
Transcriptomic landscape of transposable elements reveals LTR7-PLAAT4 as a potential oncogene and therapeutic target in pancreatic adenocarcinoma
- Meilong Shi1,10,
- Chuanqi Teng1,10,
- Shan Zhang2,10,
- Xiaobo He3,10,
- Lingyun Xu4,5,
- Fengxian Han4,5,
- Rongqi Wen5,
- Ganjun Yu3,
- Jingwen Liu6,
- Yang Feng7,
- Yanfeng Wu3,11,
- Yan Ren6,8,11,
- Gang Jin1,11 and
- Jing Li4,5,9,11
- 1Department of Hepatobiliary Pancreatic Surgery, Changhai Hospital, Second Military Medical University (Naval Medical University), Shanghai 200433, China;
- 2Center for Translational Medicine, Second Military Medical University (Naval Medical University), Shanghai 200433, China;
- 3National Key Laboratory of Immunity and Inflammation, Institute of Immunology, Naval Medical University, Shanghai 200433, China;
- 4School of Health Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China;
- 5Department of Precision Medicine, Changhai Hospital, Second Military Medical University (Naval Medical University), Shanghai 200433, China;
- 6Experiment Center for Science and Technology, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China;
- 7College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China;
- 8HIM-BGI Omics Center, Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Zhejiang, Hangzhou 310022, China;
- 9State Key Laboratory for Macromolecule Drugs and Large-scale Manufacturing, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325030, China
-
↵10 These authors contributed equally to this work.
Abstract
Eukaryotic genomes contain numerous transposable elements (TEs), whose dysregulation threatens genome stability and may contribute to cancer. Pancreatic adenocarcinoma (PAAD) is among the deadliest cancers, marked by abundant stroma that obscures tumor-specific molecular signals, complicating bulk-tissue analyses. Here, using 71 patient-derived PAAD organoids, we show that TE activities may potentially promote tumorigenesis and provide a source of novel immunotherapeutic targets. We identify 16 new TE-derived transcripts fused with 15 known oncogenes, exhibiting potential oncogenic function and prognostic value. Notably, LTR7-PLAAT4, present in 29% of tumors, encodes a protein variant transcriptionally regulated by FOXM1 binding to the LTR7 promoter. LTR7-PLAAT4 isoform 2 is associated with increased cholesterol ester accumulation and lipid droplet formation mediated through BSCL2 coexpression, potentially fostering tumor progression. On the immunogenic front, HLA-I immunopeptidomics of AsPC-1 cells and DAC13 organoids identify over 11,000 peptides respectively. Althought mutation-derived neoantigens are rare, several peptides are originated from TE-chimeric transcripts, including four predicted by TEprof2. The peptide FLIQHLPLV, detected in 27% of organoids, exhibits robust immunogenicity, validated by T2 binding, mass spectrometry and ELISPOT assays with HLA-genotyped PBMCs. Together, these findings suggest that TE activities may contribute to PAAD progression and diversify its immunopeptidome, providing new opportunities for molecular subtyping and potential immunotherapeutic intervention.
Pancreatic adenocarcinoma (PAAD) is one of the most lethal malignant neoplasms across the world and a leading cause of cancer mortality, especially in developed countries (Siegel et al. 2024). By 2030, PAAD is expected to become the second most fatal cancer, following only lung cancer (Rahib et al. 2014). Unlike the majority of other cancers, five- and 10-year survivals for pancreatic cancer have not shown much improvement since the early 1970s (Dbouk et al. 2022). A hallmark of PAAD is its abundant desmoplastic stroma, which significantly complicates the isolation and analysis of molecular signals originating specifically from neoplastic epithelial cells, particularly when using bulk tumor samples. Studies have estimated the median tumor purity of bulk sequencing samples to be <40%, underscoring the limitations of this approach (Biankin et al. 2012; Bailey et al. 2016; Cao et al. 2021). Several studies have addressed this challenge using sophisticated bioinformatics techniques (Elyada et al. 2019), labor-intensive laser capture microdissection (Maurer et al. 2019), FACS cell sorting (Espinet et al. 2021), and more recently, (spatial) single-cell sequencing approaches (Moncada et al. 2020; Ma and Zhou 2022). Recent advances in organoid models represent a transformative development in cancer research. Organoids, cultured in vitro, retain self-renewal and self-organizing capabilities that mimic the structure and function of their tissue of origin. The high consistency between organoid and bulk tumor samples has been demonstrated across multiple cancer types, including glioblastoma (Jacob et al. 2020) and PAAD (Shi et al. 2022). Patient-derived organoids, which achieve nearly 100% tumor purity while preserving intratumoral heterogeneity (Shi et al. 2022), have been used to generate high-quality sequencing data and minimize confoundment by nontumor components. These organoid models also enable high-throughput drug screening, providing valuable insights into therapeutic responses for PAAD, a disease notorious for its lack of effective treatments.
Most studies on PAAD have focused on coding regions, investigating gene mutations (Hayashi et al. 2021) and coding gene expression (Moffitt et al. 2015) to elucidate oncogenic mechanisms, identify biomarkers, and guide treatment choices. In contrast, epigenetic alterations remain underexplored. One study stratified pancreatic ductal adenocarcinoma into two distinct subgroups based on specific DNA elements, such as transposable elements (TEs), although it was limited by the use of labor-intensive FACS sorting with small sample sizes (Espinet et al. 2021). TEs exhibit significant regulatory and functional potential. For instance, repetitive region transcripts can form dsRNAs, activating interferon pathways and remodeling the tumor stroma (Espinet et al. 2021). TE-derived sequences may serve as enhancers (Xie et al. 2013), promoters (Brocks et al. 2017; Jang et al. 2019), or other regulatory elements, and constitute a substantial portion of lincRNAs (Khorkova et al. 2015; Choudhary et al. 2023). Despite their potential importance, the transcriptomic landscape and clinical relevance of TE-derived elements in PAAD remain poorly characterized, partly due to limited sample sizes and the constraints of bulk sequencing.
In this study, we aim to systematically characterize the landscape of TE-chimeric transcripts and their clinical relevance in pancreatic cancer using large-scale patient-derived organoid and bulk sample cohorts. Our objective is to clarify the potential contributions of transposable elements to PAAD pathogenesis and immunogenicity and to explore new opportunities for biomarker discovery and immunotherapeutic development.
Results
Integrative analysis of transposable element activation across patient cohorts and cell line data sets
To comprehensively characterize dynamically expressed TEs in PAADs, we analyzed our in-house RNA-seq data sets from patient-derived organoids (Fig. 1A; Supplemental Fig. S1A; Supplemental Table S1; Shi et al. 2022). We conducted analysis of differentially expressed TEs (DETEs) between 66 PAADs and five normal pancreatic ductal organoids using various recommended methods for TE expression quantification (Xiang et al. 2022). We identified 7413 upregulated and 871 downregulated DETE copies in PAADs by featureCounts at a cutoff of fourfold difference and a false discovery rate (FDR) < 0.05, based on multiple previous studies (Fig. 1B; Supplemental Fig. S1A; Supplemental Table S2; Jin et al. 2015; Savytska et al. 2022; Xiang et al. 2022). To validate these findings, we used an independent patient cohort, including 41 bulk tumor and 39 paratumor samples, as well as 41 CCLE (Ghandi et al. 2019) pancreatic cancer cell lines data sets and three hTERT-HPNE pancreatic epithelial normal cell lines (Supplemental Fig. S1A). TE expression dynamics in the cell line data sets were consistent with our bulk tumor cohort (Supplemental Fig. S1B–J). We found that all major classes of TEs exhibited upregulation in PAADs (Supplemental Fig. S2). In the 7413 upregulated DETEs, 36.4% were classified as LINE elements, and 35.8% belonged to SINE, and 0.18% of the 770,551 LTR genomic copies were upregulated. Unlike previous reports linking TE expression to genome instability, we found no association with gene mutations or tumor markers (Fig. 1C; Supplemental Fig. S3). Because epigenetic activation usually leads to TE expression, we interrogated the epigenetic signals based on ATAC-seq data (Shi et al. 2022). We found that upregulated DETEs were associated with more accessible chromatin (Fig. 1D; Supplemental Fig. S4). TEs could act as enhancers and promoters to regulate gene expression. Therefore, we applied the GREAT tool (McLean et al. 2010) to our DETE regions. We found enrichment of genes related to metabolic processes, including the purine nucleotide catabolic process and hormone metabolic process (Fig. 1E).
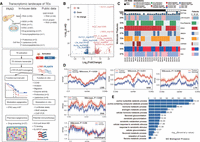
Activation of transposable elements in PAAD. (A) Overview of the data sets used in this study, including both in-house and publicly available data. Detailed numbers for each data type are provided (top panel). The schematic illustrates the activation of promoters within transposable elements (TEs) in PAAD. Upon activation, these promoters may drive onco-exaptation, resulting in the expression of chimeric transcripts that contribute to oncogenesis, or generate neoantigens that trigger an immune response (bottom panel). (B) Volcano plot showing the differentially expressed TE loci in tumor organoid and normal organoid. The TE-IDs such as “AluYm1_dup3579” were listed in Supplemental Table S2. Only genes with FDR-adjusted P-values < 0.05 and absolute log2 fold change of ≥4 are considered significantly differentially expressed. (C) Oncoprint illustrating drug sensitivity, driver gene mutations, tumor biomarker levels, and survival outcomes of patients who underwent organoid-based drug screening. (D) Comparison of ATAC-seq signals located within 3 kb upstream of and downstream from DETEs between organoid tumor and normal samples. Unchanged TE regions (2000 randomly selected) were used as a control. Statistical analysis was performed using the Wilcoxon test, with a P-value < 0.05 considered statistically significant. PAAD tumor organoid, n = 62; Normal organoid, n = 5. (E) Gene Ontology (GO) enrichment analysis of biological processes associated with DETEs in PAADs.
TE-chimeric transcripts and onco-exaptation events in the PAAD organoid cohort
To examine the products of TE activation, we utilized TEprof2 (Shah et al. 2023) to analyze TE-derived transcripts, specifically fusion transcripts between TE sequences and known genes, which we referred to as TE-chimeric transcripts. These transcripts displayed different levels in 66 tumor organoids compared to five normal organoids and 39 paratumoral samples. After rigorous filtering, we identified 469 TE-chimeric transcripts from 3513 highly confident TE transcripts as specific to PAADs, which represents a significantly higher count than previously reported (Fig. 2A; Supplemental Fig. S5A; Supplemental Table S3; Shah et al. 2023). Unlike Shah et al. (2023), we could not include GTEx data due to lack of raw RNA-seq files, which are required for splicing-aware alignment. To ensure specificity, we applied a three-step filtering strategy, including tumor enrichment scoring and removal of transcripts overlapping benign promoters from the FANTOM5 database. TE-chimeric transcripts in the organoid data sets were consistent with our bulk tumor cohort and cell line data sets (Supplemental Fig. S5B). A comparative analysis between organoid and bulk samples was conducted to assess the consistency of TE onco-exaptation events, which identified 89 concordant onco-exaptation instances (Supplemental Fig. S5B). Among these, 66% (2305/3513) were novel, presumably due to the low purity of TCGA PAAD samples used in previous studies (Supplemental Fig. S6A). Of the 469 PAAD-specific TE-chimeric transcripts, most originated from intronic (77%) or partially exonic regions (15%) (Fig. 2B). The L1PA2 subfamily was notably enriched (Fig. 2C), consistent with TCGA pan-cancer analyses. We identified 16 TE-driven fusion events involving 15 known oncogenes, spanning all major TE classes (Fig. 2D; Supplemental Fig. S6B–D; Supplemental Table S4). We assessed associations between TE-chimeric transcripts and overall survival in PAADs (Supplemental Fig. S7; Supplemental Table S4). One of the most abundant fusions, LTR7-PLAAT4, was present in 29% of PAAD organoids and correlated with poor prognosis (Figs. 2E, 3A,B; Supplemental Table S5). We found that LTR7-PLAAT4 chimeric transcripts predicted prognosis and treatment response more accurately than driver mutations or preoperative markers (e.g., CA19-9, CEA) (Fig. 1C). LTR7-PLAAT4 fusion was also detected in multiple gastrointestinal cancer cell lines, including 24% of pancreatic, 37% of esophageal, and 35% of intestinal lines (Fig. 3C,D). LTR7 was located ∼7 kb upstream of the PLAAT4 promoter and initiated two transcript isoforms in tumor organoids (Supplemental Fig. S8A–C). To verify the existence of the LTR7-PLAAT4 isoforms in pancreatic and other cancer cell lines, we conducted nested PCR and Sanger sequencing (Supplemental Fig. S8D–F). Isoform 2 was validated in Capan-1 and SNU-16 cells but absent in hTERT-HPNE normal cells (Fig. 3E; Supplemental Table S6).
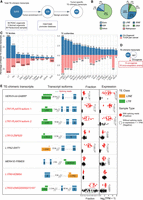
Identification of tumor-specific and clinically prognostic TE-chimeric transcripts in PAADs. (A) Diagram illustrating the process of candidate transcript filtering for tumor enrichment using normal organoids and normal bulk samples along with the FANTOM5 promoter atlas. (B) Pie charts depicting the TE location distribution of TE-chimeric transcripts (left) and their distribution across TE classes (right). IGR refers to the intergenic region. (C) Bidirectional bar graphs showing the percentage of families and subfamilies within filtered tumor-associated TE-chimeric transcripts. The upper section represents the in-house organoid cohort, whereas the lower section represents the previously reported TCGA pan-cancer data set. (D) Venn diagram illustrating the intersection between candidates identified in the organoid cohort and the 807 oncogenes based on the ONGene database and as previously reported. (E) The top eight most prevalent candidates significantly associated with patient survival. The left panel provides labels for the TE-chimeric transcripts and diagrams illustrating the transcript structure of each candidate. In the “Fraction” panel, each bar represents the proportion of total gene expression that is attributable to the TE-chimeric transcript, calculated only across the samples in which the candidate is detected. Validated candidates are denoted by red solid circles, whereas candidates lacking supporting evidence in terms of splicing reads or expression levels below 1 TPM are represented by black hollow circles. The “Expression” panel displays the expression levels of samples featuring the onco-exaptation candidate, with the candidate represented by red solid circles. Samples without supporting splicing reads or expression levels below 1 TPM are denoted by black hollow circles. Each box represents the median and interquartile range, with statistical analysis performed using t-tests. TE-chimeric transcripts labeled in red indicate the presence of coding potential.
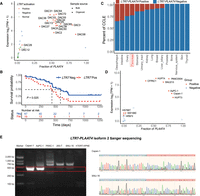
Expression profiling of LTR7-PLAAT4 across PAAD organoids and various cancer cell lines. (A) Scatterplot illustrating the expression and fraction distribution of LTR7-PLAAT4 across organoids and bulk samples (n = 110). Orange represents tumor samples positive for LTR7-PLAAT4; blue represents tumor samples negative for LTR7-PLAAT4 (lacking supporting splicing reads or with expression level <1 TPM); green represents normal samples; triangles and circles denote bulk samples and organoid samples, respectively. (B) Comparison of overall survival (OS) between the groups with and without LTR7-PLAAT4 existence in the organoid cohort (n = 66), assessed using the log-rank test. (C) Frequency distribution bar plot of the LTR7-PLAAT4 identified in CCLE cancer cell lines data sets, arranged from left to right based on the frequency of detection in cell lines (n = 685). (D) Expression and fraction distribution of LTR7-PLAAT4 across pancreatic cancer cell lines (n = 41). (E) PCR results following nested PCR were obtained for Capan-1, AsPC-1, and SNU-16 cell lines (which tested positive for LTR7-PLAAT4 isoform 2), as well as for PANC-1, 293-T, and hTERT-HPNE cell lines (which tested negative for LTR7-PLAAT4 isoform 2). Representive Sanger sequencing results of Capan-1 and SNU-16 were displayed.
LTR7 as a potential accessible TF binding site provider driving novel transcription
Next, we profiled chromatin accessibility using ATAC-seq and found that organoids positive for LTR7-PLAAT4 exhibited higher accessible signals (Fig. 4A). This suggests that an LTR7 TE is in open chromatin and could serve as an alternative promoter to drive PLAAT4 expression in pancreatic cancer organoids. Next, we used luciferase assays to test the promoter activities of LTR7 as well as MER21B, a TE fragment immediately adjacent to the 5′ end of LTR7 (Fig. 4B). MER21B alone without LTR7 displayed minimal promoter activity (Fig. 4C), but MER21B had a positive cis-regulatory effect on LTR7 transcription. We identified putative transcription factor motifs that were specific to LTR7 with PROMO (Netanely et al. 2019). Four TFs associated with these motifs, FOXM1(HNF-3alpha), TFAP2A, MAZ, and ETV4, were found to be differentially expressed between tumor and normal samples, with FOXM1(HNF-3alpha) being exclusively highly expressed in LTR7-PLAAT4-positive samples (Fig. 4D; Supplemental Fig. S9). We validated the binding of FOXM1 to LTR7 sequence using ChIP-qPCR (Fig. 4E). We then mutated each binding motif in the LTR7 sequence and performed luciferase reporter assay and found that, in both 293T and AsPC-1, mutating the FOXM1(HNF-3alpha) site significantly reduced luciferase expression (Fig. 4F,G).
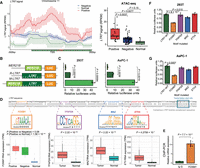
LTR7 drives PLAAT4 expression in pancreatic cancer. (A) Line plot displaying the ATAC-seq data signal within ±300-bp range upstream of and downstream from the LTR7 transcript start site. The red line represents tumor organoids with LTR7-PLAAT4 (n = 17), the blue line represents tumor organoids without LTR7-PLAAT4 (n = 45), and the green line represents normal organoids (n = 5) (left). Statistical analysis was performed using a t-test for each group comparison (right). (B,C) Luciferase assays evaluating the transcriptional activity of different TE arrangements in 293T cells (left) and AsPC-1 cells (right) (n = 3 independent experiments), with statistical analysis conducted using a t-test. (D) Schematic illustration of luciferase reporter assays measuring LTR7 promoter activity with mutated transcription factor binding motifs. (E) Quantification of FOXM1 enrichment in AsPC-1 cells by ChIP-PCR analysis. (F) Luciferase assays for promoter activity in 293T with mutagenized transcription factor motifs in LTR7 (n = 3 independent experiments). Box plots of RNA expression of transcription factor among different groups, with statistical analysis performed using t-tests. (G) Same as F but in AsPC-1 cells. All data are represented as means ± standard error (s.e.m.).
LTR7-PLAAT4 promotes tumorigenesis via lipid remodeling in PAADs
Phospholipase A and acyltransferase 4 (PLAAT4) has recently emerged as an oncogene in pancreatic cancer, in contrast to its previously reported tumor suppressor roles in other malignancies (Li et al. 2022). However, the functions of the newly discovered, TE-derived PLAAT4 isoforms are completely unknown. To address this, we individually expressed canonical PLAAT4 and two LTR7-driven isoforms (isoform 1 and 2) in PANC-1 and AsPC-1 cells. Overexpression of canonical PLAAT4 and isoform 1 promoted cell viability, whereas isoform 2 notably enhanced migration capacity (Fig. 5A–G; Supplemental Fig. S10). Sequence analysis revealed a robust alternative start codon within the LTR7 sequence, altering the N-terminus of isoform 2 (Supplemental Fig. S11A), which was validated by mass spectrometry following overexpression (Fig. 5H; Supplemental Fig. S11B; Huang et al. 2024). However, due to the low endogenous abundance of isoform 2 and its high sequence similarity to canonical PLAAT4, we were unable to confidently detect isoform 2-specific peptides under physiological conditions. LTR7-PLAAT4 isoform 2 showed a significant increase in its phospholipase activity compared to canonical PLAAT4 (Fig. 5I; Supplemental Fig. S11C). Notably, this N-terminal modification did not affect the membrane localization of the protein (Supplemental Fig. S11D). To elucidate the molecular mechanisms underlying the oncogenic effects of different LTR7-PLAAT4 transcripts, we performed correlation analysis and identified genes positively correlated with different PLAAT4 isoforms (Fig. 6A). Our results revealed that genes specifically correlated with canonical PLAAT4 were enriched in virus defense functions (Supplemental Fig. S12A), whereas those correlated with LTR7-PLAAT4 isoform 1 were enriched in apoptosis-related functions (Supplemental Fig. S12B). Notably, genes correlated with LTR7-PLAAT4 isoform 2 were enriched in lipid droplet formation (Fig. 6B), including lipid droplet-associated enzymes such as PLA2G4C and BSCL2 (Supplemental Fig. S12C). Subsequently, we investigated the impact of constitutive ectopic expression of LTR7-PLAAT4 isoforms on phospholipid composition using mass spectrometry-based lipidomics (Fig. 6C; Supplemental Table S7). Initially, we observed an increase in phosphatidylcholines (PC) and phosphatidylethanolamine (PE) levels, although the differences were not statistically significant. Consistent with previous reports (Bradley et al. 2022), overexpression of the PLAAT family gene showed a slight increase in PC and PE levels. PLAAT4 facilitates the transfer of fatty acyl groups from PC to LPC and from PE to NAPE (Golczak et al. 2012; Uyama et al. 2012b), and accordingly, we observed increased LPC and LNAPE levels in all PLAAT4-overexpressing cell lines, consistent with prior studies (Uyama et al. 2012a) and our phospholipase assay. However, no significant differences in LPC and LNAPE levels were observed among the cell lines overexpressing different PLAAT4 isoforms. The most notable differences among cell lines were in cholesteryl esters (CEs), with the cell line overexpressing PLAAT4 isoform 2 showing the highest CE levels compared to those overexpressing the other two isoforms. Meanwhile, there was a trend toward decreased fatty acid (FA) levels. As a member of the PLA1/PLA2 family, PLAAT4 releases FA from phospholipids (e.g., PC), providing FA for the esterification of cholesterol to form CEs, which are stored in lipid droplets (Tu et al. 2023). There are mainly two types of lipid droplets: CEs and triglycerides (TGs) (Fujimoto and Parton 2011). However, in these cells, PLAAT4-mediated lipid droplet formation primarily involved CE lipid droplets because TG levels decreased in the cell line overexpressing PLAAT4 isoform 2. To further elucidate the relationship between PLAAT4 isoform 2 and cholesterol ester–rich lipid droplets, we conducted in vitro induction of CE-rich lipid droplets. The results demonstrated that both the number and area of lipid droplets in cells expressing PLAAT4 isoform 2 were significantly higher compared to control cells (Fig. 6D–F). The expression of PLAAT4 isoform 2 was positively correlated with BSCL2, which was significantly upregulated in the isoform 2-expressing cells compared to control cells (Fig. 6G). Seipin, derived from the BSCL2 gene, is a crucial protein involved in forming the lipid droplet assembly complex, including CE droplets (Dumesnil et al. 2023). Lipid droplet formation is crucial for tumors, as it alleviates lipid toxicity, mitigates ER stress, provides energy, and supports cell survival (Zadoorian et al. 2023). These findings suggest that TEs contribute to the diversity of the PLAAT4 gene products and function, thereby contributing to cancer metabolism remodeling (Fig. 6H).
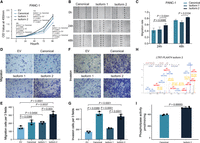
Oncogenic properties of LTR7-PLAAT4 isoform 2 in pancreatic cancer cell lines. (A) PANC-1 cell proliferation rate comparison using a CCK-8 assay after LTR7-PLAAT4 isoform 2 overexpression. (B,C) PANC-1 cell migration rate comparison using wound healing assay after different PLAAT4 isoforms overexpression. Representative images of the assay were shown at 50× magnification. Scale bar, 200 µm. (D,E) PANC-1 cell migration rate comparison using migration assay after different PLAAT4 isoforms overexpression. Scale bar, 150 µm. (F,G) Invasion assay was used to detect invasion capability after LTR7-PLAAT4 isoform 2 overexpression in PANC-1 cell lines. Scale bar, 150 µm. (H) Secondary mass spectra were obtained from 293T cells overexpressing LTR7-PLAAT4 isoform 2. (I) Phospholipase activity assay of canonical and isoform 2 LTR7-PLAAT4 proteins.
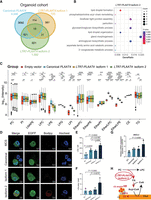
Potential oncogenic mechanism of LTR7-PLAAT4 isoform 2 in pancreatic cancer. (A) Venn diagram showing the number of genes positively correlated with different PLAAT4 isoforms in organoid cohort. The canonical PLAAT4, LTR7-PLAAT4 isoform 1, and LTR7-PLAAT4 isoform 2 are marked in light blue, yellow, and green, respectively. (B) Gene Ontology analysis of specific genes correlated with LTR7-PLAAT4 isoform 2. The size of each bubble represents the number of enriched genes, and the color indicates the P-value, with darker colors representing smaller P-values. (C) Lipidomics analysis of PANC-1 cells overexpressing LTR7-PLAAT4 isoform 1, LTR7-PLAAT4 isoform 2, and canonical PLAAT4. Box plots display the distribution of different PLAAT4 isoforms across various lipid classes. Each box plot includes the median, interquartile range, and outliers for the lipid classes measured. Statistical significance is indicated with (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001, (****) P ≤ 0.0001. The exact P-values of panel C are listed in Supplemental Table S6. (D) PANC-1 cells imaged after 24 h of methyl-ß-cyclodextrin complexed cholesterol (200 µM) feeding. BODIPY was added upon imaging for LD labeling and analysis. (E) Analysis of D. Number of LDs per cell. (F) Analysis of D. Area of LDs per pixel. The experiment was independently repeated three times with similar results. (G) Expression of BSCL2 in different PLAAT4 isoforms overexpressed pancreatic cancer cell lines (PANC-1) measured by qPCR. (H) Schematic illustrating the special oncogenic role of LTR7-PLAAT4 isoform 2 in PAAD.
Tumors positive for the LTR7-PLAAT4 oncogene exhibit multidrug resistance
To identify potential therapeutic strategies in light of LTR7-PLAAT4, we conducted a high-throughput drug screen using our organoid platform and evaluated the correlation between drug sensitivity and LTR7-PLAAT4 status in the organoids. A bespoke 64 compound library was assembled for screening, including chemotherapy (n = 14) and targeted drugs (n = 23) (Supplemental Table S8). As positive controls, one organoid (DAC78) bearing the KRAS p.G12C mutation exhibited sensitivity to AMG510 and MRTX849 (Fig. 7A). Clustering of compounds based on their AUC revealed a diverse range of sensitivities across the organoids, identifying two distinct clusters. Tumors positive for the LTR7-PLAAT4 oncogene were predominantly enriched in the resistant subgroup, exhibiting a broad spectrum of drug resistance (Fig. 7B). This was further supported by Genomics of Drug Sensitivity in Cancer (GDSC) data, where LTR7-PLAAT4–positive CCLE cell lines also showed pharmacologic resistance (Supplemental Fig. S13; The Cancer Cell Line Encyclopedia Consortium, The Genomics of Drug Sensitivity in Cancer Consortium 2015).
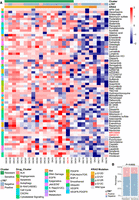
Drug sensitivity profiles of PAAD organoids. (A) Drug response of 35 PAAD organoids to 64 different drugs. Colors ranging from red to blue represent Z-scores based on the Area Under the Curve (AUC). A low Z-score (blue) indicates high drug sensitivity, whereas a high Z-score (red) indicates high resistance. The positive control is highlighted in red font for clarity. (B) Stacked bar chart displaying the distribution of LTR7 in different states across two groups with χ2 test (Bonferroni correction applied).
TE-chimeric transcript–derived peptides are validated as putative neoantigens
To overcome the resistance associated with LTR7-PLAAT4 and target lethal cancers driven by aberrant epigenetic activation of TEs, we turned to recent investigations that revealed the capacity of TEs in harboring small open reading frames (smORFs) that may serve as tumor-specific antigens (TSAs) (Yadav et al. 2014; Newey et al. 2019; Shah et al. 2023). In AsPC-1 pancreatic cancer cells, HLA-bound immunopeptidomics identified 12,234 peptides by mass spectrometry (MS), of which 7481 were predicted to bind the endogenous HLA alleles using NetMHCpan-4.1. Annotation against multiple reference databases revealed these peptides originated from benign reference immunopeptidome databases (n = 3701) (Hoenisch Gravel et al. 2023), UniProt-only entries (n = 774), a single mutation-derived ORF from CAPN2 (n = 1), locally translated TE ORFs (n = 4), TEprof2-predicted ORFs (n = 4), and unannotated genomic regions (n = 2998) (Fig. 8A; Supplemental Table S9). A similar analysis in DAC13 patient-derived pancreatic tumor organoids identified 11,380 HLA-bound peptides. Among them, the TEprof2-derived peptide FLIQHLPLV was predicted to bind HLA-A*02:01 (Fig. 8B). This peptide demonstrated strong binding in T2 binding assays (Fig. 8C) and was validated through spectral matching with its synthetic counterpart (Fig. 8D). This peptide was also identified in two additional organoid lines, DAC30 and DAC50, yielding a detection frequency of ∼27% (3/11). Two other TE-derived peptides, ETVTGVWSY and FLPDHWAVL, were similarly validated in AsPC-1 cells (Fig. 8D). To investigate their genomic and translational origins, we visualized the corresponding chimeric transcripts using the WashU Epigenome Browser. RNA-seq coverage and getorf-predicted open reading frames provided evidence of local translation from TE loci (Fig. 8E). Only one mutation-derived HLA-presented peptide (CAPN2) was identified in AsPC-1, consistent with its rare mutation status as cataloged in the TRON Cell Line Portal (TCLP). No mutation-derived neoantigens were found in DAC13, underscoring the low mutational burden of PAAD and the limited repertoire of conventional mutation-derived TSAs. These observations prompted us to investigate whether TEs might serve as an alternative antigen source. To assess immunogenicity, two TEprof2-derived peptides—FLPDHWAVL and FLIQHLPLV—were tested via ELISPOT assays using PBMCs from HLA-genotyped healthy donors. Notably, FLIQHLPLV elicited a strong IFNG response, indicating potent T cell activation (Fig. 8F). This same peptide was independently identified as a naturally presented HLA ligand in a recent study on pancreatic cancer (Ely et al. 2025), further substantiating its potential as a bona fide neoantigen candidate.
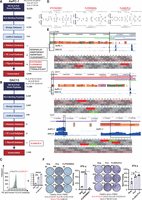
TE-chimeric transcript–derived peptides as neoantigens. (A) Annotation of 12,234 HLA-bound peptides identified by mass spectrometry (MS) from AsPC-1 cells across multiple reference databases, including benign, UniProt, mutation, locally translated TE ORFs, TEprof2, and unannotated regions. (B) Annotation of 11,380 HLA-bound peptides identified from DAC13 organoids, with one TEprof2-derived peptide (FLIQHLPLV) retained for further analysis. (C) Validation of the binding affinity of the TEprof2-derived peptide FLIQHLPLV to HLA-A*02:01 using T2 binding assay. (D) Spectral comparison between MS-identified endogenous peptides and synthetic reference peptides: ETVTGVWSY and FLPDHWAVL (AsPC-1), and FLIQHLPLV (DAC13), confirming accurate peptide identification. (E) Visualization of TE-chimeric transcripts and translation evidence using the WashU Epigenome Browser. TE loci are shown with RNA-seq read coverage and getorf-based predicted ORFs; corresponding peptide-coding regions are highlighted. (F) Immunogenicity validation of TE-derived peptides (FLPDHWAVL and FLIQHLPLV) in ELISPOT assays using HLA-genotyped healthy donor PBMCs. Significant IFNG responses indicate these peptides elicit T cell activation.
Discussion
Our study presents the first comprehensive analysis of TE-derived transcripts using a large panel of tumor organoids, providing insights into their potential oncogenic roles and immunotherapeutic relevance. Organoid models offer high tumor cell purity and retain many features of the original tumor, making them suitable platforms for studying gene function and drug response. However, limitations include loss of some tumor heterogeneity and absence of components such as immune cells and vasculature.
LTR7-PLAAT4 emerged as a top TE-oncogene candidate in PAAD and other cancers. LTR7, regulated by HNF4A, may interact with m6A-modified HERV-H transcripts and TET1 to evade epigenetic silencing (Sun et al. 2023). Functionally, PLAAT4 participates in lipid metabolism, and our data suggest isoform 2 overexpression promotes cholesteryl ester accumulation—an established metabolic signature in pancreatic cancer (Li et al. 2016). These findings point to a potential role of TE-driven isoform expression in lipid droplet remodeling.
Whereas our overexpression studies in PLAAT4-null cells highlight isoform-specific effects, further validation is needed. Isoform-specific loss-of-function studies are technically challenging due to repetitive sequences and shared promoters. Tools like long-read isoform-specific RNA-seq, CRISPRa screening, or bespoke antibodies will be required to test these hypotheses.
We also identified TE-derived peptides that function as neoantigens. In PAAD models with low mutational burden, these TE antigens could represent alternative immunotherapeutic targets. One peptide, FLIQHLPLV, showed strong MHC binding and T cell activation (Chiappinelli et al. 2015; Roulois et al. 2015; Attermann et al. 2018) and was independently validated in a recent report (Shah et al. 2023).
Additional limitations of our study include lack of in vivo validation, incomplete antigen universality due to MHC diversity, and absence of TCR sequencing. Our RNA-seq approach also excluded non-polyadenylated TE transcripts, underscoring the need for deeper profiling.
In summary, our study provides evidence that TEs can function as both drivers and immunological targets in pancreatic cancer. These findings lay the groundwork for future TE-targeted strategies in oncology.
Methods
Study design
The in-house data set comprised two cohorts: the organoid discovery cohort (Shi et al. 2022) and a new bulk validation cohort. The organoid discovery cohort included 71 patient-derived organoids (PDOs) from pancreatic ductal adenocarcinoma, with 66 samples obtained from tumor tissues and five from paratumoral samples. Whole-genome sequencing (WGS) was performed on tumor and paired blood/normal samples (n = 29), and RNA sequencing was conducted on all 71 organoids. Additionally, 62 tumor organoids and five normal organoids underwent ATAC-seq experiment. Public RNA-seq data from 685 cancer cell lines (CCLE), including 41 PDAC cell lines, and hTERT-HPNE cell lines were retrieved from public repositories (see Supplemental Material).
RNA-seq and DNA-seq library preparation and sequencing
RNA-seq libraries were generated using the NEBNext Ultra RNA Library Prep kit for Illumina, followed by sequencing on the Illumina HiSeq X TEN platform (150-bp paired-end reads). DNA libraries were prepared from tumor and matched normal samples and sequenced using WGS protocols. Standard protocols for library preparation, sequencing, and QC are provided in the Supplemental Material.
ATAC-seq library preparation and processing
ATAC-seq was performed on 67 PDOs (62 tumor, five normal) following a modified protocol to remove adapters and transposase sequences before alignment to GRCh38 using BWA-MEM (Jung et al. 2022). Peak calling was performed using MACS2 (Zhang et al. 2008). Full details are available in the Supplemental Material.
RNA-seq and DNA-seq data processing
Raw FASTQ files were processed using fastp v0.23.2 (Chen et al. 2018), aligned to GRCh38 using STAR v2.7.7a (Dobin et al. 2013) for RNA-seq and BWA v.0.7.17 (Li and Durbin 2009) for WGS data. Somatic variants were called using GATK v.4.1.9 (McKenna et al. 2010) MuTect2 with panel-of-normal filtering and annotated using ANNOVAR (Wang et al. 2010). Standard processing pipelines and parameters are outlined in the Supplemental Material.
TE quantification and differential analysis
TE expression was quantified using featureCounts v2.0.6 (Liao et al. 2014) and TElocal v1.1.1 (Jin et al. 2015) with RepeatMasker annotations. Differentially expressed TEs were identified using DESeq2 (RRID:SCR_000154) v1.40.2 (Love et al. 2014), with a cutoff of fold-change ≥ 4 and FDR < 0.05.
Identification and quantification of TE-chimeric transcripts
TE-chimeric transcripts were identified using TEProf2 (Shah et al. 2023), which integrates StringTie v1.3.3 (Pertea et al. 2015) for de novo assembly and transcript quantification. Transcripts overlapping TEs and splicing into downstream genes were filtered using expression (TPM > 1) and splice-junction read support. Tumor-specific fusions were prioritized by ≥eightfold enrichment in tumor versus normal samples. Expression specificity was further refined by filtering against FANTOM5 (Lizio et al. 2015) CAGE peaks, excluding overlapping peaks on the same strand. Fusions were annotated with the ONGene database (Liu et al. 2017) and known onco-exaptation events.
Possible open reading frame predictions
To evaluate coding potential, we applied CPC2 (Kang et al. 2017) and searched for the longest ORFs starting with Kozak-compatible ATG codons (Kozak 2005). Additional predictions were performed using EMBOSS getorf (Rice et al. 2000) (available at ftp://emboss.open-bio.org/pub/EMBOSS/), retaining sequences with ATG and ≥8 amino acids. Resulting ORFs were categorized into original, truncated, chimeric-original, or chimeric-truncated. These were forwarded for immunogenicity prediction (see Supplemental Material).
Functional association of genes with DETEs
GREAT (McLean et al. 2010) (http://great.stanford.edu/) was used to map DETEs to nearby genes (5 kb upstream of to 1 kb downstream from the TSS, with distal extension up to 1000 kb). Enriched GO terms were extracted from GREAT output.
Visualization of TE-chimeric transcripts
bigWig files were generated from BAM files using deepTools v3.5.6 (Ramírez et al. 2014) and visualized on the WashU Epigenome Browser (Zhuo et al. 2023). Public ChIP-seq data were sourced from CistromeDB (Zheng et al. 2019) (http://cistrome.org/db/).
Sanger sequencing
Nested PCR gene-specific primers (GSPs) were designed to target the fusion sequence of the predicted LTR7-PLAAT4 isoform 2 and were provided in Supplemental Table S10. Amplified products from two positive (Capan-1, SNU-16) and one negative (hTERT-HPNE) cell line were sequenced using ABI 3730XL. Sequences were aligned using SnapGene v6.0.2 to confirm fusion junctions.
Survival analysis
Overall survival was analyzed using the Kaplan-Meier method and log-rank tests. Follow-up data were obtained from institutional records and LinkDoc Technology Co., Ltd. Detailed definitions and statistical tools are listed in the Supplemental Material.
Cell line and cell culture
AsPC-1, Capan-1, PANC-1, hTERT-HPNE, 293T, and SNU-16 cell lines (RRIDs listed in Supplemental Material) were obtained from authenticated sources and tested negative for mycoplasma. Cell lines were cultured in medium conditions optimized per cell type, with fetal bovine serum and penicillin/streptomycin supplementation.
Construction of LTR7-PLAAT4 overexpressing cell lines
The predicted sequences of LTR7-PLAAT4 isoform 1 and LTR7-PLAAT4 isoform 2 were amplified and cloned into pSLenti-EF1-EGFP-P2A-Puro-CMV-3xFLAG-WPRE plasmids. Lentiviral vectors (OBiO Technology) were then utilized to generate various recombinant vectors for overexpressing LTR7-PLAAT4 isoform 1, LTR7-PLAAT4 isoform 2, Canonical PLAAT4, and an empty plasmid serving as a negative control (EV). Stable clones were selected with puromycin.
RT-qPCR
Gene expression was quantified using SYBR Green-based RT-qPCR on an Applied Biosystems QuantStudio 7 Flex system. Amplification specificity was confirmed by melting curve analysis. Data were normalized using the ΔΔCt method with three biological and three technical replicates per group. Primer sequences are listed in Supplemental Table S10.
Western blot
Cells were lysed by sonication in buffer containing protease inhibitors. Proteins (20 µg) were resolved by SDS-polyacrylamide gel, specifically a Bis-Tris gel (GenScript, #M00930), transferred to a nitrocellulose membrane (Millipore), and probed with anti-FLAG (Sigma-Aldrich, #F1804, RRID:AB_262044, 1:1000) and anti–β-actin antibodies (66009-1-Ig). Detection was performed with HRP-conjugated secondary antibodies (goat anti-mouse IgG [H+L]) (Thermo Fisher Scientific, #31430, RRID: AB_228307, 1:10,000) and quantified using ImageJ.
ChIP-PCR
ChIP–PCR was performed using the CUT&RUN approach with the Hyperactive pG-MNase CUT&RUN kit (Vazyme, #HD101-01) in AsPC-1 cells. Briefly, 1 × 105 cells were bound to ConA beads, incubated overnight with FOXM1 antibody (Proteintech, #13147-1-AP, RRID:AB_2106213) or IgG control (Proteintech, #30000-0-AP, RRID: AB_2819035), and digested with MNase. DNA was extracted and analyzed by RT-qPCR following the manufacturer's protocol.
Immunofluorescence staining
PANC-1 cells were seeded on glass coverslips (Genecom, #WHB-12-CS-LC) and fixed with 4% paraformaldehyde. Cells were incubated with 5 µg/mL WGA conjugates (Thermo Fisher Scientific, #W32464; diluted in HBSS, #14175095), permeabilized using 0.2% Triton X-100 (Sangon Biotech, #9002-93-1), and blocked with 2.5% Normal Goat Serum (Thermo Fisher Scientific, #R37624). After overnight incubation at 4°C with anti-FLAG antibody (Sigma-Aldrich, #F1804, 1:500), samples were stained with Cy5-conjugated secondary antibody (Abcam, #ab6563, 1:1000) and Hoechst 33342 (Biosharp, #BL803A). Images were acquired using a Zeiss LSM 900 confocal microscope under consistent settings.
Enzyme activity
A liposome substrate mixture was prepared by combining DOPC (30 µL, 10 mM), DOPG (30 µL, 10 mM), and Red/Green BODIPY PC-A2 (30 µL, 1.5 mM, Thermo Fisher Scientific, #E10217) in DMSO. The mixture (10 µL) was added dropwise into 1 mL reaction buffer under vortexing. Protein solutions (0.3 mg/mL, 200 µL) were diluted 1:1 with reaction buffer. PLA2 activity was assessed using a standard curve (0–0.5 U/mL) and measured in 96-well microplates (Thermo Fisher Scientific, #M33089). Each well received 50 µL enzyme or standard solution and 50 µL liposome substrate. Fluorescence was recorded every 2 min for 30 min using a SYNERGY H1 plate reader (Ex: 460 nm; Em: 515 nm).
LC-MS/MS analysis
For proteomic analysis, 293T cells overexpressing LTR7-PLAAT4 isoform 2 or canonical PLAAT4 (2–5 × 107 cells) were lysed in ice-cold Cell Lysis Reagent (Sigma-Aldrich, #C2978) supplemented with protease inhibitor cocktail (Bimake, #B14002). Lysates were sonicated every 10 min on ice and centrifuged at 21,000g for 1 h at 4°C. FLAG-tagged proteins were immunoprecipitated using magnetic beads coupled with anti-FLAG antibody (Sigma-Aldrich, #F1804, RRID:AB_262044).
Immunoprecipitated proteins (100 µg) were digested with either Endoproteinase Glu-C or α-Chymotrypsin (Sigma) at a 1:30 enzyme-to-substrate ratio (w/w) for 12 h at 37°C or 25°C, respectively, in 100 mM sodium bicarbonate (pH 8.0). Peptides were desalted using MonoSpin C18 columns (GL Sciences) and dried using a Speed Vacuum Concentrator (Labogene Aps).
Desalted peptides were loaded onto a PepMap Neo C18 trap column (300 µm i.d. × 5 mm, 5 µm, 100 Å; Thermo Fisher Scientific) and separated on a house-packed C18 analytical column (75 µm i.d. × 25 cm, 1.9 µm, 100 Å) using the NanoElute system (Bruker Daltonics) at 50°C. Peptides were eluted over a 60-min gradient with buffer B (0.1% formic acid in acetonitrile) as follows: 2%–22% (0–45 min), 22%–35% (45–50 min), 35%–80% (50–55 min), and held at 80% (55–60 min). Buffer A consisted of 0.1% formic acid in water.
Lipid manipulations and image analysis
PANC-1 cells were delipidated in serum-free medium containing 5% lipoprotein-deficient serum (LPDS) for 2.5 days. For enhanced lipid depletion, DGAT1 and DGAT2 inhibitors (5 µM each) were added during the final 18 h. To induce cholesterol ester accumulation, cells were loaded with methyl-β-cyclodextrin-complexed cholesterol (20:1 molar ratio) in 5% LPDS for 24 h.
Cells were fixed with 4% paraformaldehyde in 250 mM Hepes buffer (pH 7.4) containing 100 µM CaCl2 and 100 µM MgCl2 and stained with BODIPY 576/589 (10 µM; MCE, #HY-D1118) and Hoechst 33342 (5 µg/mL; MCE, #HY-15559) for lipid droplet and nuclear visualization. Confocal images were acquired using a Zeiss LSM 900 microscope under consistent settings.
Image analysis was performed in ImageJ (v1.54f). TIFF images were converted to 8-bit grayscale, denoised using a Gaussian blur (sigma = 2.0), and thresholded (Otsu or manual). Particles (10–5000 pixels, circularity 0.5–1.0) were detected using the Analyze Particles function. Edge particles were excluded, and extracted parameters (area, perimeter, circularity) were analyzed in R.
Immunopeptidomics
Peptides were analyzed on a timsTOF Pro2 system (Bruker Daltonik) in DDA mode. Each acquisition cycle included one MS1 TIMS-MS and 10 PASEF MS/MS scans. Ion accumulation and ramp time were set to 100 msec; ion mobility range was 1/K0 = 0.75–1.30 Vs·cm−2. MS/MS precursor ions were measured in the m/z range 100–1700 using CID fragmentation. Dynamic exclusion was set to 30 sec.
Raw data analysis
MaxQuant (v2.0.1.3; RRID:SCR_014485) (Tyanova et al. 2016) was used for data analysis with default settings unless otherwise noted. Chymotrypsin or Glu-C was set as the digestion enzyme, with specificity enabled. The minimum peptide length was seven amino acids, allowing up to two missed cleavages. The false discovery rate was controlled using a target-decoy approach at <1% for both peptide spectrum matches and protein groups.
Promoter and mutagenesis luciferase assay
Based on the results from RNA-seq, ATAC-seq, and ChIP-seq, various promoter sequences derived from TEs were amplified. The minimal promoter sequence of the pGL3-basic luciferase plasmid was removed with Kpnl and Xhol restriction enzyme digestion, and the TE-derived promoters were cloned into the pGL3-basic plasmid via Gibson Assembly following the manufacturer's protocol (New England Biolabs, #E2661S). For the mutagenesis assay, we first used PROMO v 3.0.2 (Netanely et al. 2019) to predict the transcription factor (TF) binding sites within the sequences, and then compared the expression levels of these TFs between groups. We selected four TFs for mutation: FOXM1 (HNF-3alpha), TFAP2A, MAZ, and ETV4. Specific motifs within the LTR7 promoter-luciferase vector were mutated using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent, #210518). We used the Neon transfection system (MPK5000) to deliver 400 ng of the promoter-luciferase vector and 200 ng of the pRL-TK Renilla vector (Addgene, #E2241) into 3 × 104 AsPC-1 cells and 3 × 104 293T cells. Luciferase levels were measured after 24 h of incubation using the Dual-Glo Luciferase Assay System (Promega, #E2940).
Lipid metabolomics mass spectrometry analysis
PANC-1 cells (2–5 × 107) overexpressing different PLAAT4 isoforms were extracted with prechilled HPLC-grade methanol (−80°C), dried, and reconstituted in 100 µL isopropanol:methanol (1:1). Lipids were analyzed using a Waters ACQUITY UPLC BEH C18 column (2.1 × 100 mm, 1.7 µm) on a UPLC system coupled to an Orbitrap-LTQ-Elite MS (Thermo Fisher Scientific). Gradient and MS parameters are detailed in Supplemental Methods. Data were converted with ProteoWizard v3.0.18178 (Kessner et al. 2008), features extracted using XCMS v3.9.3 (Domingo-Almenara and Siuzdak 2020), and identified by matching to LipidBlast v68 (Tsugawa et al. 2020), with MS1 and MS2 mass tolerances of 0.01 and 0.05 Da, respectively.
Functional assays
Cell viability was assessed at 0, 24, 48, 72, and 96 h using the CCK-8 kit (Beyotime Biotechnology) and read at 450 nm (Epoch microplate reader, Biotek US). Migration and invasion assays were performed using Transwell inserts (Corning) with or without Matrigel (BD Biosciences) following 24-h serum starvation. Cells were stained with 0.2% crystal violet and quantified in triplicate experiments.
High-throughput drug screening of patient-derived organoids
Patient-derived organoids were dissociated and seeded (3000 cells/well) into collagen-precoated 384-well plates. After 24 h, 64 compounds (Selleck) were added in a seven-point dilution series (see Supplemental Table S8) using a Bravo robotic system. Viability was measured after 5 days using CellTiter-Glo (Promega) and read with an Envision plate reader. AUCs were computed using GraphPad Prism 9 and normalized. Clustering was performed with ConsensusClusterPlus v1.64.0 (Wilkerson and Hayes 2010) using parameters: reps = 1000, pItem = 0.8, pFeature = 1, algorithm = “km”.
HLA class I antigen isolation from AsPC-1 and organoids
HLA-I peptides were enriched from AsPC-1 cells and PDOs (DAC13) using the NEO Discovery HLA-I Peptide Enrichment kit (Baizhen Biotech), following a modified protocol (Chong et al. 2022). Eluted peptides were analyzed using a timsTOF Pro2 mass spectrometer (Bruker Daltonik) with a 120-min DDA method (MS1 range: m/z 100–1700; PASEF with 10 MS/MS scans, cycle time: 2.27 sec, ion threshold: 1000).
HLA mass spectrometry analysis
Raw data (.d files) were analyzed using PEAKS DeepNovo Peptidome (Bioinformatics Solutions, Inc.) against the UniProt human-reviewed + NGS libraries. De novo peptides (DeepNovo) were filtered with average local confidence (ALC) > 50%, no enzyme digestion, 20 ppm precursor and 0.05 Da fragment tolerance, and FDR < 1%. Peptides of 7–16 aa were retained and compared to databases: (1) Human Benign Proteome (PXD03878); (2) UniProt (https://www.uniprot.org/); and (3) De novo ORF peptide libraries generated using EMBOSS getorf and TEProf2 references. Selected peptides were synthesized, and MS/MS spectra were validated by comparison with eluted HLA peptides.
HLA typing, binding prediction, and validation
HLA allele types were predicted from RNA-seq using seq2HLA v2.2 (Boegel et al. 2013). Binding affinities of TE-derived peptides were predicted using NetMHCPan-4.1 (Nielsen et al. 2020). Peptides with strong predicted binding (EL-Rank < 0.5) to HLA-A*02:01 were synthesized for validation.
Binding was tested in T2 cell assays (Tang et al. 2017): cells were incubated with peptide (50 µM) and β2-microglobulin (1 µM; Sigma) for 18 h, then stained with PE-labeled anti-HLA-A2.1 (BD Biosciences, #551230, RRID:AB_394106) and analyzed by flow cytometry (Sony ID7000). Fluorescence intensity (FI) was calculated; FI > 1 indicated strong binding.
ELISPOT assay
PBMCs from HLA-typed donors were plated (2.5 × 105/well) on precoated ELISPOT plates (Mabtech, #3420-4APW-2) and stimulated with 20 µg/mL peptide for 24 h. After washing, anti-human IFNG antibody (Mabtech) and SA-ALP were applied sequentially (1:1000 dilution). Spots were developed using BCIP substrate and counted with AID EliSPOT Reader and software v7.0.
Statistical analysis
Two group statistical analysis was performed using the independent samples Student's t-test (two-sided) or the Wilcoxon/Fisher's exact test. Multiple groups statistical analysis was performed using the two-way ANOVA. Data were presented as mean ± standard error of mean (SEM); (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Sample sizes for each experiment were indicated in the relevant results section or figure legends. All statistical analyses were conducted using R (version 4.3.2) (R Core Team 2024) and GraphPad Prism v8 software.
Code availability
All source code and custom scripts used in this study are available at GitHub (https://github.com/MaxlogiLee/PLAAT4) and as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the Genome Sequence Archive (https://ngdc.cncb.ac.cn/gsa-human) with the accession number HRA007136.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank our patients and their families. This study was supported by the “National Key R&D Program of China” fund (2022YFA1305700, J.Li) granted by the Chinese Ministry of Science and Technology and granted by National Natural Science Foundation of China grant 82372719 (J.Li). We thank Jihang Yuan for experimental assistance.
Author contributions: J.Li and G.J. conceived and led the study. Y.W. and Y.R. provided supervision for the wet lab and mass spectrometry, respectively. M.S. conducted data analyses. C.T. and S.Z. carried out the majority of wet lab experiments. X.H. contributed to the T2 binding and immunogeneity experiments. G.Y. checked the analysis of immune peptide experiment results. F.H., R.W., and L.X. provided support for basic statistics, proofreading, data storage, and experiment preparation. Y.F. stored samples in liquid nitrogen. J.Liu conducted the MS experiment. J.Li prepared the manuscript with input from all authors. All authors reviewed the manuscript.
Footnotes
-
↵11 These authors jointly supervised this work.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280528.125.
-
Freely available online through the Genome Research Open Access option.
- Received February 9, 2025.
- Accepted November 24, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








