
Chimeric mitochondrial RNA transcripts predict mitochondrial genome deletion mutations in mitochondrial genetic diseases and aging
- 1Department of Medicine, Division of Dermatology, University of California, Los Angeles, Los Angeles, California 90095, USA;
- 2Veterans Administration Greater Los Angeles Healthcare System, Los Angeles, California 90073, USA;
- 3Department of Agricultural, Food, and Nutritional Science, University of Alberta, Edmonton, Alberta T6G 2R3, Canada;
- 4Department of Medicine, Division of Geriatrics, University of California, Los Angeles, Los Angeles, California 90095, USA
Abstract
Although it is well understood that mitochondrial DNA (mtDNA) deletion mutations cause incurable diseases and contribute to aging, little is known about the transcriptional products that arise from these DNA structural variants. We hypothesized that mitochondrial genomes containing deletion mutations express chimeric mitochondrial RNAs. To test this, we analyzed human and rat RNA sequencing data to identify, quantitate, and characterize chimeric mitochondrial RNAs. We observe increased chimeric mitochondrial RNA frequency in samples from patients with mitochondrial genetic diseases and in samples from aged humans. The spectrum of chimeric mitochondrial transcripts reflects the known pattern of mtDNA deletion mutations. To test the hypothesis that mtDNA deletions induce chimeric RNA transcripts, we treated 18 month old and 34 month old rats with guanidinopropionic acid to induce high levels of skeletal muscle mtDNA deletion mutations. With mtDNA deletion induction, we demonstrate that the chimeric mitochondrial transcript frequency also increases and correlates strongly with an orthogonal DNA-based mutation assay performed on identical samples. Further, we show that the frequency of chimeric mitochondrial transcripts predicts expression of both nuclear and mitochondrial genes central to mitochondrial function, demonstrating the utility of these events as metrics of age-induced metabolic change. Mapping and quantitation of chimeric mitochondrial RNAs provide an accessible, orthogonal approach to DNA-based mutation assays, offer a potential method for identifying mitochondrial pathology in widely accessible data sets, and open a new area of study in mitochondrial genetics and transcriptomics.
Mitochondrial DNA (mtDNA) deletion mutation containing genomes contribute to disease and aging by causing mitochondrial dysfunction and cell death (Pak et al. 2003; Bua et al. 2006; Herbst et al. 2007, 2013, 2016, 2021b; Cheema et al. 2015) Mitochondrial genetic diseases affect approximately one in 5000 individuals (Ng and Turnbull 2016) with a smaller prevalence of about one in 100,000 for individuals with large-scale mtDNA deletion syndromes, most commonly Kearns–Sayre syndrome, Pearson syndrome, and chronic progressive external ophthalmoplegia (Goldstein and Falk 1993). Disease progression in these individuals correlates with deletion size, deletion frequency within vulnerable tissues (e.g., brain, heart, skeletal muscle), and location of the deletion within the mitochondrial genome (Grady et al. 2014). Exciting potential therapies are being developed and tested (Ng and Turnbull 2016; Pirinen et al. 2020; Shoop et al. 2023), but none yet are disease modifying or available for routine use. Although mitochondrial genetic diseases often present in children and young adults and progress to premature mortality (García-Cazorla et al. 2005; Barends et al. 2016), mtDNA deletion mutations are also implicated in the aging process (Pak et al. 2003; Bua et al. 2006; Herbst et al. 2007, 2013, 2016, 2021b; Cheema et al. 2015). mtDNA deletion mutations are directly linked to multiple hallmarks of aging (López-Otín et al. 2013), including mitochondrial dysfunction, genomic instability, and cellular senescence. The stochastic development of mtDNA deletion mutations has implications for the role of these mutations in somatic mosaicism (Campbell et al. 2015) and aging in postmitotic tissues. The relative lack of genetic tools to correct or modulate mtDNA deletion mutations and a lack of understanding regarding the mechanisms of mutation accumulation are driving research in mitochondrial genetics to reveal new opportunities for treatment or prevention.
Current methods for mtDNA deletion mutation detection, mapping, and quantitation are exclusively DNA-based, and few studies explored the effects on mitochondrial transcription (Herbst et al. 2013; Lee et al. 2020). Work done prior to the widespread use of RNA sequencing observed chimeric mitochondrial transcripts associated with single mtDNA deletions in disease settings (Shoubridge et al. 1990; Savre-Train et al. 1992). These data suggested to us that unique, chimeric mitochondrial RNA (mtRNA) transcripts would be formed by additional mtDNA deletion events (Fig. 1). We hypothesized that these chimeric transcripts would be readily observable in RNA-seq data from diverse tissues and that the chimeric mtRNA frequency would be higher in individuals with known mitochondrial genetic diseases and in older individuals.
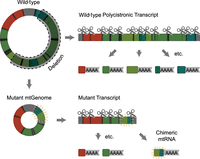
Diagram outlining the process of mitochondrial transcript processing in wild-type (top) versus mtDNA deletion mutation (bottom) genomes. Different colors denote different electron transport chain complexes or the mitochondrial ribosomal RNAs.
Chimeric RNAs in the nucleus are transcripts formed by gene fusion or intergenic splicing events and have been studied extensively in cancer and rare nuclear genetic diseases (Sun and Li 2022). Numerous unique chimeric RNAs have been found in these disorders and are often diagnostic biomarkers and therapeutic targets. For example, discovery of the BCR–ABL gene fusion (Rowley 1973) and corresponding chimeric transcript (Shtivelman et al. 1985) found in chronic myelogenous leukemia led to the development of BCR–ABL inhibitors that are the mainstay of treatment (Wong and Witte 2004). More recently, with the advent of RNA-seq and transcriptomic analyses, nuclear chimeric RNAs are being found in normal cells (Singh et al. 2020). In contrast, mtRNA transcripts are typically excluded in silico from RNA-seq analyses to allow better detection of lower abundance nuclear transcripts, and chimeric mtRNAs have not previously been reported from such data. For RNA-seq data, numerous prediction algorithms have been devised to identify chimeric RNAs (Sun and Li 2022).
In this study, we aimed to deploy a well-established chimeric RNA prediction algorithm on publicly available and made-for-purpose RNA-seq data sets representing a range of conditions in which mtDNA deletion mutations are pathogenic. We further aimed to relate the chimeric mtRNAs to age, disease state, gene expression, and, when possible, the underlying mtDNA deletion mutation frequency.
Results
Chimeric mtRNA transcripts are observed in patients with mitochondrial myopathy owing to large-scale mtDNA deletion mutations
To test whether chimeric mitochondrial transcripts corresponding to large deletion events can be found through application of established chimeric RNA detection software, we first analyzed RNA-seq of two human fibroblast lines previously documented to contain low levels of a single clonal genomic deletion (Majora et al. 2009) and a control neonatal dermal fibroblast cell line. Chimeric mtRNA transcripts were identified using STAR-fusion, using the most sensitive parameters to understand the breadth of events present. In control cells, no fusion was identified more than seven times. In contrast, RNA sequencing of disease fibroblasts demonstrated abundant transcripts (2248 transcripts and 287 transcripts) with fusion sites corresponding to the breakpoints of the mitochondrial “common deletion.” Paired long-read sequencing of the same samples demonstrated the presence of a single clonal deletion at this location (Supplemental Fig. 1).
To test whether chimeric mtRNA transcripts exist in human transcriptomic data, we next analyzed skeletal muscle RNA-seq data obtained from adult patients with a nuclear mutation that is known to induce high levels of mtDNA deletion mutations (Pirinen et al. 2020). Some of these patients have mutations in the nuclear encoded gene for Twinkle, a mtDNA helicase. Disruption of Twinkle is known to induce a wide range of mtDNA deletion mutations that, following our hypothesis, would be prone to generating chimeric mtRNAs (Young and Copeland 2016). When our approach was used to quantify chimeric mtRNA in this patient population, we observed chimeric mtRNAs in all samples. The two subjects with the Twinkle mutations had a 2.4-fold higher chimeric mtRNA frequency in their skeletal muscle compared with healthy control subjects, consistent with the known increase in deletion mutations (Table 1). When specific fusion locations were considered, a 4.7-fold increase in chimeric mtRNA frequency was observed in major-arc fusions between cytochrome c oxidase subunits and cytochrome b in Twinkle subjects.
Chimeric mtRNA frequency in patients with mitochondrial genetic disease
The same RNA-seq data set also contained three individuals (patients 1, 4, and 5), without Twinkle mutations, who have a single mtDNA deletion mutation species observed on long-extension PCR and agarose gel electrophoresis. The breakpoints of the mtDNA mutation in these subjects had not been sequenced (A. Suomalainen, pers. comm.). In patient 1, we identified a single chimeric mtRNA (MT:9189–MT:14,990) at levels 10-fold to 20-fold higher than chimeras in the Twinkle subjects. This particular chimeric mtRNA was present in all biopsy samples from patient 1. We did not find similarly expanded chimeras in the other two single-deletion mutation subjects, possibly because their mtDNA deletion mutations do not create a detectable chimeric mtRNA, for example, an mt-mRNA fusion between two mt-tRNA genes. Principle component analysis of the chimeric mtRNAs from control subjects and those with large-scale deletions showed clustering of the control samples separate from the Twinkle subjects (Fig. 2A). The most influential loadings in this analysis patients included MT-ND6—MT-TE, MT-RNR2—MT-TL2, and MT-TL1—MT-TL2.
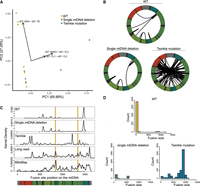
Chimeric mitochondrial RNAs (mtRNAs) in patients with mitochondrial genetic diseases. (A) Principal component analysis of human chimeric mtRNA data. (B) Distribution of chimeric mtRNAs detected more than seven times in patients with single large deletions, patients with heterogeneous mtDNA deletions owing to nuclear gene Twinkle mutations, and control subjects. (C) Location of fusion sites across the human mitochondrial genome and comparison to known deletion breakpoint distributions. Vertical yellow bars denote the location of the human 4977 or “common” deletion. (D) Histogram of fusion event sizes of chimeric mtRNAs from patients with single large deletions, Twinkle patients, and control subjects.
We next analyzed the characteristics of fusion events represented by at least seven transcripts, the cut off established by our control data. The fusion sites from control subjects and single-deletion subjects clustered in the major arc (Fig. 2B,C). The fusion sites in the Twinkle subjects were more evenly distributed across the mitochondrial genome. The distribution of fusion event locations was distinct from the distribution of mtDNA deletion mutations breakpoints reported in long-read sequencing of normal aging muscle and in available online mtDNA mutation databases (Fig. 2C). The fusion event sizes (i.e., the base pairs of DNA between the fusion sites) varied greatly between the subject groups. Control subjects had fusion sizes ranging from <1 kb–13 kb, whereas the fusion sizes for the single-deletion subjects predominated in the location of the presumed deletion mutation. The fusion sizes in the Twinkle subjects ranged from 1 kb to 11 kb with most being 7–9 kb (Fig. 2D).
Chimeric mtRNA transcripts increase with age in human skeletal muscle and brain
Our interest in mtDNA deletion mutations focuses on their contribution to morbidity and mortality associated with mammalian aging. Aging induces an exponential increase in deletion frequency across a number of tissues but especially pronounced is the increase in human skeletal muscle (Herbst et al. 2021a,b), where clonal expansion of somatically derived deletions causes metabolic dysfunction and triggers cell death (Pak et al. 2003; Bua et al. 2006; Herbst et al. 2007, 2016, 2021b; Cheema et al. 2015). To determine the relevance of chimeric mtRNA in this setting, we applied our analytical approach to an RNA-seq database from a study of skeletal muscle biopsies from individuals <30 years old and >65 years old (Kulkarni et al. 2020). We observed a 1.8-fold increase in average chimeric mtRNA frequency in skeletal muscle from the older individuals (Table 2). mtDNA deletions characterized in single muscle fibers from aged humans often identify deletion breakpoints joining the mtDNA cytochrome c oxidase (COX) subunits 1, 2, or 3 and cytochrome b (Bua et al. 2006). When we examined a subset of MT-CO1, MT-CO2, MT-CO3 to MT-CYB chimeric mtRNAs, we found a 2.4-fold increase with age (Table 2).
Chimeric mtRNA frequency in human aging muscle samples
When considering events sequenced more than seven times, the chimeric mtRNA fusion sites clustered primarily in the mitochondrial major arc in both young and older humans (Fig. 3A,B); 1.8% of chimeric mtRNA reads had fusion sites corresponding to the large direct repeats that flank the human common mtDNA deletion mutation (mtDNAdel4977) (Fig. 3C), the most commonly reported mtDNA deletion event in aging human tissue. Fifty-eight percent of chimeric mitochondrial reads found in muscle data had fusion sites within the mtDNA “contact zone,” observed to enrich for mtDNA deletion breakpoints (Shamanskiy et al. 2023). The chimeric mtRNA sizes averaged ∼6 kb with a greater mean width in the older subjects (Welch two-sample t-test, P = 0.005) (Fig. 3D).
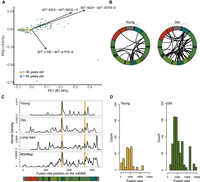
Chimeric mtRNAs in aging human skeletal muscle. (A) PCA plot of human skeletal muscle chimeric mtRNA data. (B) Distribution of chimeric mtRNA breakpoints in 14 younger (left) and 14 older (right) subjects for fusions detected more than seven times. (C) Location of chimeric ends across the human mitochondrial genome and comparison to known deletion breakpoint distributions. Vertical yellow bars denote the location of the human 4977 or common deletion. (D) Histogram of fusion event sizes of chimeric mtRNAs from <30- and >65-year-old subjects.
We next examined an RNA-seq data set from a study of male human brain samples ages 52 to 93 taken at the time of death with or without a diagnosis of Parkinson's disease (Table 3; Dumitriu et al. 2016). In the human brain samples, the chimeric mtRNA fusion sites are distributed throughout the mitochondrial genome in contrast to human skeletal muscle, where they are primarily localized to the major arc (Fig. 4A,B). The size distribution of chimeric mtRNA fusion events also differs from skeletal muscle with smaller-sized fusions than what we observed in skeletal muscle (median 3627 vs. 5150 bp, Welch's t-test P < 0.001) (Fig. 4C). Many fusion sites overlap with deletion mutations observed with nanopore sequencing of substantia nigra samples (Fig. 4B) with the exception of breakpoints in the minor arc that are more numerous in the chimeric mtRNA data. Sixty-four percent of fusion sites found in brain data are within the mtDNA “contact zone.”
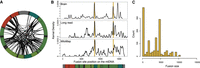
Chimeric mtRNAs in aging human brain. (A) Distribution of chimeric mtRNA fusion sites for fusions detected more than seven times in human brain. (B) Location of fusion sites across the human mitochondrial genome and comparison to known mtDNA deletion mutation breakpoint distributions in human brain. (C) Histogram of fusion event sizes in human brain.
Chimeric mtRNA frequency in human aging brain samples
Chimeric mitochondrial transcripts are induced with induction of mtDNA deletions
It is established that treatment with guanidinopropionic acid (GPA), a drug known to worsen muscle aging, increases the frequency of age-related mtDNA deletions in skeletal muscle (Herbst et al. 2013). To test the hypothesis that chimeric mitochondrial transcripts are products of mtDNA deletions, we analyzed RNA-seq data obtained from skeletal muscle from rats aged 18 months and 34 months and rats we treated with GPA. When STAR-fusion was used to identify chimeric transcripts in these data obtained from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA793055, we observed numerous chimeric transcripts with both breakpoints on the mitochondrial genome. This analysis demonstrated a 4.4-fold age-induced increase in chimeric mtRNA frequency in control mice with a further 2.2-fold increase with GPA treatment in aged mice (Table 4), corresponding closely to the previously reported magnitude of mtDNA deletion mutation change measured by DNA-based methods (Herbst et al. 2013).
Chimeric mtRNA frequency in rat muscle samples
In the same rat skeletal muscle samples used to generate the RNA-seq data, we measured mtDNA deletion frequency using a DNA-based digital PCR approach that targets the large deletion events that occur in aging (Herbst et al. 2017, 2022). We found a strong correlation between mtDNA deletion mutation frequency and chimeric mtRNA frequency (linear regression, R2= 0.86, P < 0.0001) (Fig. 5).
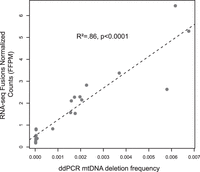
mtDNA deletion mutation frequency predicts chimeric mtRNA frequency in rat skeletal muscle.
Principle component analysis of chimeric mtRNA event frequency (Fig. 6A) demonstrates increased variability of chimeric mtRNA events in 34-month-old rats with and without GPA treatment. The most influential loadings in the PCA include chimeric mtRNAs involving MT-CO1 or MT-CO2 fused with MT-CYB. The fusion sites are primarily within the major arc of the mitochondrial genome (Fig. 6B,C). For 90% of fusion events, the distance between break points is between 1 kb and 10 kb with 66% of events being in the range of 6–9 kb (Fig. 6D).
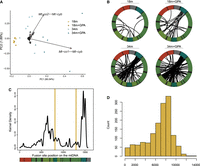
Mapping of age- and drug-induced mtRNA fusion events in rats. (A) PCA plot of rat chimeric mtRNA data. (B) Distribution of chimeric mtRNA breakpoints for representative sample from each category mapped onto a diagram of the wild-type mitochondrial genome. (C) Location of chimeric ends across the rat mitochondrial genome. Vertical yellow bars denote the location of the longest direct repeats (16 bp) in the rat mtDNA reference genome (located at positions 8102 and 12,936). (D) Histogram of fusion event sizes for the chimeric mtRNAs.
Chimeric mtRNA frequency predicts expression of genes involved in mitochondrial dysfunction in an aging rat model
Having observed chimeric mtRNA transcripts in multiple models associated with mitochondrial pathology, we next asked whether the presence of the chimeric transcripts would predict specific gene expression changes involved in disease pathogenesis. For an initial test, we evaluated gene expression data from aging rat skeletal muscle from which we observed an increase in chimeric mtRNA associated with aging (PRJNA793055). When we compared each samples’ sum total of chimeric mtRNA transcripts for all rat samples to the global transcriptome, we identified 6104 differentially expressed genes associated with the chimeric mtRNA frequency. Gene set enrichment analysis of these genes identified significant enrichment for downregulation of multiple pathways involving mitochondrial function, including electron transport chain and tricarboxylic acid cycle as well as mitochondrial translation, whereas upregulated pathways focused on cell communication and immunoregulatory pathways (Fig. 7A). When differential gene expression analysis was adjusted for the age and GPA treatment status of the animal, these findings persisted (Supplemental Fig. 2). Although mtDNA encoded genes are seen to be differentially expressed (Fig. 7B,D), the vast majority (99.8%) of differentially expressed genes identified are encoded in the nuclear genome (Fig. 7C,E), suggesting that chimeric mtRNA frequency predicts transcriptomic changes outside the mitochondria.
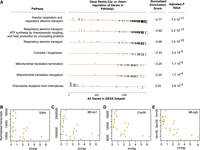
Chimeric mtRNA frequency predicts altered transcription of cellular pathways. (A) Gene set enrichment analysis of differentially expressed genes associated with chimeric mtRNA frequency in rat skeletal muscle aging. Negative correlations of FFPM to gene expression for nuclear-encoded SDHB (B) and COX5b (D) and mitochondrially encoded Mt-co1 (C) and Mt-cyb (E).
Discussion
We report a class of previously undocumented chimeric mtRNAs, which increase in multiple conditions associated with mitochondrial pathology and predict mtDNA deletion frequency and altered transcription of genes essential for mitochondrial function. The new class of chimeric mtRNAs we have discovered provides an exciting window into mitochondrial genomics. The distribution of chimeric mtRNA breakpoints closely matches the breakpoints identified previously by DNA sequencing approaches, whereas the frequency of chimeric mtRNAs predicts mtDNA deletion mutation frequency measured by PCR-based approaches. These relationships strongly suggest that the chimeric mtRNAs are transcriptional products of the mtDNA deletion events.
Many aspects of the chimeric mtRNAs strengthen what is known about mtDNA deletion mutations from DNA-based assays such as Southern blot (Corral-Debrinski et al. 1992), whole-genome PCR (Tengan and Moraes 1996), high-throughput sequencing (Lujan et al. 2020), digital PCR (Herbst et al. 2017), and long-read mtDNA sequencing (Vandiver et al. 2022). The chimeric mtRNAs predominate in the mitochondrial major arc, span large portions of the mitochondrial genome (i.e., 6–8 kb), and increase with age in human and rat skeletal muscle and human brain. In the human muscle and brain RNA-seq databases, we observed chimeric mtRNAs that correspond to the frequently reported mitochondrial “common” or “4977” deletion. The chimeric mtRNA frequency in the rat skeletal muscle correlates strongly with a DNA-based digital PCR assay, an orthogonal approach, supporting the validity of both approaches and the observed age-induced increases. This newly identified ability to identify pathogenic mtDNA variants in patients from available RNA-seq data is in line with evolving strategies to leverage transcriptomic data to better identify and understand undiagnosed and rare diseases (Lee et al. 2020) and greatly extends the ability to identify these events in difficult to obtain specimens. Using an RNA-based approach extends prior DNA-based findings by indicating a potential postgenomic role for mtDNA deletions that may uncover new mechanisms or points for intervention.
An extensive literature on chimeric RNAs from the nuclear genome in cancer suggests many possible roles for chimeric mtRNAs (Sun and Li 2022). Chimeric mtRNAs could interfere with mtDNA replication, transcription, and translation. mtDNA deletion mutations reach very high levels within individual cells (Pak et al. 2003; Bua et al. 2006; Herbst et al. 2007, 2013; Cheema et al. 2015), and in such cells, the chimeric mtRNA would be expected to also accumulate to very high levels in which it could deplete nucleotide pools, disrupt mt-mRNA processing, or impair mt-mRNA degradation/turnover pathways. Effects of chimeric mtRNAs on protein translation could include the sequestration of ribosomes and tRNAs on these aberrant transcripts. Chimeric mtRNAs could also bind other mtRNA transcripts or even leak into the cytoplasm (Kim et al. 2017) and interfere with nuclear transcriptional and translation machinery or trigger inflammatory responses (Hooftman et al. 2023; Zecchini et al. 2023). For example, when we previously examined the GPA and aging gene expression data, we found limited correlates (Herbst et al. 2023). By using the embedded chimeric mtRNA frequency, we observed additional correlations that point to disruption of mitochondrial function and metabolism and identified upregulation of inflammatory responses.
Chimeric mtRNA analyses are complimentary to the extensive literature on mtDNA deletions using DNA-based approaches. The age-induced increases in chimeric mtRNAs add rigor to DNA-based findings that mtDNA deletions increase with age in mammalian skeletal muscle and brain (Taylor et al. 2014; Herbst et al. 2017; Vandiver et al. 2022, 2023). In addition to verifying prior findings, the use of chimeric mtRNA to query mitochondrial genome rearrangements offers several advantages to DNA-based methods. The use of RNA transcripts to uncover mutation events leverages the transcriptional amplification of these genomic events that may improve detection of rare events. Finally, it would seem reasonable to exploit all of the biological inferences available from an RNA-seq experiment, especially when the signals are interpretable.
The potential to use RNA sequencing data for identification of mtDNA structural variation is particularly robust owing to the abundance of mitochondrial reads in standard RNA sequencing data sets. Mitochondrial transcripts make up 30%–40% of reads in standard RNA-seq libraries (Mercer et al. 2011; Yang et al. 2014) and as high as 70%–80% in single-cell RNA sequencing experiments (Yang et al. 2014). RNA-seq experiments are often designed to maximize nuclear gene expression data and avoid the sequencing of RNA species such as mitochondrial transcripts that are considered nonrelevant, unimportant, or redundant. In single-cell sequencing data, a preponderance of mitochondrial transcripts is used to exclude apoptotic cells (Yang et al. 2014). Although it may be acceptable to discard or ignore some data that truly have no relevance or possible interpretation, the detection of chimeric mtRNAs in RNA-seq data sets cautions against this bias and demonstrates the importance of sharing and depositing the raw data.
We focused our efforts on interrogating RNA-seq databases that extended our previous studies on aging skeletal muscle. The identification and quantitation of chimeric mtRNAs should be generalizable across organisms, making it potentially useful in comparative biology studies of aging for any organism for which there are annotated mitochondrial genomes or methods developed to search for chimeric mtRNAs in de novo transcriptomic data. Single-cell or single-nucleus RNA-seq characterization of chimeric mtRNAs could be used to define tissue and cell distributions of chimeric mtRNAs and the underlying mtDNA deletion mutations, and these could be combined with nuclear gene expression data to identify cellular impacts.
Although we demonstrate a strong correspondence between chimeric mtRNA and mtDNA mutations, there remain many open questions as to the specific relationship. We observe a strong linear relationship between the frequency of chimeric RNAs and mtDNA mutations in our rat model; however, this is not sufficient to suggest that the frequency of chimeric RNAs is a direct report of the frequency of mtDNA deletions. Transcript frequency is a reflection not only of DNA abundance but also transcription, transcript maturation, and transcript stability. It is possible that the presence of mtDNA deletions and chimeric transcripts could interfere with any of these processes. As such, inferences cannot yet be made as to how chimeric mtRNA frequency relates to well-defined characteristics of mtDNA deletions, such as a phenotypic threshold or cellular heteroplasmy. Further, our analysis indicates that a significant level of chimeric mtRNA is detected even in younger tissues, which are not frequently reported to contain abundant mtDNA deletions. One potential explanation is that low levels of mtDNA deletions are present in younger tissues and are easier to detect in this setting owing to transcriptional amplification. Alternatively, there may be background chimeric mtRNA reads present in sequencing data that are not reflective of underlying mtDNA deletions. Studies of other classes of mtRNAs have called into question the potential for those mtRNAs to derive from nuclear embedded mitochondrial sequences (NUMTs) (Pozzi and Dowling 2019). The lack of chimeric RNAs observed in a control cell line argues against NUMTs as a primary source of chimeric mtRNAs. In addition, NUMT origination cannot be accurately excluded using short-read sequencing (Albayrak et al. 2016). These questions and more will be important to address as these transcripts are examined in more data sets.
The location of chimeric fusion locations observed is intriguing. Although age-associated mtDNA deletions are most frequently observed to occur within the mitochondrial major arc, recent studies have demonstrated deletion events involving other areas of the genome (Bua et al. 2006; Lujan et al. 2020; Vandiver et al. 2023). The tissue specificity of this finding remains an open question. In this initial analysis of chimeric mitochondrial transcripts, fusion sites found in muscle data were primarily localized within the major arc, whereas brain data showed a broader distribution, suggesting potential differences in the distribution of mtDNA deletions between tissues. The identification of this unstudied class of mitochondrial transcripts will facilitate further studies to clarify these differences and gain insight into tissue specificity of mitochondrial genomic variation.
Limitations of our initial chimeric mtRNA analyses suggest next steps in characterizing chimeric mtRNAs. Addressing these limitations should include additional definition of the RNA-seq characteristics that affect chimeric mtRNA detection and quantitation. Factors such as RNA isolation method, mRNA enrichment, library preparation, and read depth need to be validated to enable sensitive detection of chimeric mtRNA as has been done for general RNA-seq approaches (Conesa et al. 2016). Methods that preserve biological signals arising from the mitochondria (rather than the standard approaches that discard them) will increase the sensitivity of the RNA-seq approach for detecting chimeric mtRNAs and improve chimera quantitation.
Alternative explanations for our findings could include age-related disruption of mitochondrial transcription, leading to aberrant RNA splicing events. However, this would not be consistent with the occurrence of the chimeric mtRNA corresponding to the well-characterized human “common” deletion. We adapted computational methods that were designed for detection of chimeric RNAs in cancer that may need to be further customized for chimeric mtRNAs. Further, we utilized short-read sequencing data, which has limitations for study of fusion events owing to mapping challenges; thus, long-read RNA-seq may be particularly well suited to find these previously undocumented RNA mutants (Workman et al. 2019).
We have begun identifying, mapping, and quantitating a new class of mitochondrial transcripts that appear to be the direct result of age-induced and disease-associated mtDNA deletion mutations. This approach provides orthogonal validation to prior studies of mtDNA deletion mutations and extends the ability to identify such events across a broader range of samples. Further study of these chimeric mtRNAs will expand our understanding of the underlying mutational events and open new avenues for disease prevention or therapeutic intervention.
Methods
RNA-seq databases
Publicly available RNA-seq data were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE129811, GSE157585, and GSE68719 and from the BioProject database under accession number PRJNA793055.
Analysis of RNA sequencing data for mitochondrial fusion transcripts
Gene fusion identification
STAR-fusion version 1.10.0 (Haas et al. 2019) was used to identify candidate fusion transcripts within RNA-seq data sets. For the rat study, a “CTAT genome lib” reference package was prepared for STAR-fusion using sequence and feature information for assembly Rnor6.0 downloaded from Ensembl version 104 (Harrison et al. 2024). For the human studies, reference files for assembly GRCh38 were used, also from Ensembl version 104. For both the rat and human, the Ensembl gene transfer format (GTF) files were manually modified prior to running the “CTAT genome lib” building script, in order to convey that the MT-ATP8 and MT-ATP6 genes are encoded within a single overlapping transcript that, although detected by STAR-fusion, does not represent a chimeric mtRNA. Similarly, GTF information for the MT-ND4 l and MT-ND4 genes was updated to reflect that they are normally expressed as a single transcript. Single-end reads were aligned to the rat or human reference genomes using STAR v2.7.11a (Dobin et al. 2013), and STAR-fusion v1.13.0 was then run on the STAR output to identify fusion candidates. Default STAR-fusion parameters were used with the exception of the following settings: ‐‐min_FFPM 0 was used to prevent filtering of fusion transcripts based on abundance, and ‐‐no_remove_dups was used to prevent removal of duplicate reads. The ‐‐alignIntronMax 100 option was used with STAR, which we have found to improve fusion detection for some deletions.
Comparing gene fusions among samples
Further analysis of fusion locations was done within the R environment (R Core Team 2023). The STAR-fusion output files describing the candidate fusions (star-fusion.fusion_candidates.preliminary files) were used in downstream analyses. R scripts were used to parse the files and to enumerate mitochondrial gene fusions within each sample. Specifically, for each observed fusion type (based on genes involved and ignoring the precise boundaries of the fusion), the total number of supporting reads was calculated, using values extracted from the JunctionReadCount column. Subsequently, for each data set, a table termed “raw counts” was generated, consisting of samples (rows) and fusion types (columns) with cells containing the summation of the JunctionReadCount values. A second table, termed “FFPM” for “fusion fragments per million total RNA-seq fragments,” was generated from the first table by dividing each raw count by the total number of sequenced fragments (in millions) in the corresponding sample. Sample meta data were programmatically added to each table as additional columns to facilitate further analyses. PCA plots were produced from FFPM tables using the ggfortify R package (Tang et al. 2016). Plots visualizing or comparing fusion site locations were generated from the star-fusion.fusion_candidates.preliminary files, using the reported breakpoint locations. Kernel density of break points was calculated and plotted using base R density function. Histograms were plotted using base R hist function. For plots examining gene fusion location and fusion sizes, events detected in more than seven transcripts were considered.
Gene set enrichment analysis
Publicly available FASTQ files were quasi-mapped and quantified to the Rattus norvegicus, Ensembl version 109 all cDNA reference transcriptome using Salmon (v1.9.0) (Patro et al. 2017) in Python 3.9.15. Differential gene expression analysis and gene set enrichment analysis were conducted in the R environment (R v4.2). FFPM counts were extracted, and differential gene expression analysis was performed using DESeq2 (v1.38.1) (Love et al. 2014). For differential gene identification, a sum of FFPM for all mitochondrial chimeric transcripts was calculated. Regression was then conducted using DESeq2 using both a model considering only this sum and a model considering this sum and adjusting for age and GPA treatment status. Significance testing was performed using a Wald test, and the resulting P-values were adjusted for multiple testing using package default. Gene set enrichment analysis was performed on resultant differentially expressed genes using the FGSEA package (v1.24.0) (Korotkevich et al. 2016) and reactome pathywas from reactome.db. Pathways with NES magnitude greater than two and at least 10 included transcripts were considered.
Fibroblast RNA preparation
Dermal fibroblasts containing the mitochondrial common deletion were obtained as a gift from Dr. Jean Kruttman (Majora et al. 2009). DNA was extracted using Qiagen DNeasy according to the manufacturer's instructions. Control dermal fibroblasts were purchased from Thermo Fisher Scientific (004-5C). RNA was extracted with TRIzol (Thermo Fisher Scientific) followed by RNeasy (Qiagen) clean-up, in accordance with the manufacturer's instructions; 150 bp paired-end sequencing was performed on Illumina NextSeq 500 through the UCLA Technology Center for Genomics and Bioinformatics.
Data access
The fibroblast sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1164295. All analysis code is available at GitHub (https://github.com/paulstothard/chimeric-mitochondrial-RNA-analysis) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Greater Los Angeles Veterans Healthcare System. Scientific illustration assistance was provided by Kate Baldwin, PhD (https://www.k8baldwin.com). The authors were supported by the National Institutes of Health (National Institute on Aging) R01AG055518 (J.W.), R01AG069924 (J.W.), and K02AG059847 (J.W.); the Dermatology Foundation (A.R.V.); and the Melanoma Research Alliance (A.R.V.). This research was enabled in part by support provided by the Digital Research Alliance of Canada (alliancecan.ca).
Author contributions: A.H., P.S., and J.W. conceived and designed the experiments. P.S. developed the algorithms. A.R.V. and P.S. conducted the bioinformatic analyses. All authors interpreted the data and wrote and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279072.124.
- Received February 5, 2024.
- Accepted November 25, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








