reflecting the dominance of B6 alleles. The distribution of the F2 generation deviated significantly from normal, skewing toward the B6 phenotype (Fig. 3B). One hundred sixteen out of 196 mice (60%) were categorized as d = 0. There were only four mice categorized as d = 5. This suggests that multiple QTL are involved in this trait and the effect of each alone may be very weak.
As we anticipated, the single marker genome scan (Fig.8A) on the trait dissociation shows no significant peaks. However the pairwise genome scan (Fig. 8B, Table 2) identifies the marker pair D12Mit251.1 and D15Mit28(F-all = 5.90) as being significant at the genome-wide 0.1 level. The genome-wide critical values are 5.5 and 6.0 for alpha = 0.1 and 0.05, respectively. There is a significant interaction between these loci (F-int = 8.80, 1.5 × 10−6). The interaction effect is summarized in Figure 8C,D. This shows that high values of dissociation are associated with a homozygous BALB genotype (C/C) atD15Mit28 in combination with homozygous B6 genotype (B/B) atD12Mit251.1. Only seven mice in the F2 population had this genotype combination. All of these mice showed some degree of dissociation (none had a score of d = 0). We named the locus on chromosome 12 Dissociation (Disso)-1 and the locus on chromosome 15 Disso-2. These two loci explain 20% of phenotypic variation of the F2 population (Table 4).
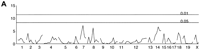
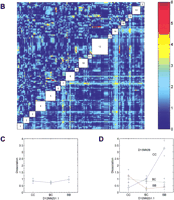
Genome-wide scan for dissociation phenotype. (See Fig. 4 for plotting details.) (A) Single marker regression genome scans for dissociation. (B) Pairwise marker regression genome scans. (C) Main effect of Disso-1. (D) Pairwise interaction of Disso-1 and Disso-2.
Mapping Candidate Clock Genes
To assess whether the loci mapped in this study correspond to known clock candidate genes, we genetically mapped five of the proposed clock genes (mPer1, mPer2, mPer3, mCry1, and mCry2) (Fig. 9). The locations of the remaining four genes have been reported previously and are shown in Figure 9 (King et al. 1997; Sangoram et al. 1998; Wolting and McGlade 1998; Lowrey et al. 2000). We identified polymorphisms, typically within intronic sequence or closely flanking genomic sequence, that allowed us to map these genes either in our own mapping crosses, or in the B6 × SPRET/Ei reciprocal backcrosses available from Jackson Laboratories (Rowe et al. 1994) (see Methods for complete mapping descriptions).
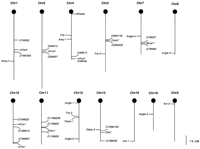
Chromosomal locations of mapped loci and clock genes. Each vertical bar represents a mouse chromosome, with the centromere noted by the black circle. Loci to the left of each chromosome are those identified in this study and loci to the right are the previously cloned clock genes likely to form the core of the central pacemaker mechanism, with flanking SSLP markers included. Also noted is the map position of thewheels locus, identified by a mutation believed to have an effect on circadian rhythms in mammals (Nolan et al. 1995). Genes noted by an asterisk are core clock genes mapped previously (King et al. 1997; Sangoram et al. 1998; Wolting and McGlade 1998; Lowrey et al. 2000).
mPer1
We identified linkage between mPer1 and SSLPs on mouse chromosome 11. mPer1 was subsequently mapped to chromosome 11, 1.0 cM (1/103) distal of D11Mit29 and 10.7 cM (11/103) proximal of D11Mit39.
mPer2
We identified linkage between mPer2 and SSLPs on mouse chromosome 1, leading to our mapping the gene to mouse chromosome 1, 14 cM (67/482) distal of D1Mit22 and 5.5 cM (26/482) proximal ofD1Mit308.
mPer3
We identified linkage between mPer3 and SSLPs on the distal end of mouse chromosome 4 resulting in the mapping of mPer3 to the region of D4Mit42 (0/94 recombination events).
mCry1
mCry1 was linked strongly to loci on mouse chromosome 10; the mCry1 genetic locus was mapped 8.7 cM (8/92) distal ofD10Mit20 and 5.4 cM (5/92) proximal of D10Mit11.
mCry2
The mCry2 locus showed a strong linkage to loci on mouse chromosome 2 and mapped 2.1 cM (2/94) distal of D2Mit15 and 4.3 cM (4/94) proximal of D2Mit97.
DISCUSSION
We have applied novel whole-genome techniques, complex trait (quantitative trait locus), and genetic interaction analyses in a segregating intercross (F2) population, to characterize genetic polymorphisms affecting circadian rhythms in the mouse. Prior to our experiments, two general strategies have been used for clock gene identification investigations in mammals: analysis of single gene mutations, and characterization of genes identified through cross-species homology. These experiments, although they have laid an essential groundwork, do not include a more thorough examination of the breadth and complexity of genetic influences on circadian behavior. Our effort has led to the identification of 14 loci, based on the analysis of five discrete aspects of circadian behavior, that make significant contributions to the variation of this behavior in mice. We have identified several loci that have significant main effects on quantitative circadian behaviors, including free-running period, phase angle of entrainment, and amplitude of the circadian rhythm. Many of these 14 loci, however, were identified primarily, or solely, from strong epistatic interactions detected among specific alleles at different genetic loci. Indeed, a fundamental conclusion to be drawn from these data is that circadian phenotypes can be significantly affected by complex genetic interactions at multiple loci. This introduces a new, previously undescribed, aspect to the analysis of circadian behavior. The genome-wide interaction analysis identified numerous pairs of loci that, in particular allelic combinations, had epistatic effects on circadian behavior much greater than either of the loci alone. (Additional higher order interactions may also be present; however, our current sample size is not large enough to reliably detect these.) This epistasis is most clearly seen in the analysis of the amplitude of the circadian rhythm, in which particular alleles of the loci, Amp-1 and Amp-2, each had relatively little effect on the phenotype individually, but in specific allelic combination produced a very significant effect (Fig. 6). Results such as these underscore the complexity of this behavior and argue against any approach that devotes all analysis of the circadian clock to single gene effects. A better understanding of such epistatic interactions will be essential to fully understanding the complex nature of circadian rhythms, and complex behaviors in general.
We have identified five discrete measures from the records of locomotor activity in the pedigree described here. However, these five phenotypes are not likely to be completely independent of each other. For example, extensive consideration has been given to models that integrate the phase angle of entrainment with the endogenous period and to suggestions that period itself can be modified by a feedback effect of prolonged or intense activity. With these types of phenotypic interactions in mind, it is interesting to compare the map positions of the loci we have identified here to see if variation in these five phenotypic measures might arise from polymorphisms in similar loci. Interestingly, in most cases, the loci are not the same; variation in each of the five measures is affected by a unique set of underlying genes (Fig. 9). There are two possible exceptions to the apparent independence of these phenotypic measures. Frp-1 (35.5 cM) andAmp-1 (42.5 cM) map to similar positions on chromosome 4, andFrp-3 (22 cM) and Disso-1 (29 cM) map to similar positions on chromosome 12. The phenotypes that we would have predicted, a priori, to be the result of similar genetic loci, namely period, phase, and activity level, shared no loci in common in this analysis.
Similarly, one might ask how these loci compare to the map positions of the genes proposed to form the core of the mammalian circadian clock. Again, as with the different loci arising from multiple phenotypic measures, the map positions of these genes generally differ from the loci identified here (Fig. 9). A possible exception is CKIɛ, which maps to within 10 cM of the Disso-2 locus. Four additional clock genes map to the same chromosomes as loci identified here (Clock, Bmal1, Per2, Per3), although the genetic distances are considerably larger in these cases. It is perhaps not surprising that our genome scans did not point to the proposed clock genes. The clock genes thus far identified are likely to form the core of the circadian feedback loop. As such, stringent evolutionary demands may have been placed on these genes in order for the clock to function precisely, so that few polymorphisms are tolerated in these genes. Such conservation is suggested by the significant similarity present in clock genes when comparing mouse and human sequences (Steeves et al. 1999; Hida et al. 2000). In any case, the clock genes would need to be polymorphic between the two particular mouse strains used (B6 and BALB) to be detected here. This is, of course, a caveat that extends to our analysis in general. We do not claim to have made a comprehensive scan of all loci affecting circadian rhythms in the mouse. Rather, we have characterized loci that are polymorphic relative to the strains and phenotypes we selected, and that have detectable effects. As mapping strategies become more efficient, we envisage examining the genetics of these and similar circadian phenotypes in different F2 hybrids, that may lead to the identification of additional loci or corroborate and refine the map positions of the loci identified here. An additional strategy for genetic analysis is also available, similar to a modifier mutagenesis screen, in which modifier loci are analyzed in an F2population carrying a mutation affecting one of the proposed clock genes, such as the Clock mutation. This may strengthen our ability to detect and refine loci affecting circadian rhythms in the mouse genome.
An additional comparative mapping question to consider is how our loci compare to other complex loci determined previously, in both circadian and potentially related behaviors. There have been attempts made to identify circadian rhythm QTLs in B6 × DBA recombinant inbred strains of mice (Hofstetter et al. 1995, 1999; Mayeda et al. 1996;Hofstetter and Mayeda 1998). These efforts have characterized several provisional (or “hypothetical”) loci. None of these loci has exceeded standard measures of significance, thus it is difficult to compare them to the loci identified here. Some of the provisional loci are found on chromosomes near some of the loci identified here, including Amp-2, Amp-1, Frp-1, Angle-1, and Angle-3. Two other studies examining quantitative traits in mice deserve mention. Attempts have been made to identify loci affecting aspects of sleep in B6 × BALB recombinant inbred lines (Tafti et al. 1997). Although the statistical strength of these conclusions is not powerful (owing to the limited number of RI lines examined), loci were identified on chromosomes 5, 7, and 12 in regions roughly similar to loci identified here (Frp-2, Angle-4, Angle-1, Frp-3, Disso-1; see Fig. 9). Flint et al. (1995) examined a large B6 × BALB F2 population (like the pedigree described here) to identify loci affecting emotionality scores of mice, which are believed to correlate with human affective disorders. Some of these measures are based on activity levels in various environments and situations. Interestingly, this study identified loci on chromosomes 1, 12, and 15, again in regions that correspond to loci identified here (Amp-2, Angle-1, Frp-3, and Disso-1).
If the underlying genes affected by the loci reported here are unlikely to be among the proposed clock genes (based on map positions), then what genes might be affected? There are multiple levels at which these loci may interact with the clock mechanism and its outputs to affect behavior. For example, although the molecular clock itself is likely to be cell autonomous, there are important integrations that result from interactions among such individual ‘clock cells‘ within the pacemaker tissue (in mammals, the hypothalamic SCN) (Liu et al. 1997; Herzog et al. 1998; Low-Zeddies and Takahashi 2001). Furthermore, the pathways from this central pacemaker to its output are poorly understood and the recorded activity itself can feed back to affect the circadian clock. Because so many aspects of these pathways remain unclear, it is premature to predict precisely where within the system the genes underlying these loci may be exerting their effect. In addition, the loci characterized here, although significant, are not yet precise in their location. Thus, predicting candidate genes (which would amount to little more than cataloging apparently interesting genes near these loci) is likewise premature. However, as more is learned about the circadian clock, our understanding of the underlying processes likely to be affected by these interstrain polymorphisms should provide insights into the types of genes to be expected. This, in turn, would then better inform the prioritization of candidate genes, as they become available.
Although the power of the genetic analyses described extends beyond gene discovery, including revealing genetic interactions among different loci and providing a landscape of the genes underlying a given complex phenotype, identification of the genes themselves is still of great interest. Although cloning is difficult when beginning with quantitative loci such as these, it is not impossible (Nadeau and Frankel 2000). Towards this end, we have begun developing congenic lines carrying several of the loci described here. From these congenic mice, we can determine if each locus has main effects in isolation (or effects in interacting pairs) and if so, begin to isolate the genetic interval responsible for the effect, with the eventual target being the identification and testing of candidate genes that might be responsible for the phenotypic difference. The likely availability of mouse genomic sequence (from several strains) in the near future brightens this prospect considerably.
Strategies for characterizing loci underlying complex or quantitative phenotypes usually begin with the identification of strains that show considerable divergence of the phenotype(s) in question. However, an interesting result that arose from our analysis of these circadian data is that the relationship between a particular allele and the way it affects the phenotype can be counterintuitive. By this, we mean that the phenotypic characteristics associated with the B6 strain were sometimes found to be linked to BALB alleles in our F2hybrids, and vice versa. Thus, for example, a high amplitude rhythm was linked to BALB alleles at Amp-2 and a long free-running period was linked to BALB alleles at Frp-1. One must conclude that each strain includes alleles, or combinations of alleles that are epistatic to alleles at different loci. In an F2 cross, the epistatic effects become unlinked and the counterintuitive alleles can be detected. This result brings into question the general strategy used when developing mapping crosses to analyze a particular phenotype. If each of the two strains chosen for analysis harbors alleles that tend to produce the opposing strain’s phenotype, then it becomes difficult to predict the result of these hybrid matings. Strains that have similar phenotypes may produce an equally interesting variety of hybrids because of the expression of otherwise recessive or repressed alleles; this was seen in an analysis of circadian period inArabidopsis (Swarup et al. 1999). Likewise, divergent strains may not produce extreme phenotypes in hybrid offspring.
This quantitative analysis of circadian rhythms opens a new avenue to the understanding of this behavior. This analysis, in combination with a genome-wide interaction analysis, has allowed us to uncover new loci involved in the genetics of circadian rhythms in the mouse. We have identified multiple loci affecting several aspects of circadian behavior, both as main effects and in combination with other loci in the mouse genome. This approach should provide a window to the analysis of disorders of such phenotypes in humans. Indeed, a common recognition emerging from the analysis of multiple human disorders with hard-to-assess genetic determinants, including heart disease, affective and neurodegenerative disorders, asthma, diabetes, and hypertension, is that disease susceptibility is often determined by numerous modest susceptibility alleles present at multiple loci. These alleles are likely to have very limited effects in isolation but in combination can have complicated interactions that produce phenotypic extremes not predicted by an additive model (Risch 2000). The interaction analysis described here provides a different model by which to address these nonadditive effects in such complex phenotypes.
This genetic analysis has revealed previously undetected complexity in the circadian system and points to the presence of many as yet undiscovered genes that contribute to the expression of this behavior in mice. Although severe disruption of circadian rhythms may be caused by mutations in core clock genes, it is possible that the broad variety of circadian behavior observed in mammalian species, including humans, is the result of polymorphisms in multiple, interacting loci such as the ones described here.
METHODS
Animals
(BALB/cJ X C57BL/6J)F2 mice were bred from (BALB/cJ X C57BL/6J)F1 parents that were obtained from the Jackson Laboratory. F1 animals were the result of mating BALB/cJ females with C57BL/6J males. F2 animals were raised in a 12 h light:12 h dark cycle (LD12:12) from birth. After weaning, they were group housed (1–5 mice/cage). At 8–12 wk of age, they were transferred into individual cages equipped with running wheels in LD12:12. After a minimum of 7 d, animals were transferred into DD for three weeks. Wheel-running activity was recorded from a total of 196 F2 generation mice (98 females and 98 males).
Analysis of Circadian Behavior
Free-Running Period of Activity Rhythm
We calculated the free-running period for 15 d (day 1–15 in DD) using a χ2 periodogram with 1-min resolution between 20–26 h (The Chronobiology Kit; Stanford Software System).
Phase Angle of Entrainment
The phase angle of entrainment is defined as the time difference between activity onset and the predicted time of dark onset, on the first day in constant darkness. The phase angle of entrainment was calculated by extrapolating the estimated activity onset in the last day of the LD cycle and the time of lights off. An activity onset regression line was drawn based on five consecutive activity onsets (day 1–day 5 of constant darkness). When activity starts before the predicted time of lights-off, it is defined as a negative phase angle, whereas a later activity onset is defined as a positive phase angle.
Circadian Rhythm Amplitude
The amplitude of the Fourier periodogram around 24 h, for 15 d in DD, was used as an indicator of circadian rhythm amplitude (Clock Lab, Actimetrics). Original data were collected at 1-min intervals. To prepare the power spectra, data points were first binned into 6-min intervals, for a total of 3600 bins for the 15-d period to be analyzed. A 4th order Blackman-Har window was then applied to these points prior to calculating the power spectrum using a fast Fourier transform. The spectrum was normalized to an integral of one by dividing each of its elements by the sum of all elements. For each animal the frequency of the spectrum peak is reported, along with the amplitude of the peak, which represents the relative power within a frequency band of 0.0028.
Activity Level
The total number of running-wheel revolutions was counted from days 1–15 in DD and then averaged to determine the daily activity level.
Dissociation
The degree of activity rhythm dissociation (splitting) was scored using six different levels (0–5), with 0 corresponding to the most intact rhythms and 5 corresponding to the most dissociated rhythm (see Results and Fig. 3 for additional details). The scoring was performed by two people (K.S. and J.S.T.) independently.
Analysis of Genotype
Genomic DNA was prepared by phenol-chloroform extraction from tail biopsies. SSLP markers that were polymorphic for our cross (C57BL/6J x BALB/cJ) were chosen from the MIT Whitehead Institute database and purchased from Research Genetics. For each SSLP, one primer was end-labeled with γ-32p-ATP using T4 kinase according to standard protocols. PCR reaction mixes (5 or 10 μL) contained 12.5 or 25 ng genomic DNA, 16 μM of primers, 0.2 mM each dNTP, 50 mM KCl, 10 mM Tris-HCl (pH 9.0), 0.1% Triton-X, 1mg/mL BSA, 1.25 mM MgCl2, and 0.025 U Taq polymerase (Perkin Elmer) and were arrayed in 96- or 192-well plates, overlaid with 10 μL mineral oil. Thermal cycler conditions, in 96- or 192-well PTC-100 thermocyclers (MJ Research), were 95°C for 4 min followed by 27–35 cycles at 95°C for 15 s, 30 s at 55°C, 15 s at 72°C. After 27–35 cycles, samples were held at 72°C for 5 min and then stored at 4°C until use. An equal volume of loading buffer (0.25% xylene cyanol and 0.25% bromphenol blue in 50% formamide) was added to the amplification products, which were denatured for 5 min at 95°C, then separated on 7% polyacrylamide gels (30 cm × 40 cm) at 100 W for ∼2 h. We used 4× combs (Owl Scientific) that allowed us to load 96 lanes per gel. Most of the PCR reactions included two primer pairs in one reaction (duplex reaction). Typically, two duplex reactions were loaded onto each lane (double loading) so that we obtained four genotype scores/lane and a total of 384 genotype scores were generated from each gel. The duplex primer pairs and combinations of double loading pairs used for the initial QTL search are listed in Table 2. Gels were wrapped in plastic wrap and exposed to autoradiographic film at −80°C for 16–48 h. Genotypes were scored visually from autoradiographs.
Single Marker Genome Scans
We used the Mapmaker/QTL analysis software for our initial genome scan (Lander et al. 1987). Subsequently, we implemented marker regression-based genome scans in Matlab Software (Mathworks, Inc.). Source code is available from G.A.C. on request. A single marker genome scan consists of computing the regression one-way ANOVA F-statistic (which has 2, n-3 df for an F2 population of size n) at each marker in the genotyping array. Statistical significance is assessed by permutation testing (Churchill and Doerge 1994). This approach takes into account the multiple testing implicit in a genome-wide search.
Pairwise Genome Scans
As some loci may affect a phenotype primarily through interaction effects, we have developed an approach and implemented software (Matlab source code available from G.A.C. upon request) to conduct a simultaneous search for pairs of interacting loci. The method of simultaneous search (Dupuis et al. 1995) examines all pairs of marker loci for association with the trait in a two-dimensional genome scan. In our procedure, the likelihood under the full regression model (with two main effects and an interaction) is compared to that under the null model of no genetic effects. This comparison generates an F-statistic (with 8 and n-9 d.f.) for every marker pair. We refer to this statistic as F-all. It is our primary screen for identifying interesting locus pairs. Significance of F-all is assessed by permutation analysis, which accounts for the large number of pairs tested in the genome-wide search. If the F-all statistic for a locus pair is significant (P< 0.05 genome-wide corrected), we carry out several additional tests to determine if the pair represents a genuine interaction, two additive main effects, or is merely an artifact resulting from a strong association with only one of the two loci. A second F-statistic (F-int, which has 4 and n-9 d.f.) is computed to test for interaction effects. F-int compares the full model (with interaction) to an additive model with two main effects but no interaction. If this test is significant at a nominal alpha = 0.01 level, we conclude that the two loci interact and stop. If F-int is not significant, we proceed to the next level of testing. Two additional F-statistics (F-add1 and F-add2, each with 2 and n-5 d.f.) are computed to compare each of the single locus models to the two locus additive model. Both of these tests should be significant (at alpha = 0.01 level), to indicate that both loci contribute to the effect. If one of the loci in the pair fails to achieve significance this suggests that it may not be contributing to the observed association and simply carried along by the strong effect of the other locus. Each of these secondary tests is carried out using nominal P values because pairs have been selected with a genome-wide criterion using F-all. The choice of alpha = 0.01 is stringent but somewhat arbitrary.
Multiple Regression Modeling
All loci and interactions that are detected by genome scans (single marker and pairwise) are entered into a multiple regression model. Regressions were fit using Minitab software (Minitab, Inc.). The adjusted sums of squares from the multiple regression models provides a means to assess the contribution of a given locus (or an interaction) in combination with all other loci.
Genetic Mapping of Clock Genes
mPer1
We identified several single nucleotide polymorphisms (SNPs) by sequence analysis of the mPer1 locus that led to the development of a single stranded conformational polymorphism (SSCP) between the B6 and C3H strains in mPer1 intronic sequence. We designed primers to amplify a 263 fragment to detect this SSCP. A mapping panel of 103 [(C3H/eJ X C57BL/6J)F1 X C57BL/6J]N2 mice, generated at Northwestern University, that had been genotyped using ∼80 SSLP markers was used to map this SSCP. Linkage analysis was performed with the program Map Manager QTb2868K (Manly and Olson 1999).
Primer Sequences
5′-GAG GAC ACT ACA TAA CTC ATC CTT T-3′ 5′-TGC TCT TCT TCC TCC ATG GCA GAG TC-3′
PCR Conditions
PCR conditions were 95°C for 4 min followed by 35 cycles at 95°C for 15 s, 15 s at 55°C, 15 s at 72°C. After 35 cycles, samples were held at 72°C for 5 min and then stored at 4°C until use.
Allele Detection
PCR primers were end-labeled with γ-32p-ATP using T4 kinase according to standard protocols. Following amplification, PCR products were separated on 7% polyacrylamide gels without urea for 2 h at 4°C, according to standard SSCP protocols. The C3H PCR product migrated through the gel faster than the B6 PCR product in this nondenaturing gel. Gels were processed and scored as described above for SSLP genotyping analysis.
mPer2
In an effort to identify polymorphisms within the mPer2gene, we amplified brain cDNA from B6 and BALB using 14 PCR primer pairs designed to amplify ∼500 bp fragments with ∼100 bp overlaps. After PCR the products of each primer pair for the two mouse strains were mixed together and heated to 95°C for 10 min, then slowly (>45 min) cooled to 25°C. This process allows the formation of heteroduplexes as well as homoduplexes, if polymorphisms exist between the B6 and BALB PCR products. We used a temperature-modulated heteroduplex HPLC according to the manufacture's protocols (Transgenomic WAVE) to detect SNP. We identified a C to G transversion that allowed us to discriminate the B6 and BALB alleles byNcoI digestion. Design of a new primer pair led to the amplification of a 620 bp segment of genomic DNA. Treatment withNcoI digested the BALB product into two fragments (561 bp and 59 bp), whereas the B6 product remained undigested. This locus was placed on a mapping panel of 223 DNA samples from a (BALB X C57BL/6J)F2 intercross that had been typed using ∼90 SSLPs.
Primer Sequences
5′-TAC CAG CTG CTA ATG TCC AGT G-3′, 5′-TAC ATT GGG CTA GCC TTC CTA AG-3′
PCR Conditions
PCR conditions were 95°C for 4 min followed by 32 cycles at 95°C for 30 s, 30 s at 55°C, 30 s at 72°C. After 32 cycles, samples were held at 72°C for 5 min and then stored at 4°C until use.
Allele Detection
Following amplification, PCR products were digested with 2 units ofNcoI for ⩾2 h at 37°C. After restriction digestion, the B6 allele size was 620 bp, in contrast with 561 bp and 59 bp for the BALB/c allele. Digested PCR products were separated on a 2% agarose gel.
mPer3
To identify interstrain polymorphisms in the mPer3 gene, we amplified the 3′ untranslated region of the B6 and SPRET/Ei (SPRET) mouse strains. We designed two primer pairs, each producing PCR fragments ∼500 bp long, with ∼100 bp overlap between the two products. Genomic DNA was PCR-amplified, and assayed for the presence of polymorphisms using the Transgenomic WAVE system. A polymorphism was found in one of these products. Linkage analysis was then performed using the BSB Backcross DNA panel containing samples from 94 ([C57BL/6J X SPRET/Ei] F1 X C57BL/6J)N2 backcross progeny, each of which carries a least one SPRET allele (The Jackson Laboratory) (Rowe et al. 1994). We used the temperature-modulated heteroduplex HPLC according to manufacture's protocols (Transgenomic) to detect the presence of a B6 allele in each sample, which is sufficient to determine the mPer3 genotype.
Primer Sequences
5′-TCC TGA TGG TAA GAC ATT CCA G-3′, 5′-GCG TGA ACA ATC ACA CTC ACT T-3′
PCR Conditions
PCR conditions were 95°C for 4 min followed by 32 cycles at 95°C for 30 s, 30 s at 55°C, 30 s at 72°C. After 32 cycles, samples were held at 72°C for 5 min and then stored at 4°C until use.
Allele Detection
Samples were heated at 95°C for 10 min then gradually cooled to 25°C over 45 min. Amplified PCR products from the mapping cross were analyzed by temperature-modulated heteroduplex HPLC according to manufacture's protocols (Transgenomics).
mCry1
We identified a simple sequence repeat (SSR) within a λ genomic clone containing the mCry1 locus, kindly provided by Dr. Aziz Sancar (Univ. of North Carolina). This SSR is polymorphic between the B6 and SPRET strains. The SPRET allele size was found to be ∼40 bp larger than B6. Linkage analysis was then performed using the BSB Backcross DNA panel described above.
Primer Sequences
5′-GCC TGG AAA CAC AGA GAC AAT TA-3′, 5′-ATG AGT GCA TGC TGT ACA TGG-3′
PCR Conditions
95°C for 4 min followed by 32 cycles at 95°C for 30 s, 30 s at 55°C, 30 s at 72°C. After 32 cycles, samples were held at 72°C for 5 min and then stored at 4°C until use.
Allelle Detection
PCR products from genomic DNA were separated on a 2% agarose gel for 1 h.
mCry2
We designed four primer pairs based on the GenBank mCry2cDNA sequence. The expected cDNA size of each was ∼500 bp. Of the four pairs, two amplified genomic DNA from the C57BL/6J and SPRET mouse strains. The ∼800-bp and ∼1.5-kb lengths of the products suggested the presence of intronic sequence. Products of the 800-bp fragment were analyzed by Transgenomic WAVE to identify polymorphisms, which we then confirmed by DNA sequencing. Linkage analysis was performed using the BSS Backcross DNA panel containing samples from 94 ([C57BL/6J X SPRET/Ei]F1 X SPRET/Ei)N2 backcross progeny (The Jackson Laboratory) (Rowe et al. 1994).
Primer Sequences
5′-AGA AGG TGA AGA GGA ACA GCA C -3′ 5′-TAG ATG TAT CGA GAG GGG AAG C-3′
PCR Conditions
PCR conditions were 95°C for 4 min followed by 32 cycles at 95°C for 30 s, 30 s at 55°C, 60 s at 72°C. After 32 cycles, samples were held at 72°C for 5 min and then stored at 4°C until use.
Allele Detection
After PCR, samples were heated at 95°C for 10 min followed by gradual cooling to 25°C over 45 min. PCR products amplified in the mapping cross were analyzed by DNA Fragment Analysis System with DNASep Technology.
Acknowledgments
We thank Dr. Aziz Sancar for providing us with a mCry1genomic clone and Dr. Wayne Frankel for advice and discussion of QTL analysis. This study was supported by the Air Force Office of Scientific Research, the National Science Foundation Science and Technology Center for Biological Timing, an Unrestricted Grant in Neuroscience from Bristol-Myers Squibb, and NIH grant R37-MH39592. J.S.T. is an Investigator in the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL j-takahashi{at}northwestern.edu; FAX (847) 491-4600.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.171601.
-
- Received November 22, 2000.
- Accepted March 22, 2001.
- Cold Spring Harbor Laboratory Press








