
Crossover-active regions of the wheat genome are distinguished by DMC1, the chromosome axis, H3K27me3, and signatures of adaptation
- Andrew J. Tock1,6,
- Daniel M. Holland1,6,
- Wei Jiang1,
- Kim Osman2,
- Eugenio Sanchez-Moran2,
- James D. Higgins3,
- Keith J. Edwards4,
- Cristobal Uauy5,
- F. Chris H. Franklin2 and
- Ian R. Henderson1
- 1Department of Plant Sciences, University of Cambridge, Cambridge CB2 3EA, United Kingdom;
- 2School of Biosciences, University of Birmingham, Birmingham B15 2TT, United Kingdom;
- 3Department of Genetics and Genome Biology, University of Leicester, Leicester LE1 7RH, United Kingdom;
- 4School of Biological Sciences, University of Bristol, Bristol BS8 1TQ, United Kingdom;
- 5John Innes Centre, Norwich NR4 7UH, United Kingdom
-
↵6 These authors contributed equally to this work.
Abstract
The hexaploid bread wheat genome comprises over 16 gigabases of sequence across 21 chromosomes. Meiotic crossovers are highly polarized along the chromosomes, with elevation in the gene-dense distal regions and suppression in the Gypsy retrotransposon-dense centromere-proximal regions. We profiled the genomic landscapes of the meiotic recombinase DMC1 and the chromosome axis protein ASY1 in wheat and investigated their relationships with crossovers, chromatin state, and genetic diversity. DMC1 and ASY1 chromatin immunoprecipitation followed by sequencing (ChIP-seq) revealed strong co-enrichment in the distal, crossover-active regions of the wheat chromosomes. Distal ChIP-seq enrichment is consistent with spatiotemporally biased cytological immunolocalization of DMC1 and ASY1 close to the telomeres during meiotic prophase I. DMC1 and ASY1 ChIP-seq peaks show significant overlap with genes and transposable elements in the Mariner and Mutator superfamilies. However, DMC1 and ASY1 ChIP-seq peaks were detected along the length of each chromosome, including in low-crossover regions. At the fine scale, crossover elevation at DMC1 and ASY1 peaks and genes correlates with enrichment of the Polycomb histone modification H3K27me3. This indicates a role for facultative heterochromatin, coincident with high DMC1 and ASY1, in promoting crossovers in wheat and is reflected in distalized H3K27me3 enrichment observed via ChIP-seq and immunocytology. Genes with elevated crossover rates and high DMC1 and ASY1 ChIP-seq signals are overrepresented for defense-response and immunity annotations, have higher sequence polymorphism, and exhibit signatures of selection. Our findings are consistent with meiotic recombination promoting genetic diversity, shaping host–pathogen co-evolution, and accelerating adaptation by increasing the efficiency of selection.
Meiosis is a germ-line cell division whereby chromosomes undergo DNA replication followed by two rounds of segregation, producing gametes required for sexual reproduction (Villeneuve and Hillers 2001). Homologous chromosomes pair and recombine, yielding crossovers or noncrossovers (Hunter 2015). Crossovers are required for balanced segregation at the first division and generate genetic diversity. Recombination is initiated during early prophase I, when chromosomes undergo DNA double-strand breaks (DSBs) catalyzed by SPO11 enzymes (Keeney et al. 2014). DMC1 and RAD51 recombinases bind single-stranded DNA at each DSB end and promote strand invasion of homologous DNA (Hunter 2015). In plants and mammals, a minority of strand invasion events mature into crossovers (∼3%–10% of DSBs) (Cole et al. 2010; Mercier et al. 2015), with most dissociated via anticrossover pathways (Hunter 2015).
Meiotic recombination is orchestrated by the assembly of a proteinaceous axis, which underpins meiotic chromosome architecture in eukaryotes (Zickler and Kleckner 1999). Sister chromatids are organized as linear arrays of chromatin loops connected to the axis (Zickler and Kleckner 1999). In plants, the chromosome axis includes the HORMA domain protein ASY1 and its interacting partners ASY3 and ASY4, which promote DMC1-mediated interhomolog synapsis and recombination (Armstrong et al. 2002; Sanchez-Moran et al. 2007; Ferdous et al. 2012; Chambon et al. 2018). As prophase I progresses, the synaptonemal complex is installed between the axes of homologous chromosomes, promoting crossover maturation (Zickler and Kleckner 1999).
Crossovers exert a powerful influence on eukaryotic genetic variation and genome evolution (Barton and Charlesworth 1998) and are an essential plant-breeding tool. However, significant heterogeneity in crossover rates occurs along chromosomes, which can impede crop improvement (Taagen et al. 2020). Selection for useful variation can cause hitchhiking of deleterious alleles via linkage drag in low-recombination regions (Hill and Robertson 1966). This is particularly pronounced in bread wheat (Triticum aestivum), as crossovers concentrate in distal regions (Saintenac et al. 2009; Choulet et al. 2014). For example, 82% of crossovers on Chromosome 3B occur within 19% of its physical length (Darrier et al. 2017). Bread wheat is an allohexaploid, comprising homoeologous A, B, and D subgenomes of seven chromosomes each (2n = 6x = 42, AABBDD) (Marcussen et al. 2014). Wheat chromosomes are large (473–830 Mb), with high transposable element (TE) content, and comprise one of the most complex assembled genomes (IWGSC 2018).
Pronounced compartmentalization of features occurs along the wheat chromosomes. For example, crossover rate, gene density, and euchromatic (H3K4me3, H3K9ac, and H3K27ac) and facultative heterochromatic (H3K27me3) marks increase toward the telomeres, whereas Gypsy LTR retrotransposon density and constitutive heterochromatic marks (H3K9me2 and CG- and CHG-context DNA methylation) increase toward the centromeres (Choulet et al. 2014; IWGSC 2018; Li et al. 2019). Gene function, chromatin state, and expression breadth across tissues are also stratified (Ramírez-González et al. 2018). Genes with broad expression, high H3K36me3 and H3K9ac levels, and “housekeeping” functions are located predominantly in the low-recombination interstitial and centromere-proximal regions (R2 and C compartments), whereas those with tissue-specific expression, high H3K27me3 levels, and functions in development and response to environmental stimuli are enriched in the high-recombination distal regions (R1 and R3 compartments) (IWGSC 2018; Ramírez-González et al. 2018).
Chromatin state significantly influences recombination in plant genomes. For example, meiotic DSB hotspots in Arabidopsis and maize are located in nucleosome-depleted regions (NDRs) associated with genes and specific transposon families (He et al. 2017; Choi et al. 2018). Furthermore, Arabidopsis crossover hotspots can be suppressed via RNA-directed DNA methylation (Yelina et al. 2015), and mutants with reduced non-CG DNA methylation show increased pericentromeric SPO11-1-oligonucleotides and crossovers (Underwood et al. 2018). Large tracts of the crossover-suppressed compartments of the wheat genome are marked by constitutive heterochromatin (IWGSC 2018; Li et al. 2019; Concia et al. 2020). Additionally, H3K27me2 associates with crossover suppression in wheat, despite distal enrichment (Liu et al. 2021). However, fine-scale relationships between chromatin, meiotic recombination, axis occupancy, and sequence variation in wheat remain incompletely understood.
As DMC1 marks an early stage in meiotic recombination, and the axis protein ASY1 promotes synapsis and recombination, we sought to profile their localization in wheat using ChIP-seq. This allowed us to investigate the genetic and epigenetic features that associate with meiotic DSBs and axis occupancy. DMC1 binding and axis occupancy are necessary, but not sufficient, for repair of meiotic DSBs as crossovers. We therefore aimed to identify properties that distinguish crossover-active DMC1 and ASY1 ChIP-seq peaks and genes by examining chromatin state and genetic variation across crossover-rate gradients.
Results
DMC1 recombinase colocalizes with the meiotic axis protein ASY1
We used polyclonal antibodies raised against Arabidopsis DMC1 in rabbit (Sanchez-Moran et al. 2007) and the recombinant wheat ASY1 HORMA domain in guinea pig (Desjardins et al. 2020). We performed western blotting using these antibodies and detected a specific band of the expected size from wheat floral extracts in each case, following nuclei extraction (DMC1 = 38 kDa; ASY1 = 67 kDa) (Supplemental Fig. S1). For ChIP-seq, the landrace Chinese Spring was selected due to the availability of a high-quality genome assembly, extensive chromatin data sets, and a genetic map (Guo et al. 2016; IWGSC 2018; Li et al. 2019). We collected pre-emergence spikes, which were crosslinked using formaldehyde, followed by nuclei purification and ChIP-seq using α-DMC1 and α-ASY1 antibodies.
After deduplication and trimming, 185,448,819 of 218,747,974 (85%) DMC1 and 344,590,350 of 405,615,455 (85%) ASY1 ChIP-seq read pairs mapped with a best-scoring alignment to the Chinese Spring genome (Supplemental Table S1). We also performed ChIP-seq for euchromatic (H3K4me3) and constitutive heterochromatic (H3K9me2 and H3K27me1) marks and mapped nucleosome occupancy via micrococcal nuclease digestion and sequencing (MNase-seq), using Chinese Spring leaf tissue. We combined analysis of our data with published ChIP-seq data sets for H3K4me1, H3K9ac, H3K27ac, H3K27me3, H3K36me3, and CENH3, and whole-genome bisulfite sequencing maps of DNA methylation (Guo et al. 2016; IWGSC 2018; Li et al. 2019; Supplemental Table S1).
At the chromosome scale, we observed a strong positive correlation between DMC1 and ASY1 ChIP-seq signals (log2[ChIP/input]) in adjacent 1-Mb windows (genome-wide Spearman's rs = 0.97), with co-enrichment in distal compartments (Fig. 1A,B; Supplemental Figs. S2–S9). Distal ChIP-seq enrichment is consistent with immunolocalization of DMC1 and ASY1, showing telomere-biased accumulation early in prophase I (Fig. 2A,B; Osman et al. 2021). DMC1 and ASY1 chromosome-scale profiles positively correlate with crossover rate (cM/Mb) derived from a Chinese Spring × Renan F6 genetic map (genome-wide rs = 0.80 and 0.75, respectively) (IWGSC 2018), as well as with gene density and markers of euchromatin (H3K4me1, H3K4me3, and H3K27ac) and facultative heterochromatin (H3K27me3) (Fig. 1A,B; Supplemental Figs. S2, S3, S10–S12; IWGSC 2018; Li et al. 2019). These trends are also evident when distal (R1 and R3) and interstitial (R2) compartments are analyzed separately (Supplemental Fig. S12A,B). H3K27me3 marks facultative heterochromatin and mediates tissue-specific silencing of genes that control development or environmental responses (Mozgova and Hennig 2015). Consistent with distalized H3K27me3 ChIP-seq signal (Supplemental Fig. S10), immunostaining for this mark during meiotic prophase I also revealed distal enrichment (Fig. 2C,D; Osman et al. 2021).
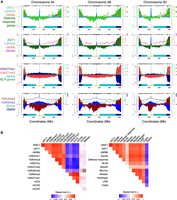
Genomic landscapes of DMC1 recombinase, the meiotic axis protein ASY1, crossovers, and chromatin states in wheat. (A) Coverage profiles along a representative set of homoeologous chromosomes (3A, 3B, and 3D) for DMC1 (light green), ASY1 (dark green), CENH3 (light blue) (Guo et al. 2016), H3K27me3 (red) (IWGSC 2018) (all log2[ChIP/input], using a Chinese Spring chromatin input sequencing library with accession SRR6350669) (IWGSC 2018), H3K27me1 (navy), H3K4me3 (dark red), and H3K9me2 (blue) (all log2[ChIP/MNase], using a Chinese Spring MNase-seq library from this study) ChIP-seq in 1-Mb adjacent windows, smoothed by applying a moving average. ChIP-seq chromosome profiles (shading) are overlaid with crossover rates (cM/Mb, derived from a Chinese Spring × Renan genetic map) (IWGSC 2018), and gene and Gypsy LTR retrotransposon frequencies in 10-Mb sliding windows with a 1-Mb step (lines). A representative set of the IWGSC RefSeq v1.1 Chinese Spring annotation of high-confidence protein-coding gene models (IWGSC 2018), genes annotated with the “defense response” Gene Ontology (GO) term (Ramírez-González et al. 2018), genes encoding nucleotide-binding and leucine-rich repeat (NLR) proteins (Steuernagel et al. 2020), and Gypsy LTR retrotransposons in the IWGSC annotation (IWGSC 2018) were plotted. Previously defined coordinates delimiting distal (R1 and R3, navy boxes), interstitial (R2a and R2b, turquoise boxes), proximal (C, cream boxes), and centromeric (vertical dashed lines) regions are indicated along the x-axis (IWGSC 2018). Gray ticks denote genomic coordinates of genetic markers used to construct the Chinese Spring × Renan genetic map. (B) Genome-wide Spearman's rank-order correlation coefficients (rs) for the indicated parameter pairs profiled in 1-Mb adjacent windows, with cells coded according to the color-gradient correlation scale. Gray boxes denote correlation coefficients that are not significant after P-value standardization.
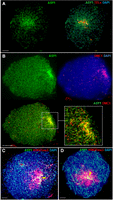
Immunolocalization of ASY1, DMC1, and H3K27me3 in pollen mother cells of wheat cultivar Cadenza during prophase I. (A) Immunolocalization of the axis protein ASY1 (green), combined with fluorescence in situ hybridization (FISH) of the telomeric repeat sequence (TELs, red) from Arabidopsis thaliana at the G2/leptotene transition. ASY1 signal is enriched in distal regions as the telomeres begin to cluster into a “bouquet” arrangement and the axis begins to linearize. (B) Leptotene-stage pollen mother cell immunostained for DMC1 foci (red) and co-immunostained for ASY1 (green), as axis linearization progresses through the nucleus. The zoomed detail of distal regions is from a single Z-frame (mid-nucleus) and shows individual DMC1 foci decorating the axis stained by ASY1. (C) Early-leptotene-stage pollen mother cell showing enrichment of H3K27me3 in distal regions, which are also marked by intense ASY1 immunostaining (yellow arrow). (D) H3K27me3 distal enrichment persists in early zygotene when chromosome axes are fully linear (yellow arrow). 4′,6-diamidino-2-phenylindole (DAPI) marks DNA. Scale bars = 10 µM. For clarity, some images are shown in several color combinations.
We examined broad-scale relationships between recombination and genes within different functional categories that are known to have contrasting distributions. DMC1, ASY1 and crossover rate each exhibit strong positive relationships with genes annotated with the “defense response” Gene Ontology (GO) term (Ramírez-González et al. 2018) and those encoding nucleotide-binding and leucine-rich repeat domain intracellular immune receptor proteins (NLR genes) (Fig. 1A,B; Supplemental Fig. S12A,B; Steuernagel et al. 2020). Defense-response and NLR genes are concentrated in H3K27me3-enriched distal compartments (Fig. 1A; Supplemental Figs. S2, S10). In contrast, orthologs of known meiotic genes and genes with meiosis-related GO annotations concentrate in interstitial and proximal compartments (Alabdullah et al. 2019) and show weaker correlations with DMC1, ASY1, and crossover rate (Fig. 1B).
DMC1 and ASY1 peaks associate with genes and TIR DNA transposons
We identified sites of local DMC1 and ASY1 enrichment by calling peaks in ChIP-seq signal. For DMC1, 101,076 peaks were identified in distal (R1 and R3) compartments and 170,649 peaks in interstitial and proximal (R2 and C) compartments (Fig. 3A). For ASY1, 185,260 peaks were detected in distal compartments, and 358,946 peaks in interstitial and proximal compartments (Fig. 3B). We observed higher DMC1 and ASY1 peak densities in distal compartments, similar to chromosome-scale profiles for crossover rate, genes, and Mariner and Mutator transposons (Fig. 3C,D).
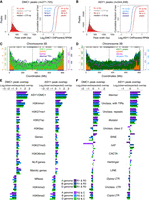
DMC1 and ASY1 ChIP-seq peaks associate with markers of euchromatin and heterochromatin. (A) Histograms of DMC1 ChIP-seq peak widths (bp) in distal compartments (R1 and R3, red) and in interstitial and proximal compartments (R2 and C, burgundy), with median widths indicated by vertical dashed lines in gray and black, respectively. Also shown are cumulative distribution curves for compartmentalized DMC1 ChIP-seq peaks and randomly positioned loci ranked by log2(ChIP/input) RPKM values. (B) As in A, but for ASY1 ChIP-seq peaks and randomly positioned loci. (C) Chromosome 3B profiles of DMC1 ChIP-seq peak frequencies (light green shading), crossover rate (cM/Mb, orange shading) (IWGSC 2018), gene frequency (magenta line), and Mariner transposon frequency (navy line) in 10-Mb sliding windows with a 1-Mb step. Previously defined coordinates delimiting distal (R1 and R3, navy boxes), interstitial (R2a and R2b, turquoise boxes), proximal (C, cream boxes), and centromeric (vertical dashed lines) regions are indicated along the x-axis (IWGSC 2018). Gray ticks denote genetic markers used to construct the Chinese Spring × Renan genetic map. (D) As in C, but showing ASY1 ChIP-seq peak frequency (dark green shading) and Mutator transposon frequency (light blue line). (E) Log2(observed/expected) overlap of compartmentalized DMC1 and ASY1 ChIP-seq peaks in each subgenome with the indicated genomic features, based on the numbers of feature-overlapping loci across 10,000 sets of randomly positioned loci. Vertical gray lines mark significance thresholds (α = 0.05). (F) As in E, but showing log2(observed/expected) overlap of DMC1 and ASY1 ChIP-seq peaks with transposon superfamilies.
To investigate DMC1 and ASY1 peak properties, we evaluated their overlap with other features using permutation tests, each utilizing 10,000 sets of randomly positioned loci. In each set of subgenomic compartments, DMC1 peaks significantly overlap ASY1 peaks and vice versa (each P = 0.0001) (Fig. 3E; Supplemental Tables S2, S3). DMC1 and ASY1 peaks also significantly overlap ChIP-seq peaks for euchromatic histone modifications (H3K4me1, H3K9ac, H3K27ac, and H3K36me3) and facultative heterochromatin (H3K27me3), whereas they are largely absent from sites occupied by the constitutive heterochromatic mark H3K9me2 and LTR retrotransposons (Fig. 3E,F; Supplemental Tables S2, S3). Furthermore, DMC1 and ASY1 peaks significantly overlap DNA TEs containing terminal inverted repeats (TIRs), including the Mariner and Mutator superfamilies (each P = 0.0001) (Fig. 3F; Supplemental Tables S2, S3).
DMC1 and ASY1 peaks overlap genes more than expected and these associations are strongest in promoter regions upstream of transcriptional start sites (TSSs) and around transcriptional termination sites (TTSs) (each P = 0.0001) (Fig. 3E; Supplemental Tables S2, S3). Additionally, DMC1 and ASY1 peaks exhibit less-than-expected overlap with sites of high nucleosome occupancy (MNase-seq peaks) (Fig. 3E; Supplemental Tables S2, S3), consistent with elevated meiotic DSBs and crossovers in NDRs of Arabidopsis, tomato, and maize genes (Demirci et al. 2017; He et al. 2017; Choi et al. 2018; Fuentes et al. 2020). Multiple AT-rich sequences are overrepresented among DMC1 and ASY1 peaks (Supplemental Figs. S13, S14). AT sequence richness reduces nucleosome occupancy and promotes meiotic DSB and crossover formation in plant genomes (Shilo et al. 2015; Demirci et al. 2017; Choi et al. 2018; Fuentes et al. 2020). Despite positive chromosome-scale associations, DMC1 and ASY1 peaks overlap H3K4me3 peaks significantly less than expected (Fig. 3E; Supplemental Tables S2, S3). This is consistent with wheat meiotic DSBs and axis occupancy avoiding the +1 nucleosome at gene 5′ ends, which is modified with H3K4me3. These trends are reminiscent of Arabidopsis SPO11-1-oligos, which are also most enriched in NDRs flanking genes and depleted at the +1 nucleosome (Choi et al. 2018).
Crossover-active DMC1 and ASY1 peaks are distinguished by H3K27me3
DMC1 binding and axis occupancy are not sufficient for resolution of meiotic DSBs as crossovers, which is reflected in the detection of DMC1 and ASY1 peaks throughout all genomic compartments, including in crossover-suppressed regions. Therefore, we next sought to identify properties that associate with crossover-rate variation across DMC1 and ASY1 ChIP-seq peaks located in distal and interstitial/proximal compartments. We defined four groups of distal ChIP-seq peaks according to decreasing crossover rate, derived from the Chinese Spring × Renan genetic map (IWGSC 2018) (Quantiles 1–4; mean peaks per quantile: DMC1 = 24,634, ASY1 = 45,249) (Fig. 4A,B). Due to the smaller range of cM/Mb values in the recombination-suppressed compartments, peaks in these regions were divided into three groups (mean peaks per quantile: DMC1 = 56,883, ASY1 = 119,649) (Supplemental Fig. S15A,B).
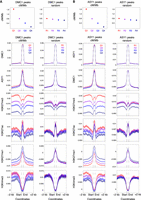
High-crossover-rate DMC1 and ASY1 ChIP-seq peaks are enriched for the Polycomb chromatin mark H3K27me3. (A) Distally located (R1 and R3) DMC1 ChIP-seq peaks were divided into four groups corresponding to those in the 100th–75th (Quantile 1), 75th–50th (Quantile 2), 50th–25th (Quantile 3), and 25th–0th (Quantile 4) percentiles with regard to their mean crossover rate (cM/Mb, derived from the Chinese Spring × Renan genetic map) between 1 kb upstream of peak start coordinates and 1 kb downstream from peak end coordinates (Q1–Q4, left), or into four randomized groups (R1–R4, right). Solid circles denote the mean crossover rate for each group of peaks (error bars = 95% confidence intervals for mean cM/Mb values). Metaprofiles show windowed mean values (solid lines) for each group of peaks and 2-kb flanking regions (transparent ribbons = 95% confidence intervals). ChIP-seq coverage metaprofiles of DMC1, ASY1, H3K27me3 (IWGSC 2018), and H3K27ac (Li et al. 2019) are derived from log2(ChIP/input) profiles, and those for H3K27me1 and H3K4me3 from log2(ChIP/MNase) profiles. (B) As in A, but for distal ASY1 ChIP-seq peaks.
DMC1 and ASY1 ChIP-seq metaprofiles show strong enrichment within peaks and a weak positive correlation with crossover rate (Fig. 4A,B; Supplemental Fig. S15A,B). H3K27me3 ChIP-seq signal correlates strongly with DMC1/ASY1 peak crossover rate, as indicated by well-separated 95% confidence intervals around per-quantile mean coverage profiles (Fig. 4A,B; Supplemental Fig. S15A,B). A positive correlation with crossover rate is also evident for H3K27ac ChIP-seq signal, which is enriched within peaks (Fig. 4A,B; Supplemental Fig. S15A,B). In contrast, the constitutive heterochromatic mark H3K27me1 is anticorrelated with crossover rate at peaks (Fig. 4A,B; Supplemental Fig. S15A,B). These findings suggest that the co-occurrence of H3K27me3-marked facultative heterochromatin with meiotic DSBs and axis occupancy promotes crossover formation in wheat.
Crossover-active genes have elevated DMC1, ASY1, H3K27me3, and polymorphisms
To investigate the properties of genes in relation to crossover rate, we divided genes into four quantiles according to decreasing crossover rate (mean genes per quantile = 25,964) (Fig. 5A). Genes with elevated crossover rates exhibit higher DMC1 and ASY1 ChIP-seq signals on average (Fig. 5A). Equivalently, genes grouped according to higher DMC1 or ASY1 signals have higher crossover rates (Fig. 5B,C). DMC1 and ASY1 signals around genes are anticorrelated with nucleosome signal (rs = −0.42 and −0.48, respectively) and are most depleted at the +1 nucleosome, where H3K4me3 ChIP-seq and MNase-seq signals are highest (Fig. 5A). Genes with higher DMC1 or ASY1 levels also have higher Mariner TE density in their promoter, transcribed, and downstream regions and higher Mutator density in their promoters (Supplemental Fig. S16A,B).
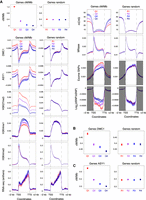
Patterns of genic recombination, chromatin, transcription, and polymorphism. (A) Genes were divided into four groups corresponding to those in the 100th–75th (Quantile 1), 75th–50th (Quantile 2), 50th–25th (Quantile 3), and 25th–0th (Quantile 4) percentiles with regard to their mean crossover rate (cM/Mb, derived from the Chinese Spring × Renan genetic map) between 1 kb upstream of transcriptional start sites (TSSs) and 1 kb downstream from transcriptional termination sites (TTSs) (Q1–Q4, left), or into four randomized groups (R1–R4, right). Solid circles denote the mean crossover rate for each group of genes (error bars = 95% confidence intervals for mean cM/Mb values). Metaprofiles show windowed mean values (solid lines) for each group of genes and 2-kb flanking regions (transparent ribbons = 95% confidence intervals). ChIP-seq coverage metaprofiles of DMC1, ASY1, H3K27me3 (IWGSC 2018), and H3K4me1 (Li et al. 2019) are derived from log2(ChIP/input) profiles, and those for H3K4me3 from log2(ChIP/MNase) profiles. Metaprofiles of RNA-seq coverage (transcripts per million, TPM) are derived from Chinese Spring anthers collected at the leptotene–zygotene transition (Martín et al. 2018). CHG-context DNA methylation (mCHG) metaprofiles are based on the proportion of cytosines that are methylated per CHG site, as detected in whole-genome bisulfite sequencing data (IWGSC 2018). Metaprofiles of exome-sequencing-derived SNPs are based on windowed SNP frequencies (He et al. 2019). Although the vast majority of SNPs detected using exome sequencing are located within gene bodies, these data also provide genotypes outside of transcribed regions. Therefore, SNPs outside of transcribed regions were retained for metaprofile calculation (gray shading). Windowed ratios of predicted deleterious SNPs to synonymous SNPs (log2[dSNP/sSNP]) were also used for metaprofile calculation. (B) Genes were divided into four quantiles according to decreasing mean DMC1 log2(ChIP/input) coverage between 1 kb upstream of TSSs and 1 kb downstream from TTSs (Q1–Q4, left), or into four randomized groups (R1–R4, right). Solid circles denote the mean crossover rate for each group of genes (error bars = 95% confidence intervals for mean cM/Mb values). (C) As in B, but with genes divided into four groups according to decreasing mean ASY1 log2(ChIP/input) coverage (Q1–Q4, left), or into four randomized groups (R1–R4, right).
Similar to DMC1 and ASY1 peaks, gene crossover rate is positively correlated with H3K27me3 (Fig. 5A). In contrast, non-CG DNA methylation is anticorrelated with crossover rate (Fig. 5A), consistent with recombination suppression by DNA methylation (Yelina et al. 2015). Metaprofiles of RNA-seq coverage derived from anthers show that genes with higher crossover rates have lower meiotic expression on average (Fig. 5A; Martín et al. 2018). Gene crossover rate is also anticorrelated with H3K4me1 (Fig. 5A), a chromatin modification deposited within the transcribed regions of ubiquitously expressed housekeeping genes and in H3K27me3-depleted regions in Arabidopsis (Zhang et al. 2009). These findings are consistent with crossovers occurring more frequently around genes that are facultatively repressed via H3K27me3 and which also have elevated meiotic DSBs and axis occupancy.
To investigate levels of genetic variation in relation to recombination, we calculated metaprofiles of single-nucleotide polymorphism (SNP) density around genes grouped by crossover rate, DMC1, or ASY1 (Fig. 5A; Supplemental Fig. S16A,B). We analyzed SNPs identified among 811 geographically diverse cultivars and landraces, profiled via exome sequencing (He et al. 2019). Per-quantile metaprofiles reveal positive relationships for genic SNP density with crossover rate, DMC1, and ASY1 (Fig. 5A; Supplemental Fig. S16A,B), consistent with the mutagenic effects of meiotic recombination (Arbeithuber et al. 2015; Halldorsson et al. 2019). Additionally, we observed a negative relationship between crossover rate and the ratio of SNPs predicted to have a deleterious impact on protein function (dSNPs) to synonymous SNPs (sSNPs) (log2[dSNP/sSNP]) (Fig. 5A). This indicates more efficient purging of deleterious mutations from loci with elevated crossover rates, which is consistent with reduced genetic load in highly recombining regions of the wheat and maize genomes (Barton and Charlesworth 1998; Rodgers-Melnick et al. 2015; Jordan et al. 2018; He et al. 2019).
Gene function and transcriptional profile correlate with crossovers, DMC1, ASY1, and H3K27me3
We examined the functional annotation of genes grouped by decreasing crossover rate, DMC1, ASY1, or H3K27me3 by applying Gene Ontology term enrichment analysis to each quantile. Genes in the upper quantile for crossover rate or H3K27me3 are overrepresented for stress response and developmental annotations (Supplemental Tables S4, S5, S10, S11). These findings agree with previously reported functional stratification along the wheat chromosomes (IWGSC 2018; Ramírez-González et al. 2018). However, genes in the top quantile for DMC1 or ASY1 ChIP-seq exhibit a more heterogeneous functional composition, including overrepresentation of terms associated with housekeeping processes and meiosis, in addition to stress response and developmental annotations (Supplemental Tables S6–S9). This is consistent with the widespread distribution of DMC1 and ASY1 peaks along the chromosomes, including in crossover-suppressed regions.
Gene recombination and chromatin quantiles were next tested for over- and underrepresentation of gene categories using the hypergeometric distribution. Meiotic genes (n = 1059) are over- and underrepresented in the upper and lower quantiles, respectively, for DMC1 or ASY1 ChIP-seq signal (Fig. 6B,C; Supplemental Tables S17, S18; Alabdullah et al. 2019). Gene quantiles for crossover rate or H3K27me3 ChIP-seq signal show trends that are opposite to those for DMC1 and ASY1 with respect to meiotic genes (Fig. 6A,D; Supplemental Tables S16, S19). This attests to the functional heterogeneity of genes enriched for DMC1 and ASY1 and is consistent with the concentration of meiotic genes in crossover-suppressed regions (Alabdullah et al. 2019).
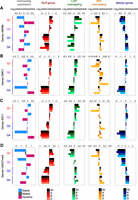
Gene expression pattern, function, and signatures of adaptation according to crossover rate, DMC1, ASY1, or H3K27me3. (A) Gene quantiles defined by decreasing mean crossover rate (Q1–Q4 = high–low cM/Mb) were evaluated for representation of (1) genes assigned to homoeolog expression categories (Homoeolog expression) (Ramírez-González et al. 2018), (2) genes encoding nucleotide-binding and leucine-rich repeat proteins (NLR genes) (Steuernagel et al. 2020), (3) genes overlapping genomic regions associated with local adaptation (LAR overlapping) (He et al. 2019), (4) genes overlapping genomic regions associated with modern wheat improvement (MIR overlapping) (He et al. 2019), and (5) genes with putative functions in meiosis (Meiotic genes) (Alabdullah et al. 2019). Log2(observed/expected) ratios (bar graphs) and significance thresholds (vertical gray lines, α = 0.05) were calculated by sampling from the hypergeometric distribution 100,000 times. Analyses were performed across all subgenomes and within each subgenome (All, A, B, and D). (B) As in A, but with genes divided into four quantiles according to decreasing mean DMC1 log2(ChIP/input) coverage. (C) As in A, but with genes divided into four quantiles according to decreasing mean ASY1 log2(ChIP/input) coverage. (D) As in A, but with genes divided into four quantiles according to decreasing mean H3K27me3 log2(ChIP/input) coverage (IWGSC 2018).
We evaluated gene quantiles for patterns of homoeologous gene expression, using previously defined categories derived from analysis of RNA-seq data from 15 tissues in Chinese Spring (Ramírez-González et al. 2018). This analysis applies to genes with a single identifiable homoeolog in each of the three subgenomes, which define gene triads (Ramírez-González et al. 2018). Stably and dynamically expressed triads are those in the bottom and top 10%, respectively, with regard to the mean of the Euclidean distances between relative transcript abundance for each tissue and the mean relative transcript abundance across all tissues (Ramírez-González et al. 2018). Balanced gene triads are those with comparable relative transcript abundances from each of the homoeologs, whereas dominant and suppressed categories refer to those with higher or lower transcript abundance from one homoeolog (Ramírez-González et al. 2018). The upper crossover-rate and H3K27me3 gene quantiles are overrepresented for dynamic and nonbalanced expression categories and underrepresented for stable and balanced categories, whereas opposite trends are observed for the lower quantiles (Fig. 6A,D; Supplemental Tables S12, S15). This is consistent with crossovers occurring preferentially in regions containing facultatively silenced genes with tissue-specific transcriptional profiles (Ramírez-González et al. 2018). DMC1 and ASY1 gene quantiles exhibit trends that are generally the inverse of those observed for crossover rate and H3K27me3 quantiles (Fig. 6B,C; Supplemental Tables S13, S14). Therefore, widespread meiotic DSB formation and axis occupancy occur beyond crossover-competent regions and around diverse gene categories.
To explore relationships between recombination and selection, we analyzed gene quantiles for overlap with genomic regions associated with local adaptation (LARs, n = 42,918, median width = 20 kb, LAR-overlapping genes = 33,247) (He et al. 2019). LARs were previously identified using a Bayesian method to test correlations between allele frequencies at 1.39 million exomic SNPs and 68 environmental and bioclimatic variables, for 26 geographically distinct wheat populations (He et al. 2019). LARs were defined as genomic regions containing locally adaptive SNPs (He et al. 2019). We observed that the upper gene quantiles for crossover rate, DMC1, and ASY1 are overrepresented for LAR overlap (Fig. 6A–C; Supplemental Tables S16–S18). We also evaluated quantiles for representation of genes overlapping genomic regions associated with the transition from landraces to modern improved cultivars (MIRs, n = 4316, median width = 350 kb, MIR-overlapping genes = 16,102), which were previously detected using nine regional populations (He et al. 2019). In contrast to genes overlapping LARs, MIR-overlapping genes are under- and overrepresented in the upper and lower quantiles for crossover rate, respectively, whereas these trends are not evident for genes ranked by DMC1 or ASY1 ChIP-seq (Fig. 6A–C; Supplemental Tables S16–S18). These relationships may reflect a role for crossovers in enabling selection for alleles conferring region-specific adaptive phenotypes, occurring since the global dissemination of hexaploid wheat. In contrast, overrepresentation of MIR-overlapping genes in the lower crossover-rate quantiles suggests that more recent agronomic gains achieved through selective breeding are associated with crossover-suppressed regions, consistent with recent analysis (Brinton et al. 2020).
NLR crossovers and clustering correlate with H3K27me3, DMC1, and ASY1
NLR genes are over- and underrepresented in the upper and lower quantiles, respectively, for crossover rate, DMC1, ASY1, and H3K27me3 (Fig. 6A–D; Supplemental Tables S16–S19). Upon recognition of pathogen virulence determinants, NLR proteins activate signal transduction pathways that precipitate hypersensitive cell death, as part of effector-triggered immunity (ETI) (Jones and Dangl 2006). NLRs are unevenly distributed throughout plant genomes and are frequently physically clustered, whereas others occur as singletons (Michelmore and Meyers 1998; Meyers et al. 2003). Among those detected in Chinese Spring (Steuernagel et al. 2020), 1233 NLRs exist in 328 physical clusters (mean per cluster = 3.76), whereas 473 are singletons. We divided NLRs into four quantiles, defined either by decreasing crossover rate (mean genes per quantile = 405) or by decreasing cluster size (Quantile 4 contains only singletons; mean genes per quantile = 427) (Fig. 7A,B). NLRs with high and low crossover rates are over- and underrepresented for clustered genes, respectively, whereas inverse trends are evident for singletons (Fig. 7C; Supplemental Table S20). We also observed a positive correlation between NLR cluster size and crossover rate (Fig. 7B), consistent with recombination promoting cluster formation. The upper and lower quantiles for ASY1 and H3K27me3 ChIP-seq signal are over- and underrepresented for clustered NLRs, respectively, whereas opposite trends are apparent for singletons (Supplemental Fig. S17B; Supplemental Tables S22, S23). However, similar correlation with clustering is not evident for NLRs grouped by DMC1 ChIP-seq signal (Supplemental Fig. S17C; Supplemental Table S21).
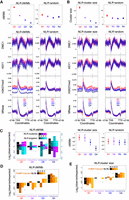
Chromatin, recombination, and clustering at nucleotide-binding and leucine-rich repeat (NLR) genes. (A) NLR genes were divided into four groups corresponding to those in the 100th–75th (Quantile 1), 75th–50th (Quantile 2), 50th–25th (Quantile 3), and 25th–0th (Quantile 4) percentiles with regard to their mean crossover rate (cM/Mb, derived from the Chinese Spring × Renan genetic map) between 1 kb upstream of transcriptional start sites and 1 kb downstream from transcriptional termination sites (Q1–Q4, left), or into four randomized groups (R1–R4, right). Solid circles denote the mean crossover rate for each group of genes (error bars = 95% confidence intervals for mean cM/Mb values). Metaprofiles show windowed mean values (solid lines) for each group of genes and 2-kb flanking regions (transparent ribbons = 95% confidence intervals). ChIP-seq coverage metaprofiles of DMC1, ASY1, and H3K27me3 (IWGSC 2018) are derived from log2(ChIP/input) profiles. (B) As in A, but with NLR genes divided into four groups according to decreasing physical cluster size (Q1–Q4, left), or into four randomized groups (R1–R4, right). Solid circles denote the mean cluster size (upper plots) or crossover rate (lower plots) for each group of genes (error bars = 95% confidence intervals for mean cluster size or cM/Mb values). (C) NLR gene quantiles defined by decreasing mean crossover rate (Q1–Q4 = high–low cM/Mb) were evaluated for representation of physically clustered or singleton NLR genes. Log2(observed/expected) ratios (bar graphs) and significance thresholds (horizontal gray lines, α = 0.05) were calculated by sampling from the hypergeometric distribution 100,000 times. Analyses were performed across all subgenomes and within each subgenome (All, A, B, and D). (D) As in C, but analyzing NLR gene quantiles defined by decreasing mean crossover rate for representation of NLR genes that were up-regulated upon challenge with pathogen-associated molecular patterns (PAMPs) (Steuernagel et al. 2020). (E) As in D, but analyzing NLR gene quantiles defined by decreasing physical cluster size.
Many NLR clusters comprise genes from the same phylogenetic clade, likely deriving from tandem duplications and intralocus recombination (Michelmore and Meyers 1998; Meyers et al. 2003). Heterogeneous NLR clusters containing genes from different clades also exist, indicative of segmental duplications and interlocus sequence exchange (Michelmore and Meyers 1998; Meyers et al. 2003). Using the wheat NLR phylogeny reported in Steuernagel et al. (2020), we found that 21% of physical clusters comprise NLRs whose most recent common ancestor is the parent node of at least one of the NLRs within their respective physical cluster. This provides a conservative estimate of NLR clusters that are monophyletic. Among NLRs in the upper and lower quantiles for crossover rate, we observed over- and underrepresentation of those that are members of monophyletic physical clusters, respectively (Supplemental Fig. S17D; Supplemental Table S20). This supports a role for intralocus crossover in the genesis of monophyletic clusters. NLRs with elevated crossover rates, or those within large physical clusters, also have higher H3K27me3 and, to lesser extents, higher DMC1 and ASY1 ChIP-seq signals (Fig. 7A,B). DMC1 and ASY1 show strongest enrichment toward the middle of NLR transcribed regions, as well as in promoters and at TTSs, patterns which are distinct from metaprofiles around all genes (Figs. 5A, 7A,B). Intragenic meiotic DSBs may provide the basis for duplications within the leucine-rich repeat (LRR)-encoding domains of plant NLRs, which have been proposed to arise from recombination (Parker et al. 1997).
Transmembrane pattern-recognition receptors (PRRs) mediate cell-surface perception of conserved microbial elicitors, termed pathogen-associated molecular patterns (PAMPs) (Jones and Dangl 2006). This process induces PAMP-triggered immunity (PTI), which can arrest infection by activating conserved plant defense pathways (Jones and Dangl 2006). Steuernagel et al. (2020) identified 266 wheat NLRs that were transcriptionally up-regulated upon challenge with PAMPs, indicating that PTI also regulates the expression of some NLRs as a primer for ETI induction. PAMP-induced NLRs are overrepresented among low-crossover-rate NLRs and singletons (Fig. 7D,E; Supplemental Tables S20, S24). Hence, NLRs that may operate at the interface between PTI and ETI concentrate in relatively crossover-suppressed regions.
DMC1 and ASY1 correlate with historical crossovers, nucleotide diversity, and signatures of selection
In view of the strong associations between contemporary crossovers, recombination initiation, polymorphism density, and genes with functional annotations related to adaptive traits, we investigated connections between DMC1, ASY1, historical crossovers, nucleotide diversity, and signatures of selection. We estimated historical recombination using per-gene RM divided by gene width. RM is the minimum number of crossovers in the history of a sample, based on the four-gamete test (Hudson and Kaplan 1985). For brevity, we present results for a population of 91 hexaploid wheat accessions sampled from Central, East, and South Asia (He et al. 2019), which are representative of findings for all regional populations. We divided genes into quantiles defined by decreasing RM (mean genes per quantile = 25,066) or by decreasing DMC1 or ASY1 ChIP-seq signal (mean genes per quantile = 26,300) (Fig. 8A,B; Supplemental Fig. S18A). Two groups were defined for RM due to the lower resolution of this statistic, which is limited by SNP density. We observed that elevated DMC1 and ASY1 levels associate with higher historical recombination (RM) and nucleotide diversity (π) within genes (Fig. 8A,B; Supplemental Fig. S18A).
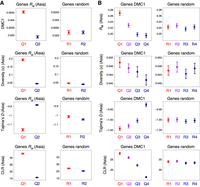
Historical recombination, DMC1, nucleotide diversity, and signatures of selection within genes. (A) Genes were divided into two groups corresponding to those in the 100th–50th (Quantile 1) and 50th–0th (Quantile 2) percentiles with regard to RM divided by gene width (Q1–Q2, left), or into two randomized groups (R1–R2, right). RM is the minimum number of crossovers that occurred in the history of a population of 91 hexaploid wheat accessions sampled from Central, East, and South Asia, inferred from genotypes at exome-sequencing-derived SNPs (He et al. 2019). Solid circles denote the mean value of the indicated parameter for each group of genes (error bars = 95% confidence intervals for mean values). Nucleotide diversity (the mean number of intrapopulation pairwise nucleotide differences per gene, π) was divided by gene width. Per-gene Tajima's D and composite likelihood ratios (CLRs) were also calculated. (B) As in A, but with genes divided into four groups according to decreasing mean DMC1 log2(ChIP/input) coverage (Q1–Q4, left), or into four randomized groups (R1–R4, right).
To investigate signatures of selection at genes as a function of recombination, we analyzed Tajima's D and composite likelihood ratios (CLRs) for each quantile (Tajima 1989; Nielsen et al. 2005). Tajima's D compares the number of segregating sites with nucleotide diversity, such that, under neutrality, D is assumed to be zero (Tajima 1989). Negative values of Tajima's D indicate directional selection or recent population expansion, whereas positive values indicate balancing selection or recent population contraction (Tajima 1989). We observed evidence for directional selection at high-recombination genes, with RM, DMC1, and ASY1 each showing a negative relationship with Tajima's D (Fig. 8A,B; Supplemental Fig. S18A). CLRs provide a measure of directional selection that is more robust to varying recombination rate and demography, with higher values indicating the effects of selective sweeps (Nielsen et al. 2005). Positive relationships were observed for RM, DMC1, and ASY1 with CLRs, indicating that signatures of selective sweeps are greater in high-recombination regions (Fig. 8A,B; Supplemental Fig. S18A). These findings suggest that loci with elevated recombination undergo more efficient selection for alleles conferring adaptive advantages.
Discussion
This work provides the first nucleotide-resolution maps of the meiosis-specific recombinase DMC1, which marks resected meiotic DSBs, and the chromosome axis protein ASY1 in wheat. Using ChIP-seq, we observed co-enrichment of DMC1 and ASY1 in the crossover-active distal regions. Consonantly, we observed cytological immunolocalization of DMC1 and ASY1 close to the telomeres during early meiotic prophase I (Osman et al. 2021). DMC1 and ASY1 ChIP-seq signals associate with gene NDRs and TIR-containing DNA transposons. TIR-containing transposons insert preferentially into gene regulatory sequences (Wicker et al. 2018) and have been widely associated with elevated meiotic recombination in plants (Yandeau-Nelson et al. 2005; Darrier et al. 2017; Marand et al. 2017; Choi et al. 2018). However, ASY1 ChIP-seq and nucleosome occupancy (MNase-seq) are positively correlated in Arabidopsis (Lambing et al. 2020). This may indicate contrasting relationships between the meiotic axis and chromatin in wheat and Arabidopsis. Alternatively, this may reflect differences in the stage of meiotic prophase analyzed by ChIP-seq.
Although DMC1 and ASY1 are distally enriched in wheat, we observed peaks along the length of each chromosome, including in crossover-suppressed compartments at lower densities. This is consistent with recent genetic maps, which revealed that gene conversion events were more evenly distributed than crossovers along the chromosomes (Gardiner et al. 2019). We therefore investigated DMC1 and ASY1 ChIP-seq peaks and genes for properties that distinguish those associated with elevated crossover rates. This revealed that the Polycomb histone modification H3K27me3, a marker of facultative heterochromatin, is one of the strongest predictors of crossover activity at these loci. Furthermore, this mark shows pronounced enrichment in the crossover-active distal regions. As H3K27me3 mediates transient and tissue-specific gene silencing during regulation of development and stress responses (Mozgova and Hennig 2015), this may reflect a coupling of crossovers to loci where elevated diversity and recombination are more likely to lead to adaptive benefits. Pedigree-based and population genetic analyses of recombination rates in human genomes have also associated H3K27me3 with higher local crossover rates (Halldorsson et al. 2019; Spence and Song 2019). However, it is important to note that differences in crossover formation between the wheat chromosome compartments are also connected to earlier homolog pairing and meiotic DSB formation that occur in cereal subtelomeric regions (Higgins et al. 2012; Osman et al. 2021). For example, crossover interference or post-translational modification of the axis proteins following crossover designation in distal regions could direct noncrossover outcomes for interstitial and centromere-proximal DSBs.
We observed positive relationships for genic sequence variation with both contemporary and historical crossover rates, as well as with DMC1 and ASY1 ChIP-seq signals. Recombination can break linkage between beneficial and deleterious mutations located on the same chromosome (Felsenstein 1974; Barton and Charlesworth 1998). For example, wheat and maize low-recombination regions carry a higher genetic load (Rodgers-Melnick et al. 2015; He et al. 2019), indicating that deleterious mutations are more efficiently removed from high-recombination regions. However, meiotic recombination is also mutagenic (Arbeithuber et al. 2015; Halldorsson et al. 2019), and positive relationships between nucleotide diversity and population-scaled recombination estimates have been observed in many plant and animal genomes (Begun and Aquadro 1992; Spencer et al. 2006; Gore et al. 2009; Cutter and Payseur 2013). Hence, elevated recombination may cause higher SNP levels due to mutational effects. Recombination also influences SNP maintenance within populations, by reducing linked selection between mutations (Hill and Robertson 1966; Barton and Charlesworth 1998). However, there is also a feedback on recombination, as very high levels of divergence, including structural changes, can strongly inhibit crossovers (Crown et al. 2018; Rowan et al. 2019). Our observations are consistent with crossovers accelerating evolution of distal loci by enabling more efficient selection for alleles conferring adaptive benefits (Hill and Robertson 1966; Barton and Charlesworth 1998). In contrast, reduced crossovers in the interstitial and centromere-proximal compartments may help preserve conserved functions of housekeeping and meiotic genes that are enriched in these regions (Akhunov et al. 2003).
We observed that genes with elevated crossover rates are overrepresented for those encoding NLR immunity proteins. Furthermore, NLRs with higher crossover rates show a greater degree of physical clustering, indicating that crossovers promote structural diversity at wheat NLR loci and thereby influence disease resistance. This is consistent with the Red Queen hypothesis that host sexual recombination provides adaptive benefits by generating novel genotypes that confer resistance to co-evolving parasites (Howard and Lively 1994; Brockhurst et al. 2014). Functional stratification of the wheat chromosomes is reminiscent of the “two-speed genome,” which describes the bipartite genome organization of some plant-pathogenic oomycetes and fungi (Frantzeskakis et al. 2019). Virulence determinants in these pathogens are concentrated within fast-evolving, repeat-rich genomic compartments, separated from widely conserved housekeeping genes (Frantzeskakis et al. 2019). The mechanisms by which wheat and pathogen genomes have become compartmentalized are likely to be distinct, although these architectures may together promote co-evolutionary adaptation.
The plant immune system consists of two major layers. PAMP-triggered immunity provides species-level (nonhost) resistance to nonadapted pathogens (Dodds and Rathjen 2010). Within effector-triggered immunity, intracellular NLR disease resistance proteins often confer race-specific resistance effective against rapidly evolving virulence effectors, providing a second layer of defense (Jones and Dangl 2006). Wheat NLR genes that are transcriptionally induced following PAMP challenge have recently been identified (Steuernagel et al. 2020), consistent with crosstalk between PTI and ETI pathways (Ngou et al. 2021; Yuan et al. 2021). We observed that PAMP-induced NLRs occur more frequently as singletons located in low-crossover regions. PAMP-induced NLRs that may operate at the interface between PTI and ETI are therefore more insulated from the diversity-generating effects of crossovers. We speculate that NLRs that are induced during ETI may concentrate in more highly recombining regions, with novel resistance specificities and physical clustering arising from elevated crossovers. Hence, crossover patterns may be further stratified within the plant immune system. However, although clustered wheat NLRs tend to occur in higher crossover-rate regions compared to singletons, very large NLR clusters may constitute structural polymorphisms that inhibit crossover formation. For example, crossovers are suppressed by structural polymorphism at the Arabidopsis RPP4–RPP5 and RPP8 loci, the Dm3 locus in lettuce, and the I locus in common bean (McDowell et al. 1998; Noël et al. 1999; Chin et al. 2001; Vallejos et al. 2006).
Our findings have relevance for wheat improvement through the deployment of genetic or epigenetic manipulations aimed at increasing crossover rates in recombination-suppressed chromosome compartments. For example, Arabidopsis DNA methyltransferase mutants show increased meiotic recombination within centromere-proximal regions (Choi et al. 2018; Underwood et al. 2018). Our work also points to the facultative heterochromatic mark H3K27me3 as a potential target for increasing crossovers. Chromatin modifications could be combined with trans-acting loci that direct crossovers to interstitial regions, which have recently been mapped in wheat (Jordan et al. 2018; Gardiner et al. 2019). Tools for promoting crossovers in these regions may have the potential to increase the efficiency of selection during breeding and accelerate adaptation to changing climates.
Methods
Western blotting and ChIP-seq
Western blotting and ChIP-seq were performed using standard protocols (Supplemental Methods). Pre-emergence spikes from wild-type Chinese Spring were crosslinked using formaldehyde, followed by nuclei purification and ChIP using α-DMC1 and α-ASY1 antibodies (Sanchez-Moran et al. 2007; Desjardins et al. 2020). Chromatin immunoprecipitation of the histone modifications H3K4me3 (Abcam ab8580), H3K9me2 (Abcam ab1220), and H3K27me1 (Sigma-Aldrich 07-448) was performed using a protocol similar to that described for DMC1 and ASY1 but using MNase digestion for chromatin fragmentation. MNase digestion was also applied without immunoprecipitation to generate a library for high-throughput sequencing (MNase-seq). DNA obtained from ChIP and MNase digestion was used to construct paired-end libraries, which were sequenced with 2 × 75-bp Illumina reads (Supplemental Methods).
Meiotic immunocytology
Chromosome spreads of pollen mother cells from the wheat cultivar Cadenza were immunostained for ASY1, DMC1, and H3K27me3 as described (Armstrong et al. 2002), with minor modifications (Supplemental Methods). Primary antibodies were used at the following dilutions: α-AtASY1 rabbit or guinea pig, 1:500 (Armstrong et al. 2002); α-AtDMC1 rabbit, 1:200 (Sanchez-Moran et al. 2007); and α-H3K27me3 rabbit, according to the manufacturer's guidelines (Diagenode C15310069 [CS-069-100]).
Short-read data alignment and processing
Deduplicated and trimmed paired-end ChIP-seq and MNase-seq reads were aligned to the RefSeq v1.0 Chinese Spring reference genome assembly (IWGSC 2018), using Bowtie 2 v2.3.4.3 (Supplemental Methods; Langmead and Salzberg 2012). Read pairs that aligned equally well to more than one genomic location, without a best-scoring alignment (with Bowtie 2-assigned MAPQ < 2), were discarded. For retained read pairs that aligned to multiple locations, with varying alignment scores, the best-scoring alignment was selected (Supplemental Table S1). Alignments with more than six mismatches or consisting of only one read in a pair were discarded. Additionally, alignment MAPQ and mismatch thresholds were varied to assess their impact on patterns of DMC1 and ASY1 ChIP-seq enrichment (Supplemental Methods; Supplemental Figs. S4–S9). Using the retained unique and multiple alignments combined in BAM format, windowed read counts per million mapped reads (CPM) were generated with the bamCoverage tool from deepTools v3.1.3 (Ramírez et al. 2014). Published bisulfite sequencing (IWGSC 2018) and RNA-seq (Martín et al. 2018) data were analyzed using standard workflows (Supplemental Methods).
ChIP-seq peak analysis
Sites of local ChIP-seq enrichment were identified by calling peaks using the ranger tool within PeakRanger v1.18 (Feng et al. 2011). Compartmentalized DMC1 and ASY1 ChIP-seq peak sets were evaluated for overlap with other genomic features using permutation tests implemented in regioneR v1.6.2, each utilizing 10,000 sets of randomly positioned loci of the same number and width distribution, and in the same genomic compartments, as the DMC1 or ASY1 peaks (Gel et al. 2016). These peak sets were also analyzed for overrepresented DNA sequence motifs using Weeder v2.0 (Supplemental Methods; Zambelli et al. 2014).
Fine-scale profiling of ChIP-seq peaks and genes
Per-feature windowed coverage profiles were calculated using the computeMatrix tool from deepTools v3.1.3 (Ramírez et al. 2014). CPM values for a ChIP input sequencing library (accessed from the NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] accession SRR6350669) (IWGSC 2018), and for the MNase-seq library generated in the current study, were used in conjunction with those for ChIP-seq libraries derived from chromatin fragmented by sonication or by MNase digestion, respectively, to calculate windowed log2([ChIP + 1]/[control + 1]) coverage ratios for each chromosome or feature. Per-feature windowed SNP (He et al. 2019) and TE (IWGSC 2018) frequency profiles were calculated using the normalizeToMatrix function from the Bioconductor package EnrichedHeatmap v1.12.0 (Supplemental Methods; Gu et al. 2018).
DMC1 and ASY1 ChIP-seq peaks, genes, and NLRs (Steuernagel et al. 2020) were divided into quantiles according to decreasing mean crossover rate between 1 kb upstream of feature start sites and 1 kb downstream from end sites, using previously calculated crossover rates derived from a Chinese Spring × Renan F6 genetic map (Supplemental Methods; IWGSC 2018). Genes were also divided into quantiles according to decreasing mean DMC1, ASY1, or H3K27me3 ChIP-seq signal (log2[(ChIP + 1)/(input + 1)]) within 1-kb-extended boundaries. Metaprofiles (windowed means and 95% confidence intervals) were calculated and plotted for each group of features in R v3.5.0 (R Core Team 2018). Gene quantiles were evaluated for over- and underrepresentation of functional and expression categories using topGO v2.32.0 (https://doi.org/10.18129/B9.bioc.topGO) and the hypergeometric distribution (Supplemental Methods). Published exome-sequencing-derived SNPs (He et al. 2019) were annotated with their predicted impact on protein function using SnpEff v4.3t (Cingolani et al. 2012) and analyzed using PopGenome v2.7.5 to compute population genetics statistics for each gene (Supplemental Methods; Pfeifer et al. 2014).
Software availability
Custom scripts used in the preparation of this paper are provided as Supplemental Code, and are also available at GitHub (at: https://ajtock.github.io/Wheat_DMC1_ASY1_paper/).
Data access
All raw sequencing data generated in this study have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession numbers E-MTAB-9632 (ChIP-seq) and E-MTAB-9633 (MNase-seq).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Ksenia Krasileva (University of California, Berkeley), Jemima Brinton (Royal Botanic Gardens, Kew), and members of the Henderson laboratory for helpful discussions. Research was supported by a European Research Council (ERC) Consolidator Grant “SynthHotSpot,” ERC Proof of Concept grant “HEIREC,” Biotechnology and Biological Sciences Research Council (BBSRC) sLola grant BB/N002628/1, Leverhulme Trust grant RPG-2019-259, Agriculture and Agri-Food Canada/BBSRC International Wheat Yield Partnership BB/T004282/1, BBSRC Industrial Partnership Award grant BB/N007557/1 with Meiogenix, and a BBSRC Doctoral Training Partnership iCASE studentship with Klein Wanzlebener Saatzucht (KWS).
Author contributions: A.J.T., D.M.H., K.O., E.S.-M., J.D.H., K.J.E., F.C.H.F., and I.R.H. designed the research. D.M.H., W.J., and K.O. performed experiments. K.O. interpreted cytological data. A.J.T. wrote software and performed computational analyses. C.U. provided advice on genetic map and polymorphism analyses. A.J.T., D.M.H., and I.R.H. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.273672.120.
- Received November 3, 2020.
- Accepted July 20, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








