
Genome-wide profiles of H3K9me3, H3K27me3 modifications, and DNA methylation during diapause of Asian corn borer (Ostrinia furnacalis)
- Department of Entomology, MOA Key Lab of Pest Monitoring and Green Management, College of Plant Protection, China Agricultural University, Beijing 100193, China
-
↵1 These authors contributed equally to this work.
Abstract
Diapause represents a crucial adaptive strategy used by insects to cope with changing environmental conditions. In North China, the Asian corn borer (Ostrinia furnacalis) enters a winter larval diapause stage. Although there is growing evidence implicating epigenetic mechanisms in diapause regulation, it remains unclear whether dynamic genome-wide profiles of epigenetic modifications exist during this process. By investigating multiple histone modifications, we have discovered the essential roles of H3K9me3 and H3K27me3 during diapause of the Asian corn borer. Building upon previous findings in vertebrates highlighting the connection between DNA methylation and repressive histone methylations, we have examined changes in the genome-wide profile of H3K9me3, H3K27me3, and DNA methylation at the nondiapause, prediapause, and diapause stages. Data analysis reveals significant alterations in these three modifications during diapause. Moreover, we observe a correlation between the H3K9me3 and H3K27me3 modification sites during diapause, whereas DNA modifications show little association with either H3K9me3 or H3K27me3. Integrative analysis of epigenome and expression data unveils the relationship between these epigenetic modifications and gene expression levels at corresponding diapause stages. Furthermore, by studying the function of histone modifications on genes known to be important in diapause, especially those involved in the juvenile pathway, we discover that the juvenile hormone pathway lies downstream from H3K9me3 and H3K27me3 histone modifications. Finally, the analysis of gene loci with modified modifications unreported in diapause uncovers novel pathways potentially crucial in diapause regulation. This study provides a valuable resource for future investigations aiming to elucidate the underlying mechanisms of diapause.
Diapause serves as a crucial strategy used by insects to adapt to changing environments (Tauber and Tauber 1976). It can manifest at various stages of development, namely, embryonic, larval, pupal, or adult stages, depending on the species (Denlinger 2002). An illustrative example is the Asian corn borer (Ostrinia furnacalis) in North China, which undergoes a winter larval diapause stage. Upon entering diapause, insects experience an internal cessation of direct morphogenesis and initiate a distinct physiological program (Koštál 2006). This program is profoundly influenced by fluctuations in environmental conditions. Prediapause, a vital process preceding diapause, involves insects responding to changes in environmental signals and adjusting their behavior accordingly (Hahn and Denlinger 2007, 2011). Although direct morphogenesis continues during this stage, the ultimate decision for individuals to enter diapause, triggering endogenous developmental arrest, is determined by specific environmental signals and conditions (Koštál 2006; Emerson et al. 2009). Gaining a comprehensive understanding of the underlying mechanisms driving this phenomenon holds immense value in devising more effective strategies for pest control.
Numerous molecular mechanisms have been unraveled in the regulation of diapause (Teets et al. 2023). The central circadian clock detects variations in temperature and daylight hours, thereby influencing the production of neurotransmitters and neuromodulators (Teets et al. 2023). Neural events then regulate the production and secretion of insulin-like peptides (ilps), which in turn regulate juvenile hormone (JH) and Forkhead box protein O (FoxO), ultimately influencing the expression of genes responsible for physiological changes (Sim et al. 2015; Nagy et al. 2019; Teets et al. 2023). Neural events also control the production and release of prothoracicotropic hormone (PTTH), as well as the production of ecdysone (Ahmadi et al. 2021). Additionally, downstream genes, such as microRNAs regulated by ecdysone titers (e.g., let-7, miR-252, and miR-8–3p) and proteins that regulate lipid metabolism, have been found to play a role in the diapause process (Reynolds 2019; Reynolds et al. 2019). Identifying the upstream regulatory mechanisms will potentially enable the perturbation of diapause process.
Significant progress has been achieved in understanding various aspects of diapause mechanisms. Notably, recent studies have elucidated the pathways connecting photoreception to the expression of the diapause phenotype (Koide et al. 2021; Takeda and Suzuki 2022). Furthermore, investigations into the involvement of transcriptional factors, including FoxO, have shed light on their role in initiating diapause (Sim et al. 2015; Batz et al. 2019; Li et al. 2022; Zhang et al. 2022). The exploration of lipid metabolism in diapause has also provided valuable insights (Xu et al. 2012; Chowański et al. 2021; Jia and Li 2023). Additionally, transcriptome analyses have been conducted on various insects, leading to important discoveries (Bresnahan et al. 2022; Cui et al. 2022; Du et al. 2022; Egi and Sakamoto 2022; Nadeau et al. 2022). Moreover, the recognition of the microbiome's significance in insect diapause has gained attention (Didion et al. 2021; Dittmer and Brucker 2021). Research in this field has also contributed to advancing knowledge in areas such as human health (Easwaran et al. 2022; Hutfilz 2022; Geng et al. 2023).
Epigenetic mechanisms were found to be involved in diapause regulation. DNA methylation in diapause induction has been studied in two insects: the silkworm, Bombyx mori (Egi et al. 2016), and the jewel wasp, Nasonia vitripennis (Pegoraro et al. 2016). In B. mori, although the methyltransferase gene is up-regulated in diapausing embryos of B. mori (Sasibhushan et al. 2013), feeding the DNA methylation inhibitor 5-azacytidine did not change the diapause incidence (Egi et al. 2016). Conversely, in N. vitripennis, down-regulation of DNA methyltransferases enhanced diapause (Pegoraro et al. 2016). Additionally, the expression of multiple genes regulating histone methylation and acetylation changed with diapause (Reynolds et al. 2013, 2016). In Helicoverpa armigera, Polycomb group (PcG) proteins regulate PTTH production and prevent diapause entry through the control of H3K27me3 (Lu et al. 2013). By ovary-specific knockdown of histone mark writers and erasers, DiVito Evans et al. (2023) showed that H3K4me3 and H3K36me1 depletion promotes reproductive diapause in Drosophila. However, the genomic distribution of these DNA and histone modifications at different stages of diapause and the dynamic changes of the distributions need to be identified to better understand the mechanism of these epigenetic modifications.
To gain a deeper understanding of the epigenetic control of diapause, we investigated the primary histone modifications associated with the diapause process of the Asian corn borer in this study. Our findings revealed that treatment with inhibitors targeting H3K9me3 and H3K27me3, both of which showed altered levels during diapause, resulted in a decreased or increased percentage of diapause occurrence, respectively. To determine the genomic distribution of DNA methylation and histone modifications (specifically H3K9me3 and H3K27me3) in the heads of the Asian corn borer, we used whole-genome bisulfite sequencing (WGBS) and ChIP-seq analysis, respectively. The analysis of these data provided insights into the genome-wide distribution and changes of these epigenetic modifications during diapause. Moreover, by clustering gene loci affected by these modifications, we highlighted the potential significance of specific gene groups in diapause regulation.
Results
H3K9me3 and H3K27me3 are involved in the regulation of diapause in the Asian corn borer
To examine the role of histone methylation and acetylation modifications in the regulation of diapause in the Asian corn borer, we conducted a comprehensive analysis of multiple modifications on histone 3. Our findings show that both H3K9me3 and H3K27me3 were significantly down-regulated in the head of diapause larvae (Fig. 1A,B). However, H3K4me2, H3K14ac, and H3K27ac remained unaffected (Fig. 1C–E). Additionally, H3K9me3 and H3K27me3 were also found to be down-regulated in the body of diapause larvae (Supplemental Fig. S1A,B). Note that relative abundance of the signals detected by different antibodies did not reflect the relative concentration of the proteins, as the signal may influenced by the antibody specificity and the stripping process (see Methods). Based on these results, we propose that specific epigenetic modifications, such as H3K9me3 and H3K27me3, may play a role in regulating diapause in the Asian corn borer.
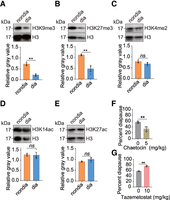
H3K9me3 and H3K27me3 are involved in diapause regulation of O. furnacalis larvae. (A–E) Western blot analysis was conducted on the head of Asian corn borer larvae to detect the levels of H3K9me3, H3K27me3, H3K4me2, H3K14ac, and H3K27ac. The antibody against total H3 was used as the loading control. The relative gray values of western blot were measured using Image J. Statistical differences were measured using an unpaired t-test: (ns) no significant difference, (**) P < 0.01. Error bars show the mean ± SEM. (F) Newly molted fourth instar larvae were fed artificial diets containing 0 mg/kg and 5 mg/kg SUV39H2-like (homologous to SU(VAR3-9) of D. melanogaster) inhibitor chaetocin, respectively. The results showed that chaetocin alleviated diapause caused by adverse environment. Statistical analysis using an unpaired t-test revealed a significant difference: (**) P < 0.01. (G) Similarly, newly molted fourth instar larvae were fed artificial diets containing 0 mg/kg and 10 mg/kg E(Z) inhibitor tazemetostat, which is homologous to E(Z) of D. melanogaster. The results indicated that tazemetostat exacerbated diapause caused by adverse environmental conditions. Statistical analysis using an unpaired t-test revealed a significant difference: (**) P < 0.01. Each point on the bar graph represents the value of an independent replicate.
To verify this hypothesis, we fed Asian corn borer larvae with chaetocin, an inhibitor of H3K9me3 methyltransferase SU(VAR)3-9, and tazemetostat, an inhibitor of H3K27me3 methyltransferase E(Z), respectively (Czermin et al. 2002; Schotta et al. 2002). Previous studies have shown the inhibitory effects of chaetocin and tazemetostat on SU(VAR)3-9 and E(Z) activities in Drosophila melanogaster, respectively (Greiner et al. 2005; Bhat et al. 2021). Through protein sequence comparison, we found that SUV39H2-like and E(Z) in the Asian corn borer share 35.92% and 62.17% homology with SU(VAR)3-9 and E(Z) in D. melanogaster, respectively. To determine the appropriate treatment concentration of these inhibitors for the Asian corn borer, we fed fourth instar Asian corn borer larvae with various concentrations of chaetocin and tazemetostat for 48 h, followed by an analysis of the H3K9me3 and H3K27me3 levels. As anticipated, increasing concentrations of chaetocin resulted in a decrease in H3K9me3 (Supplemental Fig. S1C). Similarly, tazemetostat inhibited H3K27me3 (Supplemental Fig. S1D). We did not see a dose response for tazemetostat effects on H3K27me3, suggesting a lower concentration may also be effective. As 10 mg/kg was much lower than the suggested concentration in the manufacturer's instructions, we used this concentration for future experiments. We selected relatively mild concentrations (5 mg/kg for chaetocin and 10 mg/kg for tazemetostat) to treat fourth instar larvae until their fifth instar under a 12 h light/12 h dark cycle at 20°C to evaluate the effects of these inhibitors on diapause induction. The results revealed that feeding chaetocin reduced the diapause rate, whereas feeding tazemetostat increased it (Fig. 1F,G). This suggests that the regulation of diapause by H3K9me3 differs from that by H3K27me3. In conclusion, our findings show the involvement of epigenetic modifications H3K9me3 and H3K27me3 in the regulation of diapause in the Asian corn borer.
Profiling of H3K9me3 and H3K27me3 during diapause reveals a significant correlation between their modification sites
To further investigate the role of H3K9me3 and H3K27me3 in diapause regulation in the Asian corn borer, we conducted ChIP-seq analysis to determine the genome-wide distribution of these modifications in the heads of larvae at the nondiapause, prediapause, and diapause stages (Fig. 2A). We observed that broad domains of H3K9me3 and H3K27me3 were mainly present in intergenic regions, with only ∼25% occupying the promoter region (Fig. 2B). Additionally, our analysis of binding site distribution relative to transcriptional start sites (TSSs) revealed that the majority of H3K9me3 and H3K27me3 binding sites were located within a range of 10 kb–100 kb from the TSS (Supplemental Fig. S2A). Furthermore, peak count frequency signal profiles of H3K9me3 and H3K27me3 showed increased average enrichment scores in the upstream (−1500 bp∼−750 bp) and downstream (750 bp) sequences proximal to the TSS (Supplemental Fig. S2B). These findings suggest that H3K9me3 and H3K27me3 occupy the coding and regulatory regions.
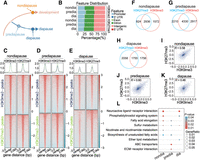
Positive correlation between H3K9me3 and H3K27me3 during different diapause stages. (A) ChIP-seq experimental design. (B) Distribution of H3K9me3 and H3K27me3 peaks across the annotated genome. (C–E) Overlapping peaks of H3K9me3 and H3K27me3 in the nondiapause (C), prediapause (D), and diapause (E) stages. (F–H) Venn diagram shows the number of genes with H3K9me3 peaks, H3K27me3 peaks, or both at the nondiapause (F), prediapause (G), and diapause (H) stages. (I–K) Genome-wide correlation plots showing correlation of the occupancies of H3K9me3 and H3K27me3 at the nondiapause (I), prediapause (J), and diapause (K) stages. The correlation coefficient R is indicated. (L) Comparative KEGG enrichment analysis of comodified genes in the nondiapause, prediapause, and diapause stages. The color shade of the dots represents the P-value, and the size of the dots represents the gene enrichment ratio.
Consistent with the levels of H3K9me3 and H3K27me3 observed in the western blots (Fig. 1), analysis of the overall levels of these two modifications indicated a positive correlation. We have three lines of evidence supporting this conclusion. First, examination of the occupancy sites of H3K9me3 and H3K27me3 revealed significant overlap and similar enrichment abundances (Fig. 2C–E). Second, the Venn diagram showed that the majority of gene loci modified by H3K9me3 and H3K27me3 overlapped, particularly in the nondiapause and prediapause stages (Fig. 2F–H). Last, to assess the relevance of H3K9me3 and H3K27me3 occupancy on the genome, we conducted a correlation analysis between the broad domains generated by these modifications. The results showed a weak positive correlation at a genome-wide scale (Fig. 2I–K). In conclusion, these findings collectively indicate a positive correlation between H3K9me3 and H3K27me3, suggesting their cooperative role in regulating gene expression during diapause in Asian corn borer larvae.
Additionally, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis on the shared broad domains of H3K9me3 and H3K27me3. The KEGG analysis revealed that pathways enriched during the nondiapause and diapause stages, but not the prediapause stage, included neuroactive ligand−receptor interaction and phosphatidylinositol signaling system. On the other hand, fatty acid metabolism pathways were enriched during the nondiapause and prediapause stages but not the diapause stage (Fig. 2L). These results suggest that H3K9me3 and H3K27me3 show distinct gene modification profiles at different stages of diapause in Asian corn borer larvae.
The enrichment analysis of genes modified by H3K9me3 and H3K27me3, with various modes of changes during diapause, reveals potential regulators of diapause
The analysis of the genome-wide landscape of H3K9me3 and H3K27me3 during diapause in Asian corn borer larvae elucidated the changes in these two modifications throughout the process. The abundance of both modifications in the positive gene locus at the nondiapause stage is down-regulated during diapause (Supplemental Fig. S3A,B), with a slight increase observed at the prediapause stage (Supplemental Fig. S3A,B). The abundance of H3K9me3 peaks, which showed a significant increase during the prediapause stage, decreased at the diapause stage (Supplemental Fig. S3C, green line). Conversely, the abundance of H3K9me3 peaks, which showed a significant decrease during the prediapause stage, increased at the diapause stage (Supplemental Fig. S3C, orange line). The abundance of H3K9me3 peaks with a significant increase at the diapause stage remained unchanged during the prediapause stage (Supplemental Fig. S3D, left and middle, green line) but showed a significant increase only upon entering diapause (Supplemental Fig. S3D, right, green line). On the other hand, the abundance of peaks with a marked decline during diapause increased during the prediapause stage and gradually decreased upon entering diapause (Supplemental Fig. S3D, orange line). Similar results were observed for H3K27me3 as for H3K9me3 (Supplemental Fig. S3E,F). In summary, both modifications showed opposite trends of changes during the prediapause and diapause stages at sites where corresponding modifications occurred.
To further investigate the correlation between the changes in H3K27me3 and H3K9me3 modifications, we conducted a correlation analysis between H3K27me3 and H3K9me3 that showed changes between stages. Under the prediapause and nondiapause conditions, the Pearson correlation coefficient (R) for H3K9me3 and H3K27me3 is 0.44 (Fig. 3A). Under prediapause and diapause conditions, the R for H3K9me3 and H3K27me3 is 0.50 (Fig. 3B). Under diapause and nondiapause conditions, the R for H3K9me3 and H3K27me3 is 0.48 (Fig. 3C). The results revealed a weak positive correlation on a genome-wide scale (Fig. 3A–C). These findings suggest that H3K9me3 and H3K27me3 undergo correlated changes during diapause in Asian corn borer larvae.
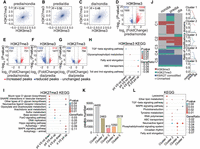
Global features of H3K9me3 and H3K27me3 modifications during diapause of Asian corn borer larvae. (A) Genome-wide correlation plots of H3K9me3 and H3K27me3 occupancies changed between the prediapause and nondiapause stages, respectively. (B) Genome-wide correlation plots of H3K9me3 and H3K27me3 occupancies changed between the prediapause and diapause stages, respectively. (C) Genome-wide correlation plots of H3K9me3 and H3K27me3 occupancies changed between the diapause and nondiapause stages, respectively. (D–G) Volcano plot of genes with statistically significant differential H3K9me3 and H3K27me3 peaks between different diapause stages. (H) Comparative KEGG enrichment analysis of genes with statistically significant differential H3K9me3 peaks. (I) Comparative KEGG enrichment analysis of genes with statistically significant differential H3K27me3 peaks. (J) Heatmaps showing the clustering of genes with H3K9me3, H3K27me3, or H3K9me3/H3K27me3 comodifications in the nondiapause, prediapause, and diapause stages (left panel). The ratios of H3K9me3, H3K27me3, and H3K9me3/H3K27me3 comodified genes are shown (right panel). Red indicates the domains that are marked only by H3K9me3; green, domains that are marked only by H3K27me3; blue, H3K9me3/H3K27me3 comodified domains; and gray, unmarked domains. (K) The number of peaks in each cluster in panel J. (L) Comparative KEGG enrichment analysis of genes in different clusters in panel J.
Analysis of the modification profiles of H3K9me3 and H3K27me3 at different diapause stages provided insights into their occupancy on gene loci. Comparing the prediapause stage with the nondiapause stage, we observed an increase in the abundance of 1558 H3K9me3 peaks and a decrease in the abundance of 372 H3K9me3 peaks (Fig. 3D). Furthermore, H3K27me3 showed significantly fewer changes in peak abundance compared with H3K9me3 (Fig. 3E). When comparing the diapause stage with the prediapause stage, we found an increase in the abundance of 314 H3K9me3 peaks and a decrease in the abundance of 1163 H3K9me3 peaks (Fig. 3F). Similarly, H3K27me3 showed significantly fewer changes in peak abundance compared with H3K9me3 (Fig. 3G). To further explore the role of H3K9me3 and H3K27me3 in the prediapause process, we performed a KEGG analysis on their unique and changed peaks. This analysis revealed the primary pathways enriched in gene profiles with increased or decreased modifications at different diapause stages (Fig. 3H,I).
Cluster analysis of the modified gene profiles of H3K9me3 and H3K27me3 at different stages provided insights into the biological features of genes showing similar trends in modification changes. We identified five types of change trends represented by clusters 1 to 5 (Fig. 3J), with variations in the number of peaks (Fig. 3K). For instance, in cluster 1 genes, the H3K9me3 ratio was down-regulated during both the prediapause and diapause stages, whereas the H3K27me3 ratio was up-regulated only during the prediapause stage. Genes with comodified H3K9me3 and H3K27me3 showed similar trends to H3K27me3. Enrichment analysis of genes belonging to different clusters revealed their functional diversity (Fig. 3L).
DNA methylation analysis in head tissues during diapause indicates a limited correlation between DNA methylation and H3K9me3 or H3K27me3 modifications
DNA methylation is an epigenetic repressive mark that cooperatively works with histone modifications in vertebrates (Xu et al. 2022). To investigate the functional role of DNA methylation in diapause, we conducted experiments to identify the DNA methylation profile of corn borer heads during different diapause stages. Analysis of the data revealed the genome-wide distribution of DNA methylation, with a notable enrichment in the gene regions (Fig. 4A). Quantification at the genome-wide level showed that total methylation levels were down-regulated during the prediapause stage and up-regulated during the diapause stage (Fig. 4B). KEGG enrichment analysis of the gene binding profile revealed distinct enriched items compared with H3K9me3 and H3K27me3 histone modifications (Fig. 4C), suggesting a different gene profile associated with DNA methylation.
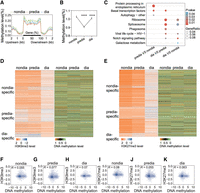
WGBS analysis reveals significant changes in DNA methylation during the diapause stages of Asian corn borer larvae but are not correlated with H3K9me3 and H3K27me3 in occupation. (A) Metaplot showing variations in DNA methylation levels upstream of and downstream from gene bodies at different developmental stages. Each region of the gene was equally divided into 50 bins, and the methylation level was defined as the average of the methylated cytosines of the bins. (B) The average levels of DNA methylation in the nondiapause, prediapause, and diapause stages displayed in a line chart showed significant changes during diapause stages. Each data point represents the mean ± SEM of the methylation level (percentage of methylated CG in all CGs) for each diapause stage. (****) P < 0.0001 (determined by unpaired t-test). (C) Comparative KEGG enrichment analysis of genes with significant changes in DNA methylation during different diapause stages. (D) Heatmaps showing the dynamics of H3K9me3 (left) and DNA methylation (right) on H3K9me3-positive genes during diapause of Asian corn borer larvae. (E) Heatmaps showing the dynamics of H3K27me3 (left) and DNA methylation (right) on H3K27me3-positive genes during diapause of Asian corn borer larvae. (F–H) Genome-wide correlation plots depicting the association between H3K9me3 and DNA methylation occupancies at the nondiapause, prediapause, and diapause stages. (I–K) Genome-wide correlation plots depicting the association between H3K27me3 and DNA methylation occupancies at the nondiapause, prediapause, and diapause stages.
To determine the potential correlation between DNA methylation and H3K9me3/H3K27me3 histone modifications, we analyzed the DNA methylation levels on gene loci bearing these modifications. Our results indicated a lack of correlation between DNA methylation levels and the levels of H3K9me3 and H3K27me3 histone modifications (Fig. 4D,E). The calculation of the Pearson correlation coefficient, R, resulted in low R-values for the correlation of DNA methylation with H3K9me3 or H3K27me3 at the three different stages (Fig. 4F–K), indicating an absence of correlation between them.
Global-level changes in three modifications and their correlations with gene expression levels during diapause
During diapause, there were significant changes in the global levels of all three modifications (Fig. 5A–K). Specifically, the global level of the H3K9me3 domains was up-regulated during prediapause and down-regulated during diapause (Fig. 5B). On the other hand, the levels of the H3K27me3-occupied domains were down-regulated during both prediapause and diapause (Fig. 5F). Furthermore, the levels of the other modification on both H3K27me3- and H3K9me3-occupied domains were down-regulated during prediapause and diapause (Fig. 5C,G). In addition, differentially methylated regions (DMRs) showed a decrease during prediapause and an increase during diapause (Fig. 5J). Similarly, the gene expressions of the domains associated with these three modifications were down-regulated during prediapause and up-regulated during diapause (Fig. 5D,H,K). In conclusion, the global levels of all three modifications changed significantly during diapause.
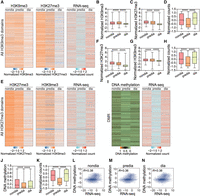
Analysis on the trend of global-level changes in three modifications and their correlations with gene expression levels during diapause. The three epigenetic modifications show significant changes during the diapause stages of Asian corn borer larvae. (A) Heatmaps illustrating the dynamics of H3K9me3 (left), H3K27me3 (middle), and gene counts (right) of all genes bearing the H3K9me3 domain in the heads of Asian corn borer larvae. (B–D) Box-plots indicating the average levels of H3K9me3, H3K27me3, and gene counts of all gene-locus-bearing H3K9me3 domains at the nondiapause, prediapause, and diapause stages. Each box represents the mean ± SEM. (****) P < 0.0001 (determined by unpaired t-test). (E) Heatmaps showing the dynamics of H3K27me3 (left), H3K9me3 (middle), and gene counts (right) of all genes bearing the H3K27me3 domain in the heads of Asian corn borer larvae. (F–H) Box-plots indicating the average levels of H3K9me3, H3K27me3, and gene counts of all gene-locus-bearing H3K27me3 domains at the nondiapause, prediapause, and diapause stages. Each box represents the mean ± SEM. (****) P < 0.0001 (determined by unpaired t-test). (I) Heatmaps showing the dynamics of DNA methylation (left) and gene counts (right) of all genes bearing DNA methylation domains in the heads of Asian corn borer larvae. (J,K) Box-plots showing the average level of DNA methylation and gene counts of all gene-locus-bearing DNA methylation domains at the nondiapause, prediapause, and diapause stages. Each box represents the mean ± SEM. (****) P < 0.0001 (determined by unpaired t-test). (L–N) Genome-wide correlation plots of DNA methylation and gene counts in the nondiapause, prediapause, and diapause stages.
Analyzing the correlation between gene expression levels and the population sites of H3K27me3/H3K9me3 modifications revealed that there was little correlation between them, regardless of the stage during diapause (Supplemental Fig. S4A–F). However, the DNA methylation displayed correlation with gene expression levels (Fig. 5L–N). These results indicated that although H3K27me3 and H3K9me3 modifications did not show significant correlations with gene expression, DNA modifications showed a stronger association with gene expression levels.
The expression levels of genes known to be important for diapause regulation are influenced by epigenetic modifications
To identify potentially important genes involved in diapause regulation, we examined three categories: diapause genes previously reported to be involved in diapause regulation, all genes with H3K27me3/H3K9me3/DNA methylation modifications, and genes changed in modifications during diapause (H3K27me3/H3K9me3/DNA methylation–changed genes). To compile a list of previously reported diapause genes, we first conducted a comprehensive search in the NCBI protein library using the keyword “diapause” to identify proteins associated with diapause across a wide range of organisms. This initial search yielded a total of 26,899 proteins. Next, we retrieved the protein sequences of these 26,899 identified proteins and compared them to the protein sequences of the Asian corn borer. Through this comparative analysis, we selected the protein with the highest alignment score and subsequently converted it into a gene. Subsequently, we meticulously removed any duplicated genes from our selection, resulting in approximately 10,592 diapause genes that remained. Analysis of these gene sets revealed overlapping profiles (Fig. 6A–C). Gene Ontology (GO) enrichment analysis highlighted specific biological processes associated with each modification (Fig. 6D). The TGF-beta signaling pathway, fatty acid elongation, ether lipid metabolism, and motor proteins were particularly enriched for H3K9me3 modification changes. On the other hand, the FoxO signaling pathway, purine metabolism, and autophagy were specifically enriched for H3K27me3 modification changes. DNA methylation changes were specifically associated with basal transcriptional factors, the Notch signaling pathway, and the spliceosome. Notably, the polycomb repressive complex was enriched for both H3K27me3 and H3K9me3 modification changes, whereas ATP-dependent chromatin remodeling was enriched for H3K27me3 and DNA methylation changes (Fig. 6D). The enrichment of these epigenetic factors across multiple modifications suggests the presence of feedback regulation.

Clustering of the epigenetically modified genes known to be important for diapause regulation. (A–C) The Venn diagram illustrates the number of overlapping genes between the known diapause-regulating gene number and the gene numbers epigenetically modified by H3K9me3 (A), H3K27me3 (B), and DNA methylation (C), with or without changes during diapause, respectively. (D) Comparative KEGG enrichment analysis of the overlapping genes shown in panels A–C for H3K9me3, H3K27me3, and DNA methylation, respectively. (E–G) Clustering of the overlapping genes shown in panels A–C for H3K9me3 (E), H3K27me3 (F), and DNA methylation (G), respectively. Heatmaps show the genes with statistically significant changes (clustered by k-means). The line displays the overall changes in genes in each cluster (left). The KEGG enrichment of genes of clusters is shown on the right panel.
To identify trends in changes among these three modifications on diapause-related gene sites, we performed clustering analysis. For H3K9me3-modified genes, the overall trend during diapause was down-regulation in the prediapause stage followed by up-regulation upon entering diapause, surpassing nondiapause levels, as depicted in clusters 2, 3, and 4 (Fig. 6E). For H3K27me3-modified genes, the overall trend during diapause was down-regulation in the prediapause stage and subsequent up-regulation upon entering diapause (although still at a lower level compared to nondiapause), as shown in clusters 1, 2, 3, 4, and 5 (Fig. 6F). However, no dominant trend was observed for DNA modification across different clusters (Fig. 6G). The enrichment annotations for genes with different modifications are displayed on the right side (Fig. 6E–G). By conducting enrichment analysis on each gene group, we can gain insight into the pathways that undergo different patterns of changes during different diapause stages. It is worth noting that some pathways displayed their own distinct changes, which deviated from the overall trends observed, such as cluster 1 in Figure 6E and cluster 2 in Figure 6G.
To evaluate the correlation between gene expression levels and DNA methylation, H3K9me3, and H3K27me3 modifications, we conducted an analysis. We examined the correlation between gene expression levels and these modifications for the genes enriched in the items depicted in Figure 6D. The strength of the correlation, ranging from −1 to 1, was color-coded, in which blue represents negative correlations, and yellow indicates positive correlations (Supplemental Fig. S5).
The results revealed that the majority of genes located on the gene locus enriched for H3K9me3 modifications displayed positive correlations with all three modifications, except for TGF-beta genes with DNA modifications, motor proteins with DNA modifications, fat acid elongation genes with H3K9me3 modifications, ether lipid metabolism with H3K9me3 modifications, certain genes involved in ether lipid metabolism with H3K27me3 modifications, and some genes involved in fat acid elongation with H3K27me3 modifications, all of which showed negative correlations (Supplemental Fig. S5A; Supplemental Table S1). Within the gene locus enriched for H3K27me3 modifications, polycomb repressive complex genes displayed positive correlations with all three modifications (Supplemental Fig. S5B; Supplemental Table S1). Conversely, the FoxO signaling pathway showed negative correlations with the three modifications (Supplemental Fig. S5B; Supplemental Table S1). Regarding the gene locus associated with DNA methylation, most of the gene loci showed positive correlations with all three modifications (Supplemental Fig. S5C; Supplemental Table S1). Regarding the gene locus related to JHs, the expression levels of most genes showed negative correlations with the three modifications (Supplemental Fig. S5D; Supplemental Table S1). However, for the gene locus associated with clock genes, the expression levels of most genes showed positive correlations with the three modifications (Supplemental Fig. S5E; Supplemental Table S1).
To validate the potential significance of H3K9me3 and H3K27me3 modifications in the diapause process, we examined the expression levels of their target genes implicated in the JH pathway. Previous studies have shown that JH plays a significant role in the diapause regulation of Lepidoptera, which has been confirmed in the southwestern corn borer and the European corn borer (Yin and Chippendale 1973, 1979; Yagi and Akaike 1976; Eizaguirre et al. 1998). The results showed that treatment with either an H3K9me3 or H3K27me3 inhibitor led to significant changes in the expression levels of some genes (Fig. 7A; Supplemental Table S1). Furthermore, when the JH analog was administered in conjunction with either an H3K9me3 or H3K27me3 inhibitor, there was no alteration in the increased diapause rate induced by the JH analog alone (Fig. 7B). These findings suggest that the JH pathway is downstream from H3K9me3 and H3K27me3 histone modifications.
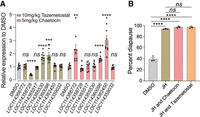
H3K9me3 and H3K27me3 regulate the expression levels of genes known to be related to diapause regulation. (A) Relative expression changes of genes in the juvenile hormone (JH) pathway after treatment with 5 mg/kg of the SUV39H2-like inhibitor chaetocin and 10 mg/kg of the E(Z) inhibitor tazemetostat compared with DMSO controls. Statistical differences were measured using an unpaired t-test: (ns) no significant difference, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, and (****) P < 0.0001. Error bars show the mean ± SEM. (B) Percentage of diapause was significantly changed by treatment of 0.01 mg/kg JH with or without 5 mg/kg of the SUV39H2-like inhibitor chaetocin or 10 mg/kg of the E(Z) inhibitor tazemetostat, respectively. Statistical differences were measured using an unpaired t-test: (ns) no significant difference, (****) P < 0.0001. Each data point represents the value of an independent replicate.
Analysis of unreported gene profiles in diapause reveals potential new regulators for diapause
We observed a significant number of gene loci that showed dynamic modifications and had not been previously implicated in diapause regulation. To gain a better understanding of the role played by these potential new participants in diapause, we conducted an enrichment analysis of these genes. Specifically, we focused on gene profiles that showed significant changes in DNA methylation, H3K9me3, or H3K27me3 during diapause but had not been previously associated with the diapause process (Fig. 8A). Gene Ontology enrichment analysis showed their involvement in various processes, including RNA binding (such as snoRNA binding and mRNA binding), glutathione transferase activity, and transcription regulation, among others (Fig. 8A). Additionally, KEGG pathway analysis revealed the enrichment of these genes in pathways related to glutathione metabolism, ribosome biosynthesis, and RNA degradation (Fig. 8A).

Analysis of previously unreported gene profiles in diapause reveals potential new regulators for diapause. (A) Comparative GO enrichment analysis and comparative KEGG enrichment analysis for the overlapping gene group shown in the upper panel (cf. with Fig. 6A–C) for H3K9me3, H3K27me3, and DNA methylation, respectively. (B,C) The top positive and top negative gene lists from the gene group indicated at the top of panel A. (D) Comparative KEGG enrichment analysis of the top positive and top negative genes. (E) Comparative GO enrichment analysis of the top positive and top negative genes.
The top-listed gene profiles also unveiled potential new players in diapause. We compiled a list of the top 10 genes from these gene profiles (Fig. 8B,C), many of which have yet to be characterized in terms of their function. Enrichment analysis of the top 30 genes shed light on the pathways or functions associated with these highly ranked genes (Fig. 8D,E). In conclusion, our study identified a collection of previously unreported, but potentially crucial, players in the control of diapause.
Discussion
In this study, we have discovered that epigenetic regulation plays crucial roles during diapause, as is the case in all developmental processes. By examining the levels of multiple histone modifications, we have identified the potential importance of H3K9me3 and H3K27me3 during diapause. Previous studies in vertebrates have established a close relationship between DNA methylation and repressive histone methylations; thus, we focused on DNA methylation, H3K9me3, and H3K27me3 in our investigation. To gain insights into the genome-wide features of these epigenetic regulations, we analyzed the changes in DNA methylation, H3K9me3, and H3K27me3 across the nondiapause, prediapause, and diapause stages. Our data analysis revealed the dynamics of these three modifications and highlighted a correlation between H3K9me3 and H3K27me3 during diapause. However, we observed little correlation between DNA modifications and either H3K9me3 or H3K27me3. Additionally, we identified the gene profile regulated by these epigenetic modifications during diapause. By integrative analysis of epigenome and expression data, we uncovered the relationship between these epigenetic expressions and gene expression levels at corresponding stages during diapause. We further investigated the function of histone modifications on genes involved in the juvenile pathway, which has been proven to be important in diapause. Our findings suggested that the JH pathway is downstream from H3K9me3 and H3K27me3 histone modifications. Finally, through the analysis of modified gene loci not previously reported in diapause, we identified potentially important new pathways in diapause regulation. This study sheds light on the repressive methylation profile of chromatin during diapause and provides valuable information on the dynamic changes in epigenetic modifications at gene loci during diapause. These findings serve as a useful resource for future studies on the potential genes responsible for regulating diapause, ultimately contributing to both a better understanding of the underlying molecular mechanisms governing diapause (analogous to hibernation in vertebrates) and improved methods for pest control.
However, it is important to acknowledge that this study has several limitations. First, diapause is a multifaceted process with distinct characteristics observed in different substages. Because we collected samples at only one time point for each stage, we cannot determine if the epigenetic features undergo changes during these substages. Additionally, the epigenetic features of the postdiapause stage were not examined in this study, which represents another limitation. To overcome these limitations, future studies should consider collecting samples at multiple time points within each diapause stage and include analysis of the postdiapause stage. Furthermore, although the application of inhibitors targeting histone modifications showed an impact on diapause progression, this study does not provide a definitive cause-and-effect relationship between these epigenetic modifications and diapause. To establish a solid understanding of the precise cause-and-effect relationship, it is necessary to develop methods for specifically manipulating these epigenetic modifications. Further research is needed in this area to fully comprehend the intricate mechanisms underlying diapause regulation.
The reason for separating heads and bodies is that the heads serve as the primary control center for diapause. Diapause in larvae can be induced by manipulating photoperiod and temperature, as evidenced by studies conducted by Mutchmor and Mazurek (1959) and Guo et al. (2013). Neurons or metabolic organs that respond promptly to changes in photoperiod and temperature are located in the head, such as the circadian system, which plays a crucial regulatory role in diapause (Cassier and Cymborowski 1993; Hartfelder et al. 1994; Shimizu et al. 1997; Barberà et al. 2017; Saunders 2020; Ahmadi et al. 2021). The larvae of the Asian corn borer display a clear anatomical division into three distinct sections: the head, thorax, and abdomen. The head, with its relatively small triangular shape, typically shows a brown or black color, whereas the body appears white. To separate the head from the body, we meticulously dissected the larvae precisely at the junction where the colors of their head and thorax meet. In our selected Asian corn borer larvae, the head consists of three key components: the brain, black compound eyes, and mouthparts. Among these components, the brain is the largest and holds primary significance in our research focus.
Note that the changes in gene expression levels caused by the inhibitors for H3K27me3 and H3K9me3 do not correlate well with the correlation of these genes with the histone modifications during diapause (cf. Fig. 7A and Supplemental Fig. S5D). This may be owing to the complexity of the gene expression regulatory network during diapause, which is not completely mimicked by the changes of histone modifications caused by the inhibitors. In addition, the difference in the diapause phenotypes caused by treatment of chaetocin, an inhibitor of H3K9me3 methyltransferase SU(VAR)3-9, and tazemetostat, an inhibitor of H3K27me3 methyltransferase E(Z), may be attributed to their different target genes. The impact of histone modification on diapause is a result of the collective effects on all of its target genes. A plausible explanation for these phenotypes is that the primary targets of H3K9me3 are genes that inhibit diapause, whereas the main targets of H3K27me3 are genes that promote diapause. Consequently, the derepressing of diapause-inhibiting genes leads to reduced diapause, whereas the derepressing of diapause-promoting genes results in increased diapause (as outlined in Fig. 1F,G, respectively). This same theory could elucidate the correlation between the results depicted in Figure 1, B and G. Previous studies have shown that PRC2, through its regulation of H3K27me3, controls PTTH expression in the insect brain and thereby influences developmental timing (Lu et al. 2013). After injection of 3-deazaneplanocin A (inhibit methyltransferase activity of the PRC2 complex) into H. armigera pupae, it was observed that the levels of H3K27me3 and PTTH in pupae decreased, resulting in an enhancement of pupal diapause. The role of H3K27me3 in diapause was found to be similar between H. armigera and O. furnacalis.
This study revealed features of H3K9me3 and H3K27me3 modifications during the diapause stage. Both of these repressive modifications showed opposite changes in the prediapause stage versus the diapause stage. There was an increase in the prediapause stage and a decrease in the diapause stage (Supplemental Fig. S3A,B). Insect diapause can be divided into the prediapause, diapause, and postdiapause stages, and insects in the prediapause stage are able to synthesize cryoprotectants and store energy for later developmental stages (Yamashita 1996; Homma et al. 2006; Koštál 2006). Histone modification shows notable changes during the prediapause stage, which likely contribute to the initiation and preparation for diapause, involving significant alterations in gene expression and regulatory mechanisms (Hahn and Denlinger 2007, 2011). The prediapause stage represents a critical period dedicated to transitioning and adapting to environmental cues before entering the diapause state. In contrast, the observed similarity in marker enrichment between the nondiapause stage and diapause stage suggests the maintenance of a shared set of molecular characteristics or regulatory pathways throughout the diapause process. This similarity may indicate the necessity to sustain certain biological processes during diapause to ensure survival and enable rapid recovery of active growth and development when favorable conditions return. This result also suggests that the main events in the prediapause stage were to halt the ongoing developmental process, whereas the main events in the diapause stage were to express the set of genes that maintain the diapause program. Although the global level of these two modifications showed significant correlations (Fig. 3A–C), on the gene sites of known key regulators of diapause, most of the genes showed a significant increase in H3K9me3 and a decrease in H3K27me3 during the diapause stage (Supplemental Fig. S5). This indicates a transition from H3K27me3 to H3K9me3 during the diapause stage. This is consistent with the fact that the coexistence of these two modifications on gene loci was decreased in the diapause stage (Fig. 2E), suggesting changes or transitions in the types of histone modifications at these gene loci.
The general distribution of H3K27me3, H3K9me3, and DNA methylation revealed in this study is consistent with that of previous publications. Our analysis revealed that DNA methylation is highest within gene regions, which is consistent with previous studies (Xiang et al. 2010). The general locations of H3K27me3 and H3K9me3 were found to be consistent with previous studies conducted on insects (Ruiz et al. 2019; Zhao et al. 2023).
The DNA methylation showed little correlation with H3K27me3 and H3K9me3, indicating evolutionary divergence from vertebrates. In early vertebrate development, high activity of DNA demethylase led to a genome-wide transition from DNA methylation to histone modifications. However, data from this study indicate no transition between DNA methylation and histone modifications during the diapause process. This could be owing to the low level of DNA methylation in insects, resulting in its less significant function. However, previous studies of DNA methylation in insects have indicated a relatively high abundance of this modification in Lepidoptera (Varma et al. 2022). Moreover, our study reveals a substantial level of correlation between changes in DNA modification and expression levels revealed by RNA-seq (Supplemental Fig. S5). Hunt et al. (2013) identified a weak correlation between DNA methylation and H3K27me3 (see Table 2 in Hunt et al. 2013). However, our data show a different scenario. Hunt et al. (2013) found no correlation between DNA methylation and H3K9me3 (see Table 2 in Hunt et al. 2013), which is consistent with our own findings. It is an intriguing question how the relationship between DNA modification and histone modification develops in evolution.
We conducted a comprehensive literature review and found that the correlation relationship between these modifications is dependent on various biological conditions, including tissue type and physiological state. We found that some of the conditions mentioned in previous literature align with our own findings. For example, analysis of data from Senaratne et al. (2021) indicated that there is no correlation between H3K9me3 and gene expression in B. mori (Supplemental Fig. S6A). This result has also been validated in the head of D. melanogaster (Yu et al. 2017). These findings are consistent with its major role in heterochromatin (Ebert et al. 2004, 2006). Regarding the correlation between H3K27me3 and gene expression, in a study by Hu et al. (2020), conducted on the African turquoise killifish (Nothobranchius furzeri), it was reported that H3K27me3 does not show a correlation with gene expression (see Supplemental Fig. S7H in Hu et al. 2020). Additionally, analysis of data from Senaratne et al. (2021) revealed a correlation between H3K27me3 and gene expression (Supplemental Fig. S6B). These findings suggest that the modification conditions vary significantly depending on biological factors such as tissue type and physiological conditions. Our results align with those of Hu et al. (2020). In most cases, there appears to be a lack of correlation between H3K27me3 or H3K9me3 and gene expression. This could be because transcription is influenced by a combination of multiple regulatory factors, including histone modifications such as H3K4me3 and H3K36me3, in addition to H3K27me3 and H3K9me3, DNA modifications, and transcriptional factors. These factors collectively contribute to the regulation of transcription, with H3K27me3 and H3K9me3 serving as upstream events that are further interpreted by downstream events depending on the specific circumstances. As a result, the repressive marks H3K27me3 and H3K9me3 function to establish a general chromosome context in most cases but show little correlation with specific transcriptional states. Regarding the correlation between DNA methylation and gene expression, in a study by Hunt et al. (2013), a weak correlation between DNA methylation and gene expression was observed (see Fig. 2 in Hunt et al. 2013). Similarly, in work by Xu et al. (2021), a correlation between DNA methylation and gene expression was also reported (see Fig. 2A in Xu et al. 2021). These findings align with our own results.
Methods
O. furnacalis strain, husbandry, maintenance, diapause induction, and sample collection
The O. furnacalis larvae used in this study were originally collected from the corn field at the Shangzhuang experimental station of China Agricultural University in Beijing, China, and maintained in the laboratory. These larvae were reared in boxes (20 × 14 × 8 cm3) and fed an artificial diet (consisting of maize flour, 150.0 g; soybean flour, 150.0 g; glucose, 75.0 g; vitamin C, 4.0 g; agar, 22.0 g; yeast power, 90.0 g; sorbic acid, 5.0 g; water 1400 mL). The adult moths were individually placed in ventilated boxes, where they laid eggs on waxed papers. All stages of O. furnacalis were maintained at a temperature of 27°C, a relative humidity of 65%, and a photoperiod of 16 h light/8 h dark.
To induce diapause in Asian corn borer larvae for western blot analysis, ChIP-seq, and WGBS, we collected 300 newly molted fourth instar larvae and placed them in a box that was 20 cm in diameter and 15 cm in height. These larvae were provided with a standard artificial diet and exposed to a temperature of 20°C, following a photoperiod of 12 h of light and 12 h of darkness, as described in a previous study (Guo et al. 2013). Throughout the photoperiod, we closely monitored the larvae's daily developmental progress, identifying those that did not undergo pupation within 45 d as diapause larvae. Additionally, after being maintained at a temperature of 20°C with a photoperiod of 12 h light/12 h dark for a duration of 20 d, these larvae entered a prediapause state (Koštál 2006). To separate the head from the body, we carefully dissected the larvae at the precise junction where the colors of their head and thorax meet.
Protein extraction and western blot analysis
To extract histones, 20 larvae heads were immersed in PBS and washed twice with ice-cold PBS supplemented with 5 mM sodium butyrate to maintain histone acetylation levels. The heads were then resuspended and homogenized in Triton extraction buffer (TEB; PBS containing 0.5% Triton X-100 [v/v], 2 mM PMSF, 0.02% [w/v] NaN3). The lysate was shaken in a rotary shaker for 1 h at 4°C and subsequently centrifuged for 10 min at 2000 rpm at 4°C. The supernatant was discarded, and the pellet was washed once with TEB. The pellet was then resuspended in 0.2 M HCl, and the histones were extracted overnight at 4°C. After extraction, samples were centrifuged at 2000 rpm for 10 min at 4°C. The resulting supernatant was transferred to a new 1.5-mL tube, and equal amounts of protein were separated on a 15% SDS-PAGE gel and transferred to a 0.22-µm PVDF membrane (Millipore ISEQ85R). The membrane was incubated with primary antibodies overnight at 4°C. HRP-labeled secondary antibodies, diluted 1:1000, were incubated with the membrane for 4 h at room temperature. The signals were detected using ECL (ABclonal RM00021P) and imaged using Amersham ImageQuant 800 (GE Healthcare). The primary antibodies used in this study were antihistone H3 (trimethyl K9; Abcam ab8898), trimethyl-histone H3-K27 mouse mAb (Abconal A16199), antidimethyl-histone H3 (Lys4; Millipore 07-030), antiacetyl-histone H3 (Lys14; Millipore 07-353), antihistone H3 (acetyl K27; Abcam ab4729), and antihistone H3 (Easy Bio BE3015). The secondary antibodies used in this study were HRP goat antirabbit IgG (H + L; ABconal AS014) and HRP goat antimouse IgG (H + L; ABconal AS003). After detecting the signal of histone modification, we used stripping buffer (0.1 L:20 mL SDS 10%; 12.5 mL Tris HCl at pH 6.8, 0.5 M; 67.5 mL distilled water; add 0.8 mL beta-mercaptoethanol under the fume hood) to remove the primary and secondary antibodies from the western blot membrane. The purified membrane was subsequently incubated with antihistone H3 and secondary antibody, resulting in the detection of the H3 signal. The signal intensity was quantified by ImageJ software (NIH). The relative levels of histone modifications were normalized to the histone H3 signal. Three biological replicates were performed for each experiment.
Drugs through oral feeding and calculation of the diapause rate
To investigate the effectiveness of inhibitors, we conducted an experiment involving oral administration of drugs to newly molted fourth instar larvae. These larvae were fed an artificial diet containing different concentrations of drugs for 48 h. The heads of the larvae were subsequently used for western blot analysis or quantitative RT–PCR (qRT-PCR). A control group was also included, in which DMSO was used instead of the drugs.
To assess the impact of drugs on the diapause of Asian corn borer larvae, the newly molted fourth instar larvae were fed with
an artificial diet containing either 0 mg/kg or 5 mg/kg of the SUV39H2-like (homologous to SU(VAR3-9) of D. melanogaster) inhibitor chaetocin (MCE, HY-N2019). These larvae were kept at a temperature of 20°C under a photoperiod of 12 h light/12
h dark for 25 d. Following this, the larvae were transferred to a temperature of 27°C under a photoperiod of 16 h light/8
h dark for 20 d. Then the diapause rate was determined using the following formula: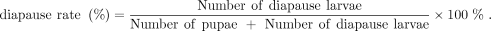 The diapause of insect individuals progresses through three stages—prediapause, diapause, and postdiapause—in a pattern observed
in the Asian corn borer as well (Koštál 2006). After entering diapause, Asian corn borer larvae transition to the postdiapause stage when encountering suitable environmental
conditions, gradually terminating diapause and resuming normal development. This transition period lasts a minimum of 20 d
for the Asian corn borer (He et al. 2018). In cases in which Asian corn borer larvae do not enter diapause, they typically undergo regular development into pupae
within 20 d upon exposure to conducive conditions. Consequently, following exposure of Asian corn borer larvae to diapause
conditions and drug treatments, we transferred the larvae to nondiapausing conditions to facilitate accurate assessment of
the diapause rate, assuming that all nondiapause larvae would pupate within this 20-d period.
The diapause of insect individuals progresses through three stages—prediapause, diapause, and postdiapause—in a pattern observed
in the Asian corn borer as well (Koštál 2006). After entering diapause, Asian corn borer larvae transition to the postdiapause stage when encountering suitable environmental
conditions, gradually terminating diapause and resuming normal development. This transition period lasts a minimum of 20 d
for the Asian corn borer (He et al. 2018). In cases in which Asian corn borer larvae do not enter diapause, they typically undergo regular development into pupae
within 20 d upon exposure to conducive conditions. Consequently, following exposure of Asian corn borer larvae to diapause
conditions and drug treatments, we transferred the larvae to nondiapausing conditions to facilitate accurate assessment of
the diapause rate, assuming that all nondiapause larvae would pupate within this 20-d period.
Each reaction used 30 to 60 larvae, and three or four biological replicates were performed for each experiment.
Similarly, newly molted fourth instar larvae were fed an artificial diet containing 0 mg/kg and 10 mg/kg E(Z) (homologous to E(Z) of D. melanogaster) inhibitor tazemetostat (MCE HY-13803), respectively. The diapause rate was then calculated following the same protocol as above. The diapause rate was calculated using a similar protocol after treating with JH (ALTER 1ST20290) with or without chaetocin or tazemetostat. Three biological replicates were performed for each experiment.
Data processing and analysis for ChIP-seq, RNA-seq, and WGBS
The ChIP protocol used in this experiment was previously described by Nègre et al. (2006). For each stage, 50 larval heads were used per reaction. In this experiment, we used antihistone H3 (trimethyl K9; Abcam ab8898) and antihistone H3 (trimethyl K27; Abcam ab6002). All steps of Illumina library preparation and sequencing were performed at Novogene's (https://cn.novogene.com/) sequencing platform. Adaptor sequence trimming, mapping to the O. furnacalis reference genome (Fang et al. 2021) using Bowtie 2 (Langmead and Salzberg 2012), and PCR duplicate removal using Picard Tools (http://broadinstitute.github.io/picard/) were performed. Broad peaks were called using MACS2 (Zhang et al. 2008). Annotation and analysis were performed using the R package ChIPseeker (Yu et al. 2015; R Core Team 2023). For RNA-seq, raw data were downloaded from the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA1026478. Two biological replicates were used for each sample group. The RNA-seq reads were mapped to the O. furnacalis reference genome using HISAT2 (Kim et al. 2019). Expression levels for all genes were quantified using featureCounts (Liao et al. 2014). In WGBS, 30 larval heads were used per reaction, and two replicates were performed for each stage. WGBS were performed at Novogene's (https://cn.novogene.com/) sequencing platform. All WGBS reads were initially processed using Trimmomatic to trim adaptor and low-quality reads (Bolger et al. 2014). Adaptor-trimmed reads were then mapped to the O. furnacalis reference genome, with lambda DNA sequence as a negative control, using Bismark (Krueger and Andrews 2011). The methylation level of each CpG site was estimated using bismark_methylation_extractor (Krueger and Andrews 2011). The deepTools computeMatrix function (Ramírez et al. 2016) was used to compute average methylation level in each bin (Schultz et al. 2012), and the methylation level distribution of genes upstream and downstream was plotted by plotProfile. DMRs were identified using R package DSS (https://www.rdocumentation.org/packages/DSS).
Heatmaps and metaplots were generated using the computeMatrix and plotHeatmap functions of deepTools, using bigWig files and scaled regions (Ramírez et al. 2016). To compute the average RPKM-normalized read count in peaks, the deepTools multiBigWigSummary function (Ramírez et al. 2016) was used. The Pearson correlation coefficient (R) between different ChIP-seq targets was calculated using ggplot2 in Rstudio. The Pearson correlation coefficient (R) was used as a measure of the degree of correlation, with values ranging from −1 to 1. A positive value indicates a positive correlation, whereas a negative value indicates a negative correlation. R-values between zero and 0.25 or between zero and −0.25 generally indicate the absence of correlation, whereas those between 0.25 and 0.50 or between −0.25 and −0.50 suggest a weak correlation. R-values ranging from 0.50 to 0.75 or −0.50 to −0.75 indicate a moderate to good correlation, and R-values from 0.75 to 1 or from −0.75 to −1 indicate a very good to excellent correlation. Data sets of interest in Supplemental Figure S6 were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE158902. Significantly differential peaks were determined using the MAnorm with P-value < 0.05 and M > 0.58 (FoldChange > 1.5) (Shao et al. 2012).
Enrichment analysis of GO and KEGG was conducted using the R package Cluster Profiler (Wu et al. 2021). To construct the GO data set for O. furnacalis, we performed protein sequence annotation using eggnog mapper V2 (Cantalapiedra et al. 2021). We then established the association and generated the GO data set using the makeOrgPackage function in the R package AnnotationForge (https://bioconductor.org/packages/AnnotationHub/).
Previously described methods were applied for the identification and clustering of H3K9me3, H3K27me3, and H3K9me3/H3K27me3 comodified genes (Xu et al. 2022). The heatmap and cluster analysis in Figure 3J was generated using the R package Complex Heatmap (Gu et al. 2016). Protocols for the clustering analysis of stage-specific H3K9me3 and H3K27me3 domains have been previously described (Xu et al. 2022). BEDTools intersect was used to construct data matrices for stage-specific H3K9me3 domains, stage-specific H3K27me3 domains, all H3K9me3 domains, and H3K27me3 domains (Quinlan and Hall 2010). The heatmap and cluster analysis in Figure 6, E through G, was generated using the R package ClusterGVis (https://github.com/junjunlab/ClusterGVis).
Construction of diapause protein library
The sequences of diapause proteins from all species and the protein sequences of Asian corn borers were downloaded from the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/?term=diapause). BLASTP was used to align diapause proteins with Asian corn borer proteins, and the protein with the highest alignment rate was extracted (https://www.ncbi.nlm.nih.gov/books/NBK279690/). Finally, the obtained diapause-related proteins were converted into gene names for data analysis.
qRT-PCR
Total RNA was isolated from approximately 10 heads of O. furnacalis using TRIzol Reagent (Vazyme R401-01), and 1 μg total RNA from each sample was used as a template for reverse transcription using HiScript III all-in-one RT supermix (Vazyme R333). The cDNA preparation was subjected to real-time qPCR using the Applied Biosystem Step-One real-time PCR system. The ChamQ SYBR qPCR master mix (high ROX premixed; Vazyme Q341) protocol was followed during the process. Data analyses were performed using Prism 8.0 (Graphpad). All primers used are listed in Supplemental Table S2. All quantitative RT-PCR analyses were conducted with three biological replicates.
Statistical analysis
For comparisons of two groups, an unpaired two-tailed Student's t-test was used. The error bars in the related types of figures represent the SEM. The precise test method used for the different figures is also specified in the corresponding figure legends. A P-value < 0.05 was considered significant.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE245333 and GSE245334. Supplemental data corresponding to the figures are available as Supplemental Data. All the code for data analysis is available as Supplemental Code and can also be accessed on GitHub (https://github.com/NGScode2/NGScode).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants no. 32122017 and no. 32070492) to J.D.
Author contributions: P.L. designed and performed the experiments, analyzed the data, and wrote the manuscript. X.Y. did bioinformatic analysis. X.Z. established the diapause induction system. Z.Z. established the facility for insect rearing. J.D. designed experiments, analyzed data, and wrote and edited the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278661.123.
- Received October 24, 2023.
- Accepted May 9, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








