
Landscape of microRNA and target expression variation and covariation in single mouse embryonic stem cells
- Marcel Tarbier1,2,
- Sebastian D. Mackowiak3,
- Vaishnovi Sekar1,
- Franziska Bonath1,
- Etka Yapar4,
- Bastian Fromm5,
- Omid R. Faridani6,7,
- Inna Biryukova1 and
- Marc R. Friedländer1
- 1Science for Life Laboratory, Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, SE 114 18 Stockholm, Sweden;
- 2Science for Life Laboratory, Department of Immunology, Genetics and Pathology, Uppsala University, SE-751 85 Uppsala, Sweden;
- 3Berlin Institute of Health at Charité–Universitätsmedizin Berlin, Center of Digital Health, 10178 Berlin, Germany;
- 4Department of Biology, Lund University, SE-221 00 Lund, Sweden;
- 5The Arctic University Museum of Norway, UiT–The Arctic University of Norway, NO-9037 Tromsø, Norway;
- 6School of Biomedical Sciences, University of New South Wales, Sydney, NSW 2052 Australia;
- 7Garvan Institute of Medical Research, Sydney, Darlinghurst NSW 2010 Australia
Abstract
microRNAs are small RNA molecules that can repress the expression of protein-coding genes post-transcriptionally. Previous studies have shown that microRNAs can also have alternative functions, including influencing target expression variation and covariation, but these observations have been limited to a few microRNAs. Here we systematically study microRNA alternative functions in mouse embryonic stem cells (mESCs) by genetically deleting Drosha, leading to global loss of microRNAs. We apply complementary single-cell RNA-seq methods to study the variation of the targets and the microRNAs themselves, and transcriptional inhibition to measure target half-lives. We find that microRNAs form four distinct coexpression groups across single cells. In particular, the mir-290 and the mir-182 genome clusters are abundantly, variably, and inversely expressed. Some cells have global biases toward specific miRNAs originating from either end of the hairpin precursor, suggesting the presence of unknown regulatory cofactors. We find that microRNAs generally increase variation and covariation of their targets at the RNA level, but we also find microRNAs such as miR-182 that appear to have opposite functions. In particular, microRNAs that are themselves variable in expression, such as miR-291a, are more likely to induce covariations. In summary, we apply genetic perturbation and multiomics to give the first global picture of microRNA dynamics at the single-cell level.
microRNAs (miRNAs) are ∼22 nucleotide (nt) small noncoding RNAs that guide Argonaute effector proteins to target mRNAs in a sequence-specific manner, leading to their translational inhibition and increased degradation through deadenylation and decapping (Bartel 2018). miRNAs group into seed families that mostly target the same pool of mRNAs through complementary base pairing of their 7-nt seed region (Lai 2002; Bartel 2009). Each family targets hundreds of mRNAs, and together, miRNAs are considered key post-transcriptional regulators (Friedman et al. 2009).
It is well established that miRNAs can repress the expression of their targets, at both the RNA and the protein level (Baek et al. 2008; Selbach et al. 2008). However, alternative functions for miRNAs have also been proposed, including buffering of gene expression variation (noise) (Hornstein and Shomron 2006; Tsang et al. 2007; Ebert and Sharp 2012; Siciliano et al. 2013). This is an attractive hypothesis because miRNAs reduce translational efficiency of their targets, which reduces the noise of their targets. Similarly, the repression allows higher transcription of the targets to reach the same final protein output, and this higher transcription again reduces noise according to theory. The hypothesis that miRNAs buffer gene expression noise fits well with observations that many mutant animals that are devoid of specific miRNAs do not display obvious phenotypes under laboratory conditions but only when subjected to stress conditions (Miska et al. 2007; Li et al. 2009).
Another alternative miRNA function that has been proposed is to induce gene expression covariances between targets (synchronize expression) (Rzepiela et al. 2018; Tarbier et al. 2020). In individual cells in which a given miRNA is highly abundant, the targets will be coordinately repressed, whereas in a cell in which the miRNA is lowly abundant, the targets will be coordinately alleviated. This would, for instance, make sense for targets that form part of the same protein complex, in which correct stoichiometry is important for correct assembly and folding (Tarbier et al. 2020; Gutiérrez-Pérez et al. 2021). However, studying these alternative functions require single-cell methods because variation and covariation across individual cells must be measured.
There is evidence for both of these alternative miRNA functions. Almost 10 years ago, a study combining fluorescent reporters bearing binding sites for select miRNAs with single-cell imaging showed that miRNAs can reduce target expression noise for lowly abundant targets, as well as increase target expression noise for highly abundant targets, at the protein level (Schmiedel et al. 2015), and more recent studies extend these observations (Wei et al. 2021). This could serve the biological function of ensuring that the exactly right amount of protein is output in each single cell, which can be particularly important for lowly abundant regulators like transcription factors.
Since then, reliable methods to measure both miRNAs and their targets in single cells using RNA sequencing have emerged. Standard single-cell RNA sequencing methods such as 10x Chromium or Smart-seq3 can detect mRNA targets with high throughout in terms of cells or with high sensitivity in detecting individual mRNA molecules, respectively (Zheng et al. 2017; Hagemann-Jensen et al. 2020). There also exist several protocols to sequence miRNAs in single cells, which are quite similar to the methods used for pooled cell (bulk) applications, but with minor modifications. These methods give representative views of the miRNA profiles of single cells, although they typically detect only a subset of the miRNA molecules in the samples (Faridani et al. 2016; Hücker et al. 2021; Isakova et al. 2021; Chen et al. 2024).
A recent study applied an innovative but labor-intensive method to split the lysate from single cells in two and sequence the small RNA complement from one half and the mRNA complement from the other half (Wang et al. 2019). This study provided evidence that miRNAs are negatively correlated with their targets in single cells and that miRNAs themselves can vary in expression between cells. Further studies have applied single-cell RNA-seq to provide evidence that miRNAs can increase or decrease expression variation of their targets (Hu et al. 2021; Liu and Shomron 2022). Another study provided evidence that miR-182 can cause its targets (including several known pluripotency factors such as NANOG and SOX2) to vary more in expression and that this increased variation is important for transitions between mouse embryonic stem cell (mESC) equilibrium states (Chakraborty et al. 2020). Two independent studies introduced a total of four miRNAs into cells and observed that their targets increased in variation but also that the targets started to covary more (Gambardella et al. 2017; Rzepiela et al. 2018). This covariation was also observed in a recent study from our group, in which we observed that expression covariations between targets disappear in mESCs that were genetically depleted of the key DROSHA biogenesis protein (Tarbier et al. 2020).
In summary, there is emerging evidence that miRNAs have other functions than simple repression of their targets, including intensifying or buffering target expression variation at the RNA or protein level. However, previous studies have focused on limited numbers of miRNAs and targets, and no systematic transcriptome-wide investigation has yet been undertaken.
Here we profile the biogenesis and function of miRNAs in single mESCs by globally profiling miRNAs in single cells, mRNA transcripts in single cells, and mRNA half-lives in the same cell type, with the purpose of studying the interaction between miRNAs and their targets at the single-cell level and yielding new insights into miRNA biogenesis and covariance patterns.
Results
Small-seq detects about 3000 distinct miRNA molecules in each mESC
We sequenced the miRNA complements of 192 individual mESCs using an optimized single-cell small RNA-seq (Small-seq) protocol (Faridani et al. 2016; Hagemann-Jensen et al. 2018) to investigate miRNA variability in these cells. The cells had been cultured in a 2i and LIF medium and sorted in the G2/M cell cycle phase to generate a homogenous population (Methods) (Kolodziejczyk et al. 2015). Following stringent quality control (Supplemental Figs. S1–S3), we found that the number of detected distinct miRNA sequences ranged from 160 to 300, with an average of about 230 distinct miRNAs (Fig. 1A; Supplemental Table S1). This number is higher than that reported in a recent benchmarking study (Hücker et al. 2021), highlighting the high quality of our data. We found that the miRNA with the highest expression was miR-290a-5p, which was detected in about 500 molecules per cell on average (Methods) (Fig. 1B). Summing over all miRNAs, we found an average of about 3000 molecules per cell. Assuming that one mESC contains about 110,000 miRNA molecules (Calabrese et al. 2007), comparable to measurements in other mammalian cells (Bissels et al. 2009), this suggests that the sensitivity of our method is ∼3%. We found that the miRNA composition is relatively similar across cells, showing mostly subtle differences (Fig. 1C). On average, the 10 most highly expressed miRNAs comprised ∼55% of the total miRNA molecules in a given cell, comparable to bulk data (Lappalainen et al. 2013). In summary, we find that miRNA complements in single mESCs can be profiled to fine resolution, even though the sequencing has relatively low sensitivity.
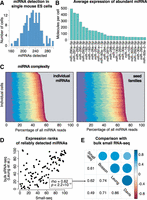
miRNA profiling in single mouse embryonic stem cells (mESCs) by Small-seq. (A) Number of distinct miRNAs detected in each single cell. (B) Top 25 miRNAs in mESCs. The number of detected molecules was estimated using unique molecular identifiers (UMIs) to resolve PCR duplicates. (C) Expression of top 10 miRNAs in 192 individual single cells. The gray area represents the contribution of all miRNAs other than the top 10 ones. (Left) Individual miRNAs: miR-290a-5p, miR-291a-5p, miR-293-3p, miR-295-3p, miR-292a-3p, miR-182-5p, miR-183-5p, miR-26a-5p, miR-294-3p, and miR-291a-3p. (Right) Families: miR-291a-3p/294-3p/295-3p/302abd-3p, miR-290a-5p/292a-5p, miR-291-5p, miR-293-3p, miR-25-3p/32-5p/92-3p/363-3p/367-3p, miR-292a-3p/467a-5p, miR-182-5p, miR-30-5p/384-5p, miR-26-5p, and miR-183-5p. (D) Expression rank of miRNAs detecting in mESCs by Small-seq and bulk small RNA-seq (Leung et al. 2011). Lower rank means higher expression. (E) Spearman's rank correlation coefficient between miRNA expression as profiled by Small-seq and three representative bulk small RNA-seq data sets (Leung et al. 2011; Davis et al. 2012; Bosson et al. 2014).
Small-seq data correlate well with bulk small RNA-seq data
The miRNA complement of mESCs has been profiled in bulk cells in several studies. We find that the expression ranks of miRNAs in bulk small RNA-seq data correlate well with Small-seq (here the data from all single cells were pooled) (Fig. 1D). The Spearman's rank correlation coefficient of miRNA expression ranks ranged from 0.49 to 0.61 compared with three representative bulk studies (Fig. 1E; also see Supplemental Figs. S4–S6). Some differences may in fact reflect biological differences in culture conditions: All available bulk sRNA sequencing data stem from mESCs cultured in serum and mLIF medium only, whereas our cell population has been grown in the 2i and LIF medium (Methods). We conclude that Small-seq overall correlates well with bulk sequencing methods, within typical variability of distinct omic methods and cell growth conditions.
miR-183 and miR-294 are abundant and variable across single cells
miRNAs show highly variable expression between tissues (Keller et al. 2022); however, little is known about the variability between individual cells belonging to the same tissues or cell culture. An advantage of single-cell sequencing over bulk sequencing is that variability of expression across cells can be measured. We first explored the cells using principal component analysis (PCA), and as expected, our homogeneous cell population groups together without clear outlier cells (Fig. 2A; Supplemental Fig. S7). However, although some cell pairs show similar expression profiles (Fig. 2B), other pairs show substantial differences (Fig. 2C). We next compared the mean expression of individual miRNAs against their expression variation across all cells (Fig. 2D). As is typical of single-cell sequencing data, the mean expression and expression variation are inversely correlated, resulting in a negative slope, because of technical sampling noise (Grün et al. 2014). However, some miRNAs were clearly more variable than expected for technical reasons, indicating biological variability (see Fig. 2D, miRNAs indicated in red). Some miRNAs, exemplified by miR-16, were confined to narrow ranges of expression, whereas others, such as miR-183 and miR-294, were variably expressed across individual cells (Fig. 2E). We next compared the mean miRNA expression against miRNA half-lives, as measured in a previous study (Kingston and Bartel 2019). We found that variable miRNAs tend to have high expression and long half-lives (Fig. 2F, top), whereas stably expressed miRNAs often have low expression and short half-lives (Fig. 2F, bottom). We would expect that miRNAs with short half-lives are stable, because fast turnover allows for rapid transcription, which in turn reduces expression variation (noise). It is surprising that highly expressed miRNAs tend to be more variable, but this could be specific to mESCs, in which cell transitions, including differentiation, might depend on induction of a few specific miRNAs. Overall, we find that most miRNAs are stably expressed in mESCs, whereas a few, such as miR-183 and miR-294, are variable.
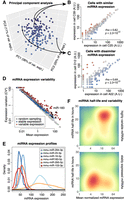
miRNA expression variation across single mESCs. (A) Principal component analysis (PCA) of 192 single cells by their miRNA expression profiles. (B) Comparison of miRNA expression between two cells with similar profiles. Each dot indicates expression of one miRNA. (C) Comparison of miRNA expression between two cells with dissimilar profiles. (D) miRNA mean expression versus expression variation. Each dot indicates one miRNA. Red color indicates variably expressed miRNAs, and blue indicates stable expressed miRNAs. The gray dots are generated by random Poisson sampling and indicate expected technical noise. Specifically, for each miRNA, the actual mean expression across cells was plotted, whereas the variation was randomly sampled from a Poisson distribution, using the mean expression as the Poisson parameter λ. (E) Expression of six select variable (red) and stable (blue) miRNAs across 192 cells. The density profiles are smoothened. (F) Heatmaps of variably expressed (top) and stably expressed (bottom) miRNAs, indicating mean expression versus miRNA half-life. Red color indicates the presence of several miRNAs with those features; blue color indicates the absence of miRNAs with those features.
miRNAs form four distinct coexpression groups across single cells
Another advantage of single-cell sequencing is that covariations of miRNAs across single cells can be detected. Grouping miRNAs and cells according to their expression revealed clear global patterns (Fig. 3A). We find several miRNAs that group together according to their expression profiles, including from top to bottom: (1) miR-26a, miR-30e, miR-182, miR-183, and miR-298; (2) miRNAs that derive mainly from the 5′ arms of the mir-290 genomic cluster; (3) miRNAs that derive mainly from the 3′ arms of the mir-290 cluster; and (4) a cluster composed of miR-16, miR-25, miR-92, miR-103, and miR-130a. Curiously, there is a group of cells that express many mature miRNAs from both the 3′ arm and the 5′ arm of the miRNA genes from the mir-290 cluster (cell group A) and a group that express few mature miRNAs from both arms of genes from this cluster (group C). At the same time, there are other groups of cells that show discordant expression of these miRNA groups. Specifically, in cell group B, miRNAs stemming from the 3′ arm of the mir-290 cluster are more abundant than those originating from the 5′ arm, whereas in group E the reverse holds. This suggests that there may be unknown global protein cofactors present in individual cells that drive preferential processing of either arm of the hairpin (see below). miR-293 seems to act contrary to all other members of the mir-290 cluster, with its 3′ arm following the expression pattern of the 5′ arm of the other mir-290 members, and its 5′ arm following the pattern of the 3′ arms of the other cluster members. In summary, we find that genomically clustered miRNAs, but also some unclustered miRNAs, follow each other in expression patterns and that strand selection of miR-293 appears to be contrary to that of the other cluster members. These observations prompted a more in-depth analysis into the expression of individual miRNAs and global processing patterns.
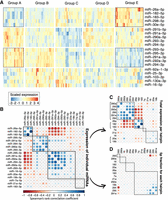
miRNA expression covariation across single mESCs. (A) Four distinct miRNA coexpression groups across 192 cells. The miRNAs (rows) and cells (columns) were grouped using unsupervised hierarchical clustering. Red color indicates high expression relative to the mean for the given miRNAs; blue color indicates low expression. (B) Correlation between expression of miRNA strands. Blue color indicates positive correlation, and red indicates negative correlation. (C) As in B, but summing expression from 5′ and 3′ arms of each miRNA hairpin. (D) As in B, but the correlation was done on the fraction of sequenced RNAs that originate from the 5′ arm of the hairpin. Blue color indicates that the two miRNA hairpins have similar arm select across the cells, and red color indicates that the two hairpins have opposite arm selection across cells.
miRNAs from the mir-182 and mir-290 clusters are anticorrelated in expression
We next changed our focus from expression patterns of groups of miRNAs to specific expression covariations of pairs of individual miRNAs (Methods). These analyses largely recapitulate three of the four groups identified above: (1) miR-26a, miR-182, and miR-183; (2) miRNAs that derive mainly from the 5′ arms of the mir-290 genomic cluster; and (3) miRNAs that derive mainly from the 3′ arms of the mir-290 cluster but also include miR-16 and miR-20a (Fig. 3B). The former is anticorrelated with the latter two, more so with the 3′ arms than with the 5′ arms. The two arms of the mir-290 cluster group more strongly among themselves but show no anticorrelation with one another. Summing up mature miRNAs from both arms to estimate overall miRNA precursor expression, we now see that all members of the mir-290 cluster group strongly together and are negatively correlated with miR-26a, miR-182, and miR-183 (Fig. 3C). We can also clearly see the coexpression within the mir-182 and mir-183 genomic cluster and within the mir-17 cluster (miR-19b and miR-20a). The covariation of miRNA originating from the same genomic clusters suggests that cotranscription is a major player in shaping mature miRNA expression profiles.
Each single mESC has biases in miRNA arm selection
We next studied arm biases of miRNA precursors, by correlating the fraction of sequenced miRNAs that originate from the 5′ arms of precursors, across cells (Fig. 3D). It appeared that arm selection is not highly correlated between miRNA precursors; however, we can clearly see that the 5′ fraction of miR-293 is strongly anticorrelated with that of other members of the mir-290 cluster, whereas these other members show positive correlations of their 5′ fractions. Further, close observation reveals that in fact the 5′ fractions of many miRNAs are subtly and positively correlated with each other and anticorrelated with the 5′ fraction of miR-293 (Supplemental Figs. S8, S9). This suggests that there may be global arm biases in certain cells, possibly mediated through the activity of a as-yet-unknown protein cofactor that impacts all miRNAs equally, with the notable exception of miR-293.
pri-miRNAs from the mir-17 and mir-290 clusters are positively correlated in expression
Although Small-seq is optimized for detecting mature miRNAs and does not detect miRNA primary transcripts (pri-miRNAs), we detect these in data from our previous study, in which we applied the Smart-seq2 method to profile polyadenylated transcripts in single cells that were also sorted in the G2/M cell phase (Supplemental Table S2; Tarbier et al. 2020). In cells genetically depleted for Drosha, we detect large parts of the pri-miRNA transcripts, which is expected because the protein product is needed for cleaving the primary transcripts to produce miRNAs (Fig. 4A, below). In control cells that express DROSHA protein, we mainly detect cleaved transcripts (Fig. 4A, above). This is consistent with previous observations that cleaved pri-miRNAs remain in the chromatin for some time after processing (Conrad et al. 2014), and also, pri-miRNAs have previously been reported in sequencing of single nuclei (Elias et al. 2023), where they are located. The paucity of full-length pri-miRNAs in the control cells further suggests that pri-miRNA are cleaved relatively fast after being transcribed in mESCs. We used the levels of pri-miRNAs in the Drosha knock-out cells as a proxy for transcription (Fig. 4B) and found that the mir-17 and mir-290 clusters were strongly covarying (correlation coefficient 0.6) (Fig. 4C,D). This suggests that the positive covariation between the miRNAs from the mir-17 cluster and the 3p-derived miRNAs from the mir-290 cluster (Fig. 3B) may be induced at the transcriptional level, whereas the negative covariation between the mir-17 cluster and the 5p-derived miRNAs from the mir-290 cluster may be induced post-transcriptionally. Unfortunately, we could not robustly profile expression of the mir-182 cluster in these data, so we cannot resolve if the anticorrelation between the mir-290 and mir-182 clusters is induced at the transcriptional level. Overall, we use single-cell data to infer expression of pri-miRNAs and infer their dynamics and interdependencies.
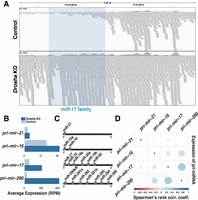
Expression covariation of miRNA primary transcripts. (A) Genome browser shot of miRNA primary transcripts from the mir-17 cluster sequenced by Smart-seq2. The clustered miRNAs (in blue shading) are cleaved out in the control cells (above) but are retained in the Drosha KO cells (below). Each gray shading indicates one sequence read; the 3′ end of the primary miRNA is to the right. (B) miRNA primary transcript expression in control and Drosha KO cells. (C) Schematic of mature miRNAs that are part of the same miRNA primary transcript (transcribed from the same genomic cluster). (D) Expression covariation of miRNA primary transcripts. The color code indicates the Spearman's rank correlation coefficient value. Blue color indicates positively covarying primary transcripts.
Most miRNA repression is conferred by miR-17, miR-291, and miR-292 seed families
We next investigated the impact of miRNAs on their target expression by comparing single-cell RNA-seq data from Drosha knockout cells with control cells (Tarbier et al. 2020). The overall correlation between expression in the knockout and control cells is comparable (Spearman's rank correlation coefficient, rho-value = 0.95). We found that computationally predicted TargetScan targets (Agarwal et al. 2015) of the 10 most highly expressed miRNAs were on average derepressed ∼15%, consistent with findings from bulk studies (Methods) (Baek et al. 2008; Selbach et al. 2008). However, this average repression effect was small relative to the overall natural variation of expression between cells (Fig. 5A), suggesting that the natural variation of expression overshadows the impact of the miRNAs at the RNA level (see Discussion section). We next looked at median changes of TargetScan targets in Drosha knockout versus control cells for the top 16 miRNA families (Fig. 5B,C). We found the miR-17, miR-291-3p, and miR-292-3p seed families to have the strongest impact on the transcriptome, consistent with their relatively high measured expression in bulk and single cells (Fig. 5B, insets). We did not observe any evidence of cell size differences between the genetically ablated and the control cells that could perturb our analyses (Supplemental Figs. S10, S11), and we observed strong repression by miR-291a-3p and miR-292a-3p even when we disregarded overlapping targets (Supplemental Fig. S12). The miR-290-5p and miR-291-5p families appear to have little impact on their targets’ expression despite being fairly abundant. This may suggest that these miRNAs are indeed just abundant by-products that are not loaded into Argonaute or that they may have functions other than simple repression (see Discussion section).

miRNA target repression in single cells. (A) Expression of miRNA targets in cells devoid of miRNAs (Drosha KO) and control cells (parental). The expression is normalized so that a cell with mean expression for the given target is assigned an expression value of one. The density plot is made of a compound of the predicted TargetScan targets for the top 10 expressed miRNAs in mESCs (Methods). The mean expression of the targets is upregulated (derepressed) by ∼15% in the Drosha KO cells. (B) Cumulative distribution function (CDF) plots of the expression of the targets of top miRNAs in mESCs. The inset boxes show miRNA expression ranks according to bulk small RNA-seq (upper left corner) and Small-seq (lower right corner). (C) Heatmap of miRNA target derepression in Drosha KO versus control cells. The color indicates the log fold-change in expression, with red indicating stronger derepression. The targets are sorted according to confidence level, as estimated by the TargetScan cumulative score. (D) As in C, but showing changes to transcript half-lives in Drosha KO versus control cells.
To gain more confidence in these observations, we estimated mRNA half-lives transcriptome-wide through a time-series experiment of transcriptionally inhibited mESCs. Specifically, we inhibited transcription with α-amanitin and measured mRNA levels at five time points following the transcriptional arrest and estimated half-lives using a linear model (Methods) (Supplemental Table S3). These experiments were conducted in control cells and cells genetically depleted for Dgcr8, which from a perspective of miRNA biogenesis and function are functionally very similar to Drosha-depleted cells (Ha and Kim 2014). We observed the same families to have strong target effects as in the previous analysis (Fig. 5D), showing the robustness of these results and confirming that expression changes reflect changes in half-life rather than indirect effects at the transcriptional level. In summary, in our single cells we find that most repression is conferred by three seed families—miR-17, miR-291, and miR-292—consistent with their high expression in bulk small RNA-seq data and also our mRNA half-life measurements.
Distinct miRNAs increase and decrease target expression variation at RNA level
It is known from studies with reporter constructs that miRNAs can buffer gene expression variation at the protein level (Schmiedel et al. 2015). Evidence from miRNA overexpression studies indicate that miRNAs may increase variation at the RNA level (Gambardella et al. 2017; Rzepiela et al. 2018); however, transcriptome-wide evidence from natural cell conditions is still lacking. We leveraged our published single-cell RNA-seq and find that miRNA targets naturally tend to be more variable in expression at the RNA level than are other genes (Fig. 6A). This particularly holds for targets of miR-291a, which is one of the most highly expressed miRNAs in mESCs (Fig. 5B). We further find that miRNA targets generally decrease in expression variation when Drosha is ablated, suggesting that they increase variation in their targets at the RNA level (Fig. 6B). This in particular holds for targets of highly expressed and strongly repressing miRNAs like miR-291a but also for miR-290a-5p, which although abundant, confers little repression (see above). The targets of let-7 and miR-182 increase in variability when Drosha is ablated, suggesting that these miRNAs naturally decrease variability of their targets at the RNA level. In summary, we provide evidence that miRNAs naturally induce expression variability of their targets at the RNA level; however, select miRNAs have the exact opposite effect.
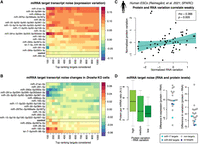
Expression variation of miRNA targets in single mESCs. (A) Heatmap of miRNA target expression variation across single cells, without perturbation and not sorted into G2/M cell phase. The color code indicates expression variation (noise) as estimated by coefficient of variation squared (CV2) residuals (Methods). Red color indicates that the targets of the miRNA are naturally more variable. (B) As in A, but showing changes in expression variation following Drosha knockout. Blue color indicates miRNAs whose targets decrease in variation upon the loss of the DROSHA biogenesis protein. (C) Expression variation for select genes at the RNA and protein level. Measurements were performed using combined single-cell RNA and protein profiling in the same single human embryonic stem cells (Reimegard et al. 2021). (Lower left) Estimated translational efficiency and expression noise. The normalized RNA variation was for select genes subtracted from the normalized protein variation, and the genes were divided into three groups: genes with higher protein variation, genes with comparable RNA and protein variation, and genes with higher RNA variation. For each group, the estimated translational efficiency (Methods) was plotted. (D) Difference between protein and RNA expression and difference between protein and RNA variation. Background nontarget genes were selected to have similar expression as the targets and are marked in gray, and miR-17 and miR-302 targets were marked in light blue and blue, respectively. Genes that are regulated by both miRNAs are marked in dark blue.
Evidence that targets of miR-17 and miR-302 are buffered at the protein level
We next investigated if RNA and protein variation correlates in single cells. In collaboration with colleagues, we recently developed SPARC, a new method to profile the whole polyadenylated transcriptome and select proteins in the same single cells (Reimegård et al. 2021). Reanalyzing SPARC data from human embryonic stem cells, we found that RNA variation is a relatively poor predictor of protein variation for the same gene, in single cells (Fig. 6C, above; Reimegård et al. 2021). We found that proteins that are less variable than their cognate mRNAs tend to have high protein-to-mRNA ratios, whereas the reverse holds for proteins that are more variable (Methods) (Fig. 6D, left). Further, when estimating the ratio of RNA to protein, we found that the targets of miR-17 and miR-302, both abundant in human embryonic stem cells, are lower than for background genes (Fig. 6D, right; Supplemental Fig. S13), suggesting translational repression. Last, we found that protein variation was lower than RNA variation for targets of miR-17 and miR-302, suggesting that the miRNA may increase RNA variation but buffer protein variation, consistent with previous studies on miRNA impact on expression noise (Fig. 6D, right). However, these observations are preliminary, and more methods development in the single-cell proteomics field will be required to elucidate this in a more systematic manner.
Variable miRNAs induce strong gene expression covariations between target transcripts
There is evidence that certain miRNAs have the potential to induce covariations within their target pool (Rzepiela et al. 2018), and we have previously provided evidence that these effects may in fact occur naturally for many miRNAs (Tarbier et al. 2020). Here we reanalyze our single-cell RNA-seq data in more detail and find that targets of several specific miRNA families naturally covary in mESCs (Fig. 7A). We find that most miRNAs that induce such covariation patterns among their targets, such as miR-294-3p, were themselves variable (cf. Figs. 2E and 7A). Abundant miRNA families with low expression variation, such as miR-16 (part of the miR-15 family), do not seem to induce target c-expression (cf. Figs. 2E and 7A). Overall, miRNA abundance does not seem to determine covariation of the targets, rather the variability of the miRNA itself influences the level of covariations (Fig. 7B). Because variation and covariation patterns within a cell population cannot be measured with bulk methods, this again highlights the importance of single-cell miRNA quantifications in understanding miRNA function. A striking outlier in these analyses is the let-7 family, whose targets are negatively covarying (asynchronous) (Fig. 7A). This miRNA family is lowly abundant in mESCs but has been included for its well-described function in exit from pluripotency (Gambardella et al. 2017). Because it is lowly abundant in mESCs and thus might not confer strong regulation of its targets, we speculate that its apparent effects in negative covariations could be linked to the function of its targets rather than direct miRNA effects. In summary, we provide evidence that several miRNA families can induce covariation between their targets and that this particularly holds for miRNA families that are variable in expression between cells.

Expression covariation of miRNA targets in single mESCs. (A) Heatmap of miRNA target covariation across single cells. The covariation enrichment was performed as previously described (Tarbier et al. 2020). Blue color indicates mRNAs that are coexpressed more frequently than expected by chance; red color indicates mRNAs that are coexpressed less frequently than expected by chance. (B) Covariance enrichment for targets of miRNAs belonging to different groups, considering the top expressed 16 miRNA families. “Variable” indicates the eight miRNA families with the highest expression variation, and “stable” indicates the eight miRNA families with the most stable expression. “Abundant” indicates the eight top expressed miRNA families, and “sparse” indicates the eight least expressed miRNA families among this set.
Discussion
Here we perform the first systematic and transcriptome-wide study of miRNA and target expression variation and covariation in single mESCs. We show that miRNA profiles in single cells largely resemble their bulk counterparts, although the single-cell measurements can be used to estimate miRNA and target expression variation and covariation. We find that the sensitivity of miRNA single-cell sequencing is ∼3%, which is lower than that of single-cell RNA-seq methods such as SmartSeq3 (Hagemann-Jensen et al. 2020). Similar to findings in bulk data, we find that the top 10 miRNAs in mESCs comprise most of the detected miRNA molecules. Most miRNAs are stably expressed in single cells, although a notable few are highly variable. In particular, miRNAs from the mir-290 cluster and the mir-182 cluster are variably and negatively correlated. We show that miRNA covariation profiles are shaped by the transcription of genomic clusters and by a tendency for each single cell to favor miRNAs originating from either end of the hairpin precursors. This bias acts in a similar fashion on all miRNAs in a given cell and suggests the existence of unknown protein cofactors whose presence or absence drives this bias. The majority of repression in mESCs is conferred by the miR-17, miR-291, and miR-292 families. Overall, we find that the natural variation of mRNA expression appears to overshadow the repressive effect of miRNAs (∼15% repression), at least on the RNA level. Most miRNAs increase noise of their targets at the RNA level, but we provide evidence from published single-cell proteomics data that they may reduce noise at the protein level. Lastly, miRNAs that are themselves highly and variably expressed tend to induce expression covariations between their targets.
In the field of sequencing mRNAs, there exists methods that have a sensitivity of >50%, meaning the probability that a given transcript molecule will be represented in the generated sequence data (Hagemann-Jensen et al. 2020). In comparison, we here estimate that our Small-seq method has a sensitivity of ∼3%, which is comparable or better than other single-cell miRNA sequencing methods tested (Hücker et al. 2021). This means that methods to sequence miRNAs in single cells are more dominated by stochastic sampling noise compared with ordinary single-cell RNA-seq and sets limits to the questions that can be quantitatively addressed. However, we here show that biological insights can be inferred from Small-seq data when we focus on the 10–20 most highly abundant miRNA molecules. Limiting analyses to the most highly abundant miRNAs may not be a major problem, because the 10 most expressed miRNAs both dominate the expression profile (Fig. 1C) and also likely confer the most of the functional repression (Mullokandov et al. 2012).
We observe that the natural variation of expression of miRNA targets appear to overshadow the relatively subtle repression conferred by miRNAs, at least at the transcript level (Fig. 5A). This could indicate that miRNAs may have important functions other than repression, such as regulating target expression variation or covariation. Some of the variation we observe in miRNA targets is also technical, although we only consider transcripts for which the biological variation substantially exceeds the measured technical noise (Tarbier et al. 2020). It has been proposed that miRNAs can influence their target expression variation in three ways (Schmiedel et al. 2015). First, by repressing their targets, the miRNAs allow the target genes to have higher transcription rates while retaining the same gene output, thus reducing transcriptional noise. Second, because the miRNAs themselves are variable, some of this variability is transferred to their targets at the RNA level. Third, by inhibiting translation, the miRNAs reduce translational noise. Because of our experimental setup, we mainly detect the second effect in our study, and therefore, it is not surprising that we find that most miRNAs increase the variability of their targets at the RNA level. Importantly, it should also be considered that the target transcripts are translated to proteins, which typically have longer half-lives than transcripts. In this way, any temporal fluctuations at the transcript level may be buffered at the more stable protein level. Also there is clear evidence for miRNA-induced noise reduction at the protein level (Schmiedel et al. 2015) and for miRNA-induced covariation for proteins that form part of the same complexes (Gutiérrez-Pérez et al. 2021).
A main finding of our miRNA covariation analyses is that the mir-290 and mir-182 clusters are variably expressed and negatively correlated, suggesting a potential “switch-like” mutually exclusive function between the two clusters. This is interesting, given that both clusters are involved in maintaining pluripotency and in stem cell self-renewal (Judson et al. 2009; Wang et al. 2017). Specifically, members of the mir-290 cluster perform this function by indirectly upregulating Myc, Lin28a, and Sall4 (Melton et al. 2010), whereas the mir-182 cluster represses Ago2 (Liu et al. 2021), a miRNA-interacting protein that can facilitate differentiation through for instance let-7 (Suh and Blelloch 2011). In addition, members of the mir-290 cluster can advance progression through the cell cycle by targeting inhibitors of Cdk2-Ccne1 (Wang et al. 2008), and the mir-182 cluster can facilitate transitions between stem cell states by increasing variability of specific target genes such as Esrrb and Sox2 (Chakraborty et al. 2020). It seems unlikely that differences between cells that have high mir-290 expression and cells with high mir-182 relate to cell cycle, given that the mESCs in our study were sorted by cell phase (Methods). However, it seems possible that any switch-like behavior relates to different stem cell states, reflecting different compositions of pluripotency factors.
We further show that miR-293 is, in many ways, an outlier in the mir-290 cluster. It is the only abundant miRNA that targets GC-rich genes (Supplemental Fig. S14) and is an outlier with regard to its expression patterns and predicted targets in mESCs (Ciaudo et al. 2009). miR-293 further appears to escape the global cellular precursor arm bias and in fact may be regulated exactly opposite from all other miRNA, including its cotranscribed clustered neighbors, by whatever factors drive said bias. This makes it an extremely interesting candidate for future research into miRNA processing.
We find that the passenger strands (5′ arms) of the mir-290 cluster are among the most abundant miRNAs in mESCs but have virtually no effect on their targets, as measured by mRNA steady-state levels and half-lives (Fig. 5C,D). This suggests they may accumulate as by-products of the mature mir-290 cluster miRNAs but are not loaded into Argonaute; however, their stability has previously been confirmed through miRNA half-life estimates (Kingston and Bartel 2019). Given that these miRNA strands have unusually many potential binding sites in the transcriptome, it is also possible that they are effectively titrated away in the large pool of binding sites (Bosson et al. 2014).
Alternative miRNA functions are still understudied. The limited evidence we have to date stems from mathematical models, reporter assays, or overexpression experiments and is often limited to few miRNA–target interactions. We here reexamine existing hypotheses and add physiological evidence for thousands of miRNA–target interactions at the RNA level. Furthermore, we show that miRNA expression variability links to target covariation, highlighting the importance of intercellular miRNA heterogeneity.
In summary, although several studies have profiled miRNAs and their targets in single cells, there is a paucity of systematic and transcriptome-wide studies in individual mESCs. We here outline the landscape of miRNA and target expression variation and covariation, and we find that most miRNAs are stably expressed, with the notable exception of a few miRNAs. The mir-290 and mir-182 clusters are variably expressed and negatively correlated, suggesting switch-like functions in defining distinct stem cell states. We further find that most miRNAs induce expression variation at the RNA level, but some may buffer variation at the protein level. miRNAs that are themselves highly and variably expressed induce covariations between their targets. Lastly, miRNA primary transcripts can be robustly detected in whole-cell single-cell RNA-seq data, opening up new possibilities in studying miRNA biogenesis at fine resolution.
Methods
Cell sorting, library preparation, and sequencing
The DroshaF E14 129Sv-derived mESC line, a kind gift of M. Chong (Chong et al. 2008), was maintained in standard 2i media containing Ndiff227 medium (Cellartis Takara Bio Y40002), 1000 U/mL ESGRO mouse LIF medium supplement (Millipore Merck ESG1107), 1 µM PD0325901 (Selleckchem S1036), 3 µM CHIR-99021 (Selleckchem S1263), and 1× penicillin-streptomycin (Gibco 15070063) onto feeder-free 0.1% EmbryoMax gelatin (Merck Millipore ES006-B)–coated flasks at 37°C, 5% CO2. Cells were routinely tested for mycoplasma contamination (Eurofins Genomics). Cells were propagated in 2i media for at least two passages prior single-cell sequencing. To harvest cells, incubation with Accutase (Sigma-Aldrich A6964) was used for 5 min at 37°C followed by centrifugation. Cells were resuspended in 2i media (about 106 cells/mL), DNA-stained with 10 µg/mL Hoechst-33342 (Sigma-Aldrich 14533) for 15 min at 37°C, and then stained with 1 µg/mL propidium iodide (Sigma-Aldrich P4864) to reveal cell viability. Single cells were sorted in G2/M using a BD Influx (BD Bioscience) into 96-well plates containing 3 µL of lysis buffer (0.13% Triton-X-100 [Sigma-Aldrich T8787], 4 U RNase inhibitor [Takara 2313A]) in each well. The single cells were distributed into four sequencing libraries pools; each pool comprised 48 distinct Illumina TruSeq small RNA barcodes. Libraries were prepared according as described previously (Faridani et al. 2016). Library pools were quantified with Qubit dsDNA high-sensitivity range assay (Invitrogen Q32854); the average library size was assessed using the Agilent DNA high-sensitivity kit (Agilent Technologies 5067-4626) and sequenced in single-end mode on a NextSeq 500 (Illumina). In total, 192 mESCs were sequenced using a 75-cycle high-output kit v2.5 (Illumina 20024906).
Sequence data preprocessing
Demultiplexing of the sequencing reads has been done with Illumina's bcl2fastq conversion tool (version 2.17), allowing zero mismatches in the cell barcode. This excludes the possibility of a false assignment of reads to a different cell. Even with one allowed mismatch, there would only be three barcodes (cells) that could get wrongly assigned reads. All 48 barcodes used have a Hamming distance of three with the exception of barcodes 11 (GGCTAC) and 41 (GACGAC; Hamming distance 2) and barcodes 31 (CACGAT) and 41 (Hamming distance 2). After demultiplexing, the 3′ adapter was identified (if present) and clipped from each read with a custom script. In particular, the first 8 nt of the adapter sequence “TGGAATTCTCGGGTGCCAAGG” was used, allowing no mismatches. Further, each read sequence is supposed to have a 10-nt-long unique molecular identifier (UMI) at its 5′ end, allowing tagging of every distinct RNA molecule in a cell. Nucleotides 9 and 10 have been fixed to be “CA,” and nucleotides 1–8 should be devoid of guanosines. Reads that have a UMI different from those or <18 nt after adapter and UMI removal have been discarded. After that, a second round of adapter removal was performed on reads for which the correct adapter sequence could not be identified in the first run. This time one mismatch or a single deletion in the first 8 nt of the adapter sequence was allowed. Further, we also checked for the presence of the adapter sequence for nucleotides 3–8 of the adapter and clipped the sequences off.
miRNA quantification
To remove PCR duplicates in a sequenced cell, we collapsed identical read sequences that have the same UMI to a single FASTA entry and noted the number of different UMIs as well as the number of all identical sequences in its ID line. The whole preprocessing of all 192 single mESCs is done with the preprocess.pl script, which also creates unique ids for each sequence and each cell. The output files were input to our custom software “quantify2” to count miRNA sequences using this command line: “quantify2 -r config_file -C -y tag_for_run -t Mouse -A -U -P -T 4 -d -S -N -F -R -B -g 0 -p mmu_hairpins_miRNAs.fa -m mmu_mature_miRNAs.fa.” The custom software is available at GitHub (https://github.com/Drmirdeep/quantify/). The annotation files used were from miRBase version 21. The hairpin sequence file was cleaned of duplicates, and only one of each hairpin was kept; for example, mmu-miR-297a has four precursors annotated in miRbase, and only miR-297a-1 is kept. miR-5099 and miR-2137 were excluded from all analysis because the former is only 1 nt different from 45S preribosomal RNA and the latter to 28S rRNA. Their high abundances are unexpected and likely stem from these ribosomal RNAs. mir-1957 family members were excluded because they reported to be fragments of tRNA sequences.
miRNA normalization
Because there were about 5000 unique reads in each cell, we scaled all miRNA reads in the same cell to add up to 5000. We thereby normalize to the respective sum of miRNA reads in each cell. Because one normalized read remains close to one actual read, this method facilitates data interpretation. We compared this normalization to normalization by size factors estimated by SCRAN (Lun et al. 2016) and size factors estimated via the median of ratios to the geometric means, mimicking DESeq2 (Anders and Huber 2010). All three normalizations yielded similar size factors. For all further analysis, we proceeded with our original normalization by the sum of miRNA reads per cell.
Excluded cells
Cells were excluded from further analysis if their mapping statistics were drastically different from all other cells. For this, we required between 2000 and 20,000 mapped miRNA reads and between 150 and 400 detected miRNA species. In a second step, we performed a PCA and removed all cells that clustered distinctly separate from the remaining cells. These two filtering steps resulted in a homogeneous cell population for downstream analyses.
miRNA complexity
To assess how many different mature miRNAs were detected in each of the sequencing libraries, all 48 mESCs were pooled together. The UMI sequences for each ID in the preprocessed files were reanalyzed and used for removing duplicates again. The maximal UMI count for a miRNA in all pooled 48 cells is thus 6561 = 38. To determine mature miRNA expression in molecules, we counted the number of distinct UMI and miRNA sequence combinations and summed them up. The 5′ end of a read needed to match exactly the annotated mature start position in a miRNA precursor, whereas the 3′ end was allowed to end up 5 nt after the annotated end position. This allows capturing also miRNA molecules with untemplated nucleotide additions or imprecise Dicer processing at the 3′ end of a read without changing the mRNA targets of a miRNA. Isomirs that are 1 or 2 nt offset of an annotated mature miRNA start position have also been kept but were not counted as read counts of their original miRNA.
Expression variation and covariation
Gene expression variation was assessed using the squared coefficient of variation. It is known that variation is dependent on mean expression. Using a least-total squared fit variation was normalized to the mean. The residuals of this fit were considered as mean-independent noise measures. Gene expression covariation was assessed using covariation enrichment score that we previously established (Tarbier et al. 2020).
miRNA seed families
For some analysis, for example, functional considerations, miRNAs that share the same seed sequence are pooled together. miRNA family annotations stem from TargetScan mouse release 7.1.
RNA half-life measurements
RNA half-lives were determined through transcriptional inhibition experiments in control cells (DroshaF) and Dgcr8−/− cells (Wang et al. 2007). The cells were treated with α-amanitin and sequenced after 0 h, 6 h, 8 h, 10 h, 12 h, and 14 h. Library were prepared using the Illumina TruSeq stranded mRNA sample preparation kit and sequenced on an Illumina NextSeq 500 platform. A standard RNA-seq analysis pipeline was applied. In brief reads were trimmed using cutadapt (Martin 2011), aligned to the RefSeq transcriptome using TopHat (Trapnell et al. 2009), and quantified using Cufflinks. Based on linear regression on the later time points, we determined the onset of inhibition to be at ∼5 h, which matches literature on α-amanitin. Using this information, we estimated decay rates and, subsequently, half-times again using a linear model. This approach led to half-live values that are in good agreement with previously published data on RNA half-lives in mESCs (Sharova et al. 2009).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1273632. Source code is available at GitHub (https://github.com/MarcelTarbier/single-cell_miRNA_variation_and_function) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the following funding sources: European Research Council (ERC) Starting Grant 758397 “miRCell,” Vetenskapsrådet (VR) Research Grant 2019-05320 “MioPec,” VR Consolidator Grant 2022-03953 “InSync,” and the Strategic Research Area (SFO) program of the Swedish Research Council (VR) through Stockholm University. We thank Claudia Kutter and Vicente Pelechano and their research groups for support and useful discussions and input. We acknowledge Jaromir Mikes and the SciLifeLab CyTOF facility for assisting with cell sorting and the SNIC/Uppsala Multidisciplinary Center for Advanced Computational Science for assistance with massively parallel sequencing and access to the UPPMAX computational infrastructure.
Author contributions: The study was conceptualized by M.T., B.F., I.B., and M.R.F. Small-seq libraries were prepared by O.R.F. The RNA sequencing and NGS raw data were generated by I.B. Sequence data quality control, preprocessing, and expression quantification were performed by S.D.M. Pri-miRNA analyses were performed by V.S. The time-series experiment was performed by F.B., and data were analyzed by E.Y. under supervision of M.T. All other computational analyses, including miRNA repression, variation, and covariation, were performed by M.T. The manuscript was written by M.T. and M.R.F., with contributions from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279914.124.
-
Freely available online through the Genome Research Open Access option.
- Received August 9, 2024.
- Accepted November 26, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








