
Nucleosomal signatures impose nucleosome positioning in coding and noncoding sequences in the genome
- Sara González1,3,
- Alicia García1,3,
- Enrique Vázquez1,
- Rebeca Serrano1,
- Mar Sánchez1,
- Luis Quintales1,2 and
- Francisco Antequera1
- 1Instituto de Biología Funcional y Genómica, Consejo Superior de Investigaciones Científicas (CSIC)/Universidad de Salamanca, 37007 Salamanca, Spain;
- 2Departamento de Informática y Automática, Universidad de Salamanca/Facultad de Ciencias, 37007 Salamanca, Spain
- Corresponding author: cpg{at}usal.es
-
↵3 These authors contributed equally to this work.
Abstract
In the yeast genome, a large proportion of nucleosomes occupy well-defined and stable positions. While the contribution of chromatin remodelers and DNA binding proteins to maintain this organization is well established, the relevance of the DNA sequence to nucleosome positioning in the genome remains controversial. Through quantitative analysis of nucleosome positioning, we show that sequence changes distort the nucleosomal pattern at the level of individual nucleosomes in three species of Schizosaccharomyces and in Saccharomyces cerevisiae. This effect is equally detected in transcribed and nontranscribed regions, suggesting the existence of sequence elements that contribute to positioning. To identify such elements, we incorporated information from nucleosomal signatures into artificial synthetic DNA molecules and found that they generated regular nucleosomal arrays indistinguishable from those of endogenous sequences. Strikingly, this information is species-specific and can be combined with coding information through the use of synonymous codons such that genes from one species can be engineered to adopt the nucleosomal organization of another. These findings open the possibility of designing coding and noncoding DNA molecules capable of directing their own nucleosomal organization.
Most nucleosomes occupy well-defined positions along the yeast genome that remain constant under many different physiological conditions (Yuan et al. 2005; Lee et al. 2007; Zhang et al. 2011a; Soriano et al. 2013). This precise positioning is essential to modulate the access of proteins to specific sites in the chromosomes to regulate transcription (Bai et al. 2010; Koster et al. 2015), replication initiation (Lipford and Bell 2001; Eaton et al. 2010; Soriano et al. 2014), and recombination (Pan et al. 2011; de Castro et al. 2012).
Nucleosomal patterns result from the combined contribution of chromatin remodelers, DNA-binding proteins, and the differential affinity of nucleosomes for different DNA sequences. Chromatin remodelers are multiprotein complexes that use ATP hydrolysis to facilitate the sliding, eviction or histone exchange of nucleosomes (Clapier and Cairns 2009). Remodelers show different specificity and directionality in their mode of action (Stockdale et al. 2006; Yen et al. 2012), and the removal of some of them, like Hrp3 in Schizosaccharomyces pombe (Hennig et al. 2012; Pointner et al. 2012; Shim et al. 2012) or Isw1 and Chd1 in Saccharomyces cerevisiae (Gkikopoulos et al. 2011), results in gross genome-wide alteration of their nucleosomal patterns.
Transcription factors contribute to chromatin organization through the recruitment of remodelers to promoters (Yudkovsky et al. 1999; Korber et al. 2004) and also through their ability to compete with nucleosomes for their binding sites (Badis et al. 2008; Hartley and Madhani 2009; Tsankov et al. 2011; Soriano et al. 2013; Koster et al. 2015). Transcriptional regulatory complexes bound at promoters could also act as physical barriers from which regular nucleosomal arrays are generated, as proposed by the statistical positioning model (Kornberg and Stryer 1988; Mavrich et al. 2008), through active ATP-dependent mechanisms (Zhang et al. 2011b).
The third element contributing to nucleosome positioning is the DNA sequence. The DNA molecule is strongly bent along its axis, and adjacent nucleotides are also under strong lateral displacement to accommodate 147 base pairs (bp) of double-stranded DNA in 1.7 turns around the histone octamer (Luger et al. 1997). Sequence motifs vary in their resistance to deformation, and therefore, different DNA molecules offer a different resistance to bending (Drew and Travers 1985; Thåström et al. 1999). Sequence analyses of aggregated nucleosomal profiles have revealed that some AT-rich dinucleotides are preferentially positioned in the minor groove of DNA facing the histone core, while GC-rich dinucleotides face outward (Satchwell et al. 1986; Ioshikhes et al. 1996; Lowary and Widom 1998; Segal et al. 2006; Albert et al. 2007). This alternating organization favors the bending of the DNA molecule and the electrostatic interaction between arginine residues and AT-rich sequences in the minor groove of DNA around the histone core (Rohs et al. 2009).
The search for sequence determinants of nucleosome positioning has led to the identification of some synthetic sequences with great affinity to form nucleosomes in vitro (Lowary and Widom 1998), among which the 601 sequence has been extensively used in many structural studies (Olson and Zhurkin 2011; Ngo et al. 2015 and references therein). Natural sequences like the 5S RNA also have strong positioning potential in vitro and in vivo (Simpson and Stafford 1983; Pennings et al. 1991).
Despite these preferences, the extent to which the DNA sequence contributes to nucleosome positioning in the genomic context remains unclear. In some cases, the nucleosomal pattern in vitro coincides with that of native chromatin in discrete genomic regions (Shen and Clark 2001; Allan et al. 2013; Beh et al. 2015). However, unlike the situation in vitro, the 601 and 603 artificial sequences do not preferentially form nucleosomes when integrated into the genome of S. cerevisiae (Gaykalova et al. 2011; Perales et al. 2011). Other studies have shown that the DNA sequence is unable to recapitulate the in vivo positioning pattern at the genomic scale in chromatin assembly assays in vitro (Zhang et al. 2009).
We have recently described that the aggregated pattern of the four nucleotides along mononucleosomal DNA follows well-defined and asymmetrical patterns that we have called nucleosomal signatures. They are present in transcribed and nontranscribed regions and vary widely even among species of the same genus (Quintales et al. 2015a). In this work, we have analyzed whether nucleosomal signatures contain information capable of targeting nucleosomes to specific positions in natural and synthetic DNA molecules integrated into the genome.
Results
Sequence changes in mononucleosomal DNA destabilize individual nucleosomes
To analyze how robust nucleosomal organization was to changes in the DNA sequence, we selected the genomic region encompassing the ura4 gene of S. pombe. This region is organized in a regular pattern of nucleosomes as shown by partial micrococcal nuclease (MNase) digestion and end-terminal hybridization (Fig. 1A,B, WT). To modify the sequence of the ura4 open reading frame (ORF), we replaced the wild-type codons (except for the START and STOP codons) by their synonymous codons such that, when possible, A or T nucleotides were changed to C or G, and vice versa. The resulting ORF (61.5% identity to the wild-type ORF but encoding the same Ura4 protein) was used to replace the wild-type ORF in its endogenous locus to generate the S. pombe ura4 1_6 strain (sequence in Supplemental Fig. S1A). Partial MNase digestion and hybridization to probe 1 revealed a gross alteration in nucleosome positioning where the sharp regular wild-type profile was replaced by a much more diffuse pattern (Fig. 1A, 1_6). This altered profile was confirmed by hybridization to probe 2 to visualize the same region from the other end of the Hind III fragment (Fig. 1B).
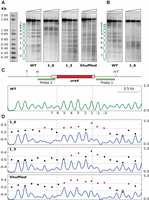
Sequence changes destabilize nucleosome positioning in the ura4 gene. (A) Chromatin from wild-type cells (WT) and from the indicated mutant strains were digested with increasing amounts of MNase (triangles) prior to digestion with HindIII or TfiI (Shuffled). Samples were electrophoresed, blotted, and hybridized to probe 1, shown in C. (B) WT and 1_6 membranes in A were stripped and rehybridized using probe 2. Some controls of naked DNA digested with MNase, and of chromatin incubated without MNase, are shown in Supplemental Figure S8. Similar controls were carried out for all the remaining MNase experiments. (C) Diagram of the analyzed region. The ura4 gene is represented by a pointed rectangle, and the coding region is shown in red. Restriction sites for HindIII (H) and TfiI (T) and the localization of the hybridization probes are indicated. The nucleosome occupancy map of wild-type cells generated by MNase-seq is shown in green. The y-axis indicates occupancy normalized to the genome average. Vertical dashed lines indicate the position of the ura4 ORF. Nucleosomes are numbered as in A. (D) Nucleosome occupancy map of the 1_6, 1_3, and Shuffled strains (blue) and DANPOS analysis. Left y-axis indicates the log2-fold difference in fuzziness in the analyzed region. Red dots represent values above or below 2σ (horizontal dotted lines) of the distribution of this difference between the three modified ura4 versions and the WT. Right y-axis indicates nucleosome occupancy as in C.
To test whether the loss of positioning along the ORF was dependent on the destabilization of the +1 nucleosome, we generated the S. pombe ura4 1_3 strain in which only codons corresponding to nucleosomes 1, 2, and 3 were replaced by the same synonymous codons as in the ura4 1_6 strain (Supplemental Fig. S1A). In this case, the impact was lower than in the 1_6 strain and affected preferentially the region encompassing these three nucleosomes, as revealed by the enhanced sensitivity to MNase of their linker regions (Fig. 1A, panel 1_3).
To quantitatively measure the differences in positioning among strains, we generated genome-wide MNase-seq maps of the wild type, 1_6, and 1_3 cells. Briefly, we digested chromatin with MNase, isolated mononucleosomal DNA, and sequenced it using the Illumina paired-end protocol (see Methods). We analyzed the MNase-seq data using the DANPOS 2 application (Chen et al. 2013a), which is widely used in the analysis of nucleosome dynamics (Chen et al. 2013b; Beh et al. 2015; Sebé-Pedrós et al. 2016). This tool allows comparing the position of the nucleosome dyad and the degree of positioning and occupancy of individual nucleosomes between different MNase-seq maps in a scale of fuzziness (Chen et al. 2013a; see Methods). The red dots in Figure 1D show that the level of log2-fold change of fuzziness, in regions encompassing the modified ura4 sequences (blue) relative to the wild-type ORF (green), was above 2σ (σ = 0.1) of the mean of the difference between the two genome-wide maps. The altered positioning was more extended to the 3′ end of the ura4 ORF in the 1_6 strain than in the 1_3 strain. The level of occupancy of nucleosomes -1 and -2 was slightly reduced in the mutant strains, probably due to the altered positioning of the nucleosomes immediately upstream of them.
Genome-wide differences between biological duplicates are very small, as shown by the overlap between the profiles of duplicated maps corresponding to Figure 1 (Supplemental Fig. S2). Accordingly, the DANPOS value of the differences between duplicates is close to zero. The same level of coincidence was found between duplicates of the remaining strains used in this work. The same quantitative analyses showed no significant differences in nucleosome positioning between different strains outside the modified regions (Supplemental Fig. S3).
It is important to note that small differences between nucleosomal profiles generated by end-labeling hybridization or by MNase-seq are likely due to the very different degrees of MNase digestion. In the first case, chromatin is very mildly digested to allow visualization of the entire nucleosomal ladders. As a consequence, most of the hybridization signal is detected at the full-length restriction fragment. In contrast, in the MNase-seq analyses, digestion proceeds until ∼80% of the chromatin is recovered as mononucleosomes prior to sequencing (Lantermann et al. 2010; Soriano et al. 2013). Despite these differences, the two approaches generate comparable results (Fig. 1).
In view of the sensitivity of positioning with regard to codon changes, we wondered whether the distribution of wild-type codons would be relevant to nucleosome positioning. To test this possibility, we generated the Shuffled ura4 strain, where we swapped the different synonymous codons for each amino acid along the wild-type ura4 ORF. The resulting ORF contained the same codons, encoded the same protein, and was 77.7% identical to the wild type (Supplemental Fig. S1A). However, end-labeling, MNase-seq, and DANPOS analyses showed that the regular wild-type nucleosomal array was also significantly disrupted in this strain, suggesting that the loss of positioning was due to the modification of the primary DNA sequence and not to changes in the overall base composition of the ORF (Fig. 1A,D, Shuffled). The distribution of MNase cutting sites along the ura4 ORF provides a higher resolution view of the differences between the wild type and the three mutant strains (Supplemental Fig. S4).
We have previously shown that nucleosome positioning in S. pombe is altered in approximately 60 genes that are highly expressed and show an elevated RNA polymerase II occupancy (Soriano et al. 2013). Chromatin immunoprecipitation and qPCR analyses showed that the occupancy of RNA polymerase II along the ura4 gene in the three mutants ranged between 1.0- and 1.5-fold relative to wild-type cells. Such a small difference is well below the level at which positioning is disrupted, indicating that the observed alterations in the nucleosomal profiles were unlikely to result from major changes in transcription (Supplemental Fig. S5).
The results obtained with the ura4 ORF raised the question of whether the link between the DNA sequence and nucleosome positioning would be exclusive to transcribed regions. To test this possibility, we selected two well-positioned nucleosomes in an intergenic region lacking any detectable transcription, as determined by microarray analysis (Soriano et al. 2013) or by RNA-seq (Rhind et al. 2011), and substituted their sequences for their randomized versions in the same genomic locus (therefore maintaining the original base composition) (Supplemental Fig. S1B). End-labeling (Fig. 2A) and DANPOS analyses (Fig. 2B) showed that the positioning of the two nucleosomes was strongly altered relative to the wild-type cells. This effect coincided with the modified sequences and extended to the immediately adjacent nucleosomes 1 and 2, probably as a consequence of the altered interaction between nucleosomes 3 and 4 with DNA (Fig. 2A,B). In any case, the additional disruption was due to the randomization of the sequences underlying nucleosomes 3 and 4, since this is the only difference relative to the wild-type control. These results indicated that modification of mononucleosomal sequences alter the positioning of nucleosomes associated with them, independently of transcription.
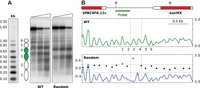
Sequence changes destabilize nucleosome positioning in nontranscribed regions. (A) MNase end-labeling analysis from wild-type cells (WT) and from the strain harboring randomized sequences underlying nucleosomes 3 and 4 (green). (B) (Top) The position of the SPAC6F6.11c gene and the kanMX marker gene is indicated. Restriction sites for NsiI (N) and the position of the hybridization probe are indicated. (Middle) Nucleosome occupancy map of the wild-type strain with nucleosomes numbered as in A. (Bottom) DANPOS analysis of nucleosome positioning of the Random strain relative to the wild type. Vertical dashed lines indicate the position of nucleosomes 3 and 4. Other symbols are as in Figure 1.
Mononucleosomal DNA encodes portable positioning information
Given the close link between nucleosomes and their underlying sequence, we asked whether mononucleosomal DNA could maintain the positioning of individual nucleosomes in ectopic genomic positions. To address this point, we tried to recapitulate the nucleosomal profile of the ura4 ORF by assembling individual mononucleosomal sequences from unrelated genomic loci. We selected six of these regions from the three chromosomes (sequences and genomic localization in Supplemental Fig. S1C) and linked them together to generate a fragment of the same size of the ura4 ORF to replace it in the endogenous locus. Since only a part of the sequences underlying nucleosomes 1 and 6 were included in the ORF of the ura4 gene, only sequences including the equivalent regions relative to the dyad position were selected from two nucleosomes located elsewhere in the genome (Fig. 3A, half-colored ovals). MNase mapping showed that the nucleosomal pattern of the chimeric construct was virtually identical to the wild-type ura4 pattern (Fig. 3B). Even nucleosomes 1 and 6 were well positioned despite the chimeric origin of their underlying sequences. As a control, we generated another strain where the sequences of the six ectopic DNA fragments were randomized individually before ligating them together in the same order. In this strain, MNase analysis generated multiple irregularly spaced bands, indicative of the absence of nucleosomal positioning (Fig. 3B, random). These results showed that individual mononucleosomal DNA sequences associated with positioned nucleosomes in the genome maintain their positioning potential when transferred to ectopic loci.
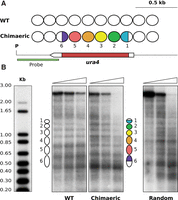
Mononucleosomal DNA encodes portable positioning information. (A) White ovals represent nucleosomes across the ura4 region as shown in Figure 1, A and C. Colored ovals 2–5 indicate regions that were replaced by nonadjacent mononucleosomal sequences from unrelated genomic regions. In nucleosomes 1 and 6, only colored regions were replaced by equivalent regions from other mononucleosomal DNA sequences. The restriction site for PsuI (P) and the hybridization probe (green) are indicated. (B) The MNase end-labeling analysis of wild-type cells (WT) and of the chimeric construct generated a comparable positioning pattern. Nucleosome positioning was lost after individual randomization of the sequences underlying the colored regions in A (Random).
Positioning information is dispersed across mononucleosomal DNA
The relevance of the DNA sequence in nucleosome positioning raised the question of whether different regions of mononucleosomal DNA would contribute differentially to it. To address this point, we generated three strains where only one third of the mononucleosomal DNA sequence associated with each of the six nucleosomes along the ura4 ORF was replaced by the same synonymous codons used in the S. pombe 1_6 strain (Supplemental Fig. S1D). In the dyad and linker strains, we replaced 51 bp centered on the midposition of mononucleosomal DNA or on the linker between adjacent nucleosomes, respectively (Fig. 4A, green and orange sections). In the third strain (int), we replaced the two remaining internal regions of each mononucleosomal DNA between positions −51 to −24 and +24 to +51 relative to the dyad (Fig. 4A, black sections). MNase end-labeling analysis showed that internucleosomal bands were slightly more diffused than in the wild type in the three cases, suggesting a small reduction in the affinity between the modified sequences and the histone octamers (Fig. 4A). These differences, however, were not detected as significant by the DANPOS analysis of this region (Fig. 4B) except in one or two nucleosomes in each strain. The comparable profile in the three mutant strains suggested that positioning information was not preferentially associated with specific regions of mononucleosomal DNA but probably depended on the collective contribution of redundant and degenerated elements dispersed along its length.
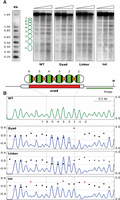
Positioning information is dispersed across mononucleosomal DNA. (A) MNase end-labeling analysis of wild-type (WT) and the dyad, linker, and internal mutant strains. Ovals represent nucleosomes across the ura4 gene. Colored sections indicate the third of the mononucleosomal DNA sequence that was replaced by synonymous codons in the dyad (green), linker (orange), or internal (black) regions of nucleosomes 1–6. The restriction site for HindIII and the hybridization probe are indicated. (B) Nucleosome occupancy maps of the four strains and DANPOS analysis of nucleosome positioning of the three mutant strains relative to the wild type. Symbols are as in Figure 1.
Engineering nucleosomal positioning on synthetic noncoding DNA sequences
If the DNA sequence plays a significant role in nucleosome positioning, we surmised that it might be possible to design synthetic DNA molecules capable of targeting nucleosomes to specific sites in the genomic context. However, it was not immediately obvious how to design such sequences given the expected degeneracy of the putative sequence determinants. Despite the large variability among the thousands of mononucleosomal sequences in the genome, their aggregated profiles generate well-defined patterns in the distribution of the four nucleotides that we have called nucleosomal signatures (Quintales et al. 2015a). We hypothesized that the information contained within these signatures could contribute to nucleosome positioning, and therefore, we used them as a starting point in the design of the synthetic DNA sequences. To extract the sequence information contained in nucleosomal signatures, we generated a position-specific weight matrix (PSWM), which incorporated the frequency of each of the 16 dinucleotides along the aggregated profiles of thousands of mononucleosomal sequences underlying well-positioned nucleosomes (see Methods). As a consequence of the species-specific nature of nucleosomal signatures, PSWMs showed different positional values in S. pombe, Schizosaccharomyces octosporus, Schizosaccharomyces japonicus, and S. cerevisiae (Fig. 5).
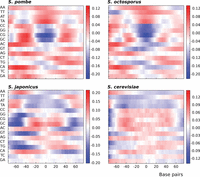
Position-specific weight matrix of nucleosomal signatures. Heat map representation of the position-specific weight matrix (PSWM) for the indicated four species. The x-axis indicates positions relative to the nucleosomal dyad; the y-axis indicates the log-odd score of the 16 dinucleotides along mononucleosomal DNA calculated as the ratio of their frequency at each position relative to their genomic frequency. Bars on the right represent a color scale associated with the log-odd score values.
Based on this information, we generated six random sequences 153 bp long (147-bp core DNA plus 6-bp linker) (Lantermann et al. 2010) and subjected them to reiterated rounds of mutation in silico to select those with a high score relative to the S. pombe PSWM (see Methods for details on the design of the synthetic sequences). The resulting 918-bp fragment (Supplemental Fig. S1E) was integrated into the intergenic, nontranscribed region between the S. pombe SPAC6F6.11c and SPAC6F6.12 genes (Fig. 6A, red arrowhead). MNase end-labeling and MNase-seq analyses across the synthetic fragment revealed a regular array of six nucleosomes, which mapped precisely to the positions predicted by the nucleosomal signatures (Fig. 6B, Seq-Sp/Sp; Fig. 6D, red trace).
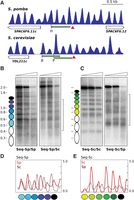
Engineering nucleosomal positioning on synthetic DNA sequences. (A) Nucleosomal profile of the genomic regions of S. pombe and S. cerevisiae, where the artificial sequences were inserted (red arrowheads). The restriction sites for HindIII (H), BsmI (B), and PagI (P) and the localization of the hybridization probes (green) are indicated. (B) The MNase end-labeling analysis of the S. pombe sequence integrated in S. pombe generates a regular nucleosomal profile as predicted (colored ovals) (Seq-Sp/Sp). Insertion of the same sequence in S. cerevisiae (bracket) generates a different pattern (Seq-Sp/Sc). (C) The S. cerevisiae sequence generates a regular profile after integration in S. cerevisiae (Seq-Sc/Sc) but fails to position nucleosomes when integrated in the S. pombe genome (Seq-Sc/Sp, bracket). (D) MNase-seq occupancy maps of the S. pombe artificial sequence integrated in S. pombe (red) or in S. cerevisiae (black). (E) MNase-seq occupancy maps of the S. cerevisiae artificial sequence integrated in S. cerevisiae (red) or in S. pombe (black).
Since nucleosomal signatures differ among species, we tested whether the same fragment would also position nucleosomes in S. cerevisiae. Insertion into the nontranscribed intergenic region upstream of the YDL211C gene in chromosome IV (Fig. 6A, red arrowhead), followed by MNase end-labeling analysis (Fig. 6B, Seq-Sp/Sc), showed a banding pattern indicative of some preferred positions but different from the pattern generated by the same artificial sequence in S. pombe. MNase-seq analysis (Fig. 6D, dotted trace) confirmed this result and showed that, in some cases, internucleosomal positions in S. cerevisiae coincided with dyad positions in S. pombe. This could be due to the fact that A + T-rich sequences are preferentially found at the dyad and linker regions in S. pombe and S. cerevisiae, respectively (Tillo and Hughes 2009; Moyle-Heyrman et al. 2013; Quintales et al. 2015a).
These results suggested that it might be possible to design sequences based on the S. cerevisiae nucleosomal signature capable of positioning nucleosomes in its own genome but, maybe, not in S. pombe. To test this possibility, we synthesized a DNA molecule of 1008 bp (Supplemental Fig. S1E) based on the S. cerevisiae PSWM capable of accommodating six nucleosomes (147-bp core plus 21-bp linker) (Lantermann et al. 2010) following the same strategy as used for S. pombe. The resulting fragment was integrated into the same two genomic positions as the previous construct (Fig. 6A), and end-labeling analysis showed that it generated a perfectly regular array of six nucleosomes at the expected positions in S. cerevisiae (Fig. 6C, Seq-Sc/Sc) but failed to do so in S. pombe (Fig. 6C, Seq-Sc/Sp). The same result was found by MNase-seq analysis (Fig. 6E). These results show that nucleosomal signatures contain positioning information capable of targeting nucleosomes to predetermined positions on synthetic artificial DNA sequences in a species-specific manner.
Nucleosome positioning is not maintained on orthologous sequences of closely related species
Given the incompatibility in positioning between the distantly related S. pombe and S. cerevisiae species within the Ascomycete lineage (Hoffman et al. 2015), we wondered whether nucleosome positioning would be maintained over orthologous sequences of closely related species. To address this question, we replaced the ORF of the S. pombe ura4 gene by the orthologous ura4 ORFs of two species of the same genus, S. octosporus and S. japonicus. The three ORFs are identical in size (795 bp) (Supplemental Fig. S1F), have a nucleotide identity of 75.0% and 70.2% relative to S. pombe, respectively, and their encoded amino acid sequences are sufficiently similar (82.6% and 74.6% identity) for them to generate functionally interchangeable Ura4 proteins. In addition, the S. octosporus and S. japonicus ura4 ORFs encompass six positioned nucleosomes in their respective genomes at positions comparable to those in S. pombe (Supplemental Fig. S6). Despite these similarities, the sharp internucleosomal bands generated by MNase in the endogenous S. pombe ura4 ORF became slightly more diffuse after its replacement by the S. octosporus ORF (Fig. 7A, Native). This suggested an increased accessibility of MNase to sequences adjacent to the linker DNA, perhaps due to a less tight interaction between nucleosomes and DNA than in the endogenous S. pombe ura4 ORF.
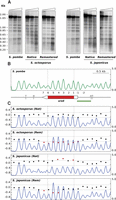
Engineering nucleosomal positioning on eukaryotic coding DNA sequences. (A) MNase end-labeling analysis of wild-type S. pombe ura4 (left panel). The pattern of internucleosomal bands becomes more diffused when the S. pombe ura4 ORF is replaced by the native S. octosporus or S. japonicus ORFs. The original sharp profile is restored when the codons of the two ORFs are replaced by synonymous codons, with the highest score in the PSWM of S. pombe (Remastered). (B) Nucleosome occupancy map of wild-type S. pombe. Restriction sites for HindIII (H) and TfiI (T) and the localization of the hybridization probe (green bar) are indicated. (C) Nucleosome occupancy maps of the strains harboring the native (Nat) and remastered (Rem) ura4 ORF of S. octosporus and S. japonicus (blue). DANPOS analysis of nucleosome positioning of the four mutant strains relative to the wild type. Symbols are as in Figure 1.
These differences fell below the level of detection by DANPOS (Fig. 7C, S. octosporus, Nat), but a wider dispersion of the MNase cutting sites was detectable at linker regions in the native S. octosporus ura4 ORF relative to S. pombe (Supplemental Fig. S7). Dispersion was much greater along the native S. japonicus ura4 ORF (Supplemental Fig. S7), and the differences in nucleosome positioning relative to S. pombe were detected by end-labeling (Fig. 7A, S. japonicus, Native) and by DANPOS analysis (Fig. 7C, S. japonicus, Nat). These results reinforce the sensitivity of nucleosome positioning to exogenous sequences even in the case of orthologous sequences integrated in the same genomic locus.
Engineering nucleosomal positioning on orthologous coding DNA sequences
Given the degenerated nature of nucleosomal signatures, we wondered whether they could be incorporated into the ura4 ORFs of S. octosporus and S. japonicus to reconstitute the S. pombe nucleosomal pattern. To maintain their native coding specificity, we took advantage of the degeneracy of the genetic code and replaced the codons along the corresponding six mononucleosomal sequences by synonymous codons with the highest possible score at each position in the S. pombe PSWM. Despite this restriction in the design of the modified sequences, the two resulting ORFs generated an MNase end-labeling nucleosomal profile virtually identical to that of the endogenous ura4 ORF of S. pombe (Fig. 7A, Remastered). DANPOS analysis of MNase-seq data revealed slightly negative score values of the remastered S. octosporus ura4 ORF (Fig. 7C, S. octosporus, Rem), indicative of a positioning profile even sharper than that of the endogenous S. pombe ura4 ORF, which was used as a reference (Fig. 7B). This effect can be more directly appreciated in the narrower distribution of MNase cutting sites at linker regions in the remastered S. octosporus ura4 ORF relative to S. pombe ORF (Supplemental Fig. S7). A similar reconstitution of a sharp positioning profile was obtained for the modified S. japonicus ura4 despite the greater differences in positioning of its native ura4 ORF relative to S. pombe (Fig. 7C; Supplemental Fig. S7). The nucleotide identity of the modified S. octosporus and S. japonicus ORF sequences relative to S. pombe was 73% and 71%, respectively, which is very close to the 75% and 70% identity of their native versions. This suggests that the overall sequence identity is not a determining factor in the specification of nucleosome positioning.
Engineering nucleosomal positioning on prokaryotic genes
To test whether ORFs completely unrelated to S. pombe or S. cerevisiae could also be engineered to position nucleosomes at predetermined positions, we selected the prokaryotic kan gene, which confers resistance to geneticin. Since no orthologs of this gene are present in the yeast genome, we replaced the endogenous SPBC16G5.03 and YKL007W ORFs of S. pombe and S. cerevisiae, respectively, by the kan ORF. We selected them because they have almost the same size (807 bp) as the kan ORF (810 bp), are dispensable for growth in the two yeasts, and have well-positioned nucleosomes along their length (Fig. 8A). Their replacement by the wild-type kan ORF generated a relatively regular profile reminiscent of that over the endogenous ORFs (Fig. 8B,C, Kan_WT). To test whether a more regular and homogeneous profile could be induced over the kan ORF, we generated two versions of it by replacing its native codons by their synonymous codons with the highest score in the S. pombe and S. cerevisiae PSWM along mononucleosomal DNA (Supplemental Fig. S1G). MNase end-labeling analysis showed that these versions generated regular nucleosomal arrays where nucleosomes occupied the expected positions as determined by the engineered nucleosomal signatures (Fig. 8B, Kan_Sp; Fig. 8C, Kan_Sc). Consistent with these results, DANPOS analysis showed a negative score of the two modified kan sequences relative to the wild-type kan (Fig. 8D, Kan_Sp; Fig. 8E, Kan_Sc).
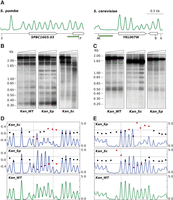
Engineering nucleosomal positioning on prokaryotic genes. (A) Nucleosomal distribution across the indicated regions in S. pombe and S. cerevisiae. Genes are represented by pointed rectangles. Restriction sites for XhoI (X), PstI (P), AvaII (A), Kpn2I (K), and BstxI (B) and the localization of the hybridization probes (green) are shown. (B) MNase end-labeling analysis of a S. pombe strain where the SPBC16G5.03 ORF has been replaced by the wild-type kanamycin ORF (Kan_WT) or a version where their codons have been replaced by the synonymous codons with the highest score in the PSWM of S. pombe (Kan_Sp). The same sequence is unable to position nucleosomes when it replaces the YKL007W ORF in the genome of S. cerevisiae (Kan_Sp in C). (C) MNase end-labeling analysis of a S. cerevisiae strain where the YKL007W ORF has been replaced by the wild-type kanamycin ORF (Kan_WT) or a version where their codons have been replaced by the synonymous codons, with the highest score in the PSWM of S. cerevisiae (Kan_Sc). The same sequence is unable to position nucleosomes when it replaces the SPBC16G5.03 ORF in the genome of S. pombe (Kan_Sc in B). (D) DANPOS analysis of modified kan ORFs in S. pombe and (E) in S. cerevisiae shows a negative score relative to the wild-type kan in each of them (middle panels). Dotted vertical lines indicate the position of the kan ORF. The positive score when the two modified sequences are swapped (top panels) indicates a lower degree of positioning than the unmodified wild-type kan ORF in the two yeasts.
To test whether this positioning information was species-specific, we swapped the modified kan versions between the two yeasts. Results showed that the regular nucleosomal array of the Kan_Sc sequence in S. cerevisiae was lost in S. pombe (Fig. 8B, Kan_Sc) and that the opposite result was obtained when the Kan_Sp sequence replaced the YKL007W ORF in S. cerevisiae (Fig. 8C, Kan_Sp). The MNase-seq occupancy pattern and DANPOS analysis confirmed a higher fuzziness relative to the wild-type kan ORF in the two yeasts (Fig. 8D, Kan_Sc; Fig. 8E, Kan_Sp). Altogether, Figures 7 and 8 show that exogenous eukaryotic or prokaryotic ORFs can be engineered in a species-specific manner to direct their packaging into regular nucleosomal arrays with the same periodicity as those of the endogenous genes of the host.
Discussion
The extent to which the DNA sequence contributes to nucleosome positioning in the genome remains controversial. As commented in the Introduction, in vitro approaches using purified components have uncovered important aspects of the interaction between DNA and histones but have the inevitable limitation that they do not always mimic the situation in vivo. This is well illustrated by the different positioning potential of the same sequences, both in vitro and in vivo, at the level of individual nucleosomes (Gaykalova et al. 2011; Perales et al. 2011) or at genome-wide scale (Zhang et al. 2009). We have focused our work on the contribution of the DNA sequence to nucleosome positioning in the genomic context through two complementary approaches: first, by modifying the sequence in discrete regions, and second, by designing DNA molecules capable of targeting nucleosomes to specific positions.
The first striking finding was the degree of sensitivity of individual nucleosomes to sequence changes, even when the modified regions span only 0.3–1.0 kb (2–6 nucleosomes, approximately). Nucleosome positioning was altered at transcribed (Fig. 1) and nontranscribed regions (Fig. 2), suggesting that transcription per se is not a requirement for nucleosome positioning. This is consistent with the similar RNA pol II occupancy of all the modified versions of the ura4 gene despite their different nucleosomal profiles (Fig. 1; Supplemental Fig. S5). Independence from transcription also agrees with positioning being maintained beyond the transcription termination sites and with the presence of regular nucleosomal arrays in active and inactive versions of many genes during mitosis or meiosis (Soriano et al. 2013).
The overlap between the length of the modified sequences and the loss of positioning suggested that sequence elements contributing to nucleosome positioning could have been disrupted. The existence of such elements was supported by the ability of individual mononucleosomal sequences to direct nucleosome positioning when transferred to ectopic regions in the genome and by the loss of this property after sequence randomization (Fig. 3). Sequence determinants, however, appeared to be species-specific as shown by the loss of the regular nucleosomal profile along the S. pombe ura4 ORF after its replacement by the orthologous ORFs of S. octosporus and S. japonicus (Fig. 7).
Additional support for the relevance of the sequence to direct nucleosome positioning came from the restoration of the endogenous S. pombe pattern after incorporating information from its nucleosomal signature through the use of synonymous codons within the S. octosporus and S. japonicus ORFs (Fig. 7) and by the generation of regular nucleosomal arrays over prokaryotic genes (Fig. 8) and even on synthetic DNA molecules (Fig. 6). These results also suggest that nucleosomal signatures in different yeast species contain positioning information that is correctly interpreted by the species from which it is derived. This species-specificity can contribute to explaining the previous observations that the same DNA sequences are packed differently by nucleosomes of phylogenetically distant species (Bernardi et al. 1992; McManus et al. 1994; Sekinger et al. 2005; Hughes et al. 2012).
As regards the origin of nucleosomal signatures, analyses of the substitution rate of mononucleosomal DNA in related species have suggested that nucleosomal positioning relative to the DNA sequence has remained stable over evolutionary timescales (Washietl et al. 2008). This long-term association between histones and DNA makes it possible that nucleosomal signatures could have emerged as a consequence of a different rate of mutation or biased repair along mononucleosomal and linker DNA, due to small differences in the structure of histone octamers; in the bias or accessibility of repair proteins (Washietl et al. 2008; Sasaki et al. 2009); or in the different impact on the sequence of chromatin remodelers or epigenetic modifications. Such a mutational scenario is compatible with the finding that sequence variation along mononucleosomal DNA is under positive selection in S. cerevisiae (Warnecke et al. 2008) and in humans (Prendergast and Semple 2011). Recent work has revealed the existence of thousands of regions ∼1 kb long in the human genome made up of intrinsic nucleosome-depleted regions flanked by two or three nucleosomes whose positioning is encoded in the GC content (Drillon et al. 2016). These results suggest that nucleosomal signatures could have a positive selective value for their contribution to nucleosome positioning. These signatures are present genome-wide and, in the case of ORFs, the finding that swapping of synonymous wild-type codons disrupts the regular nucleosomal profile of the S. pombe ura4 gene (Fig. 1, Shuffled; Supplemental Fig. S4) suggests the intriguing possibility that, in addition to modulating the stability of mRNA and its rate of translation (Plotkin and Kudla 2011; Presnyak et al. 2015), the distribution of synonymous codons might also contribute to nucleosome positioning along ORFs. Nucleosomal signatures have a strong impact in genome and protein evolution to the extent that, in the case of coding regions, they correctly predict the relative distribution of the 20 amino acids along proteins based on the position of the corresponding codons along mononucleosomal DNA (Warnecke et al. 2008; Quintales et al. 2015a).
The observation that sequences organized in positioned nucleosomes in a species do not maintain the same organization when integrated in their orthologous loci in a different species (Fig. 7) raises the question of how exogenous sequences (or endogenous randomized sequences) are distinguished from the native endogenous ones. It has been recently reported that histones from different organisms have different affinities for the same DNA molecules (Allan et al. 2013). These authors suggested that different histone octamers might have been adapted through evolution to pack genomes that differ widely in base composition, size, gene density, and other structural and functional properties. Histone amino acid identity among the yeasts that we have studied ranges from 96.5% to 99.0% in S. octosporus, 94.7% to 98.5% in S. japonicus, and 83.9% to 92.6% in S. cerevisiae, relative to S. pombe. These differences are consistent with the phylogenetic distance of 119, 221, and 350 million years of S. octosporus, S. japonicus and S. cerevisiae, respectively, relative to S. pombe (Rhind et al. 2011; Hoffman et al. 2015) and with the fact that the nucleosomal signatures are more similar between S. octosporus and S. pombe than between S. japonicus and S. pombe (Fig. 5; Quintales et al. 2015a). It is currently unknown whether these small differences in the histone octamer could be responsible for the different affinity of nucleosomes of different organisms for the same DNA sequence or whether other factors like histone concentration, the linker-to-core histone ratio, or other thermodynamic or biophysical parameters could be involved (Beshnova et al. 2014). A parameter that might affect the different nucleosomal positioning of the same sequences in S. pombe and S. cerevisae is the absence or presence of histone H1 and an internucleosomal repeat length of 154 bp or 167 bp, respectively (Lantermann et al. 2010). Different sequence requirements between the two yeasts are also present at nucleosome-depleted regions in promoters. They are enriched in poly dA:dT elements in S. cerevisiae (Iyer and Struhl 1995; Zhang et al. 2011b), whereas they are not in S. pombe (Lantermann et al. 2010). In fact, the A + T content of these regions is lower than the intergenic average in S. pombe (de Castro et al. 2012). Chromatin assembly experiments have shown that the in vivo positioning pattern at the 5′ end of genes of S. cerevisiae can be reconstituted in the presence of ATP-dependent trans-acting factors, suggesting that the DNA sequence would play a minor role in nucleosome positioning (Zhang et al. 2011b). While it is possible that ATP-dependent remodelers play a dominant role in that system, it is conceivable that the various factors contributing to positioning (see the Introduction) could have a different relative weight under diverse experimental conditions, across species, and even in different regions of the same genome (Beshnova et al. 2014).
The information contained in nucleosomal signatures is degenerated, can accommodate a great variety of sequences with similar positioning potential (Fig. 6), and is also redundantly distributed along mononucleosomal DNA (Fig. 4). The combination of degeneracy and redundancy makes it possible that a great variety of sequences can contribute to nucleosome positioning in the genome. Other matrices have been derived from mononucleosomal DNA based on a probabilistic nucleosome-DNA interaction model (Segal et al. 2006) or on the concatenation of a 12-bp consensus palindromic motif derived from Caenorhabditis elegans mononucleosomal DNA (Gabdank et al. 2009). These matrices and other computational tools have been used to predict nucleosome positioning on genomic DNA sequences (revised in Teif 2016) whose predictive power varies widely (Liu et al. 2014). We have used nucleosomal signature matrices to design sequences capable of inducing nucleosome positioning in vivo, but we have not tested their potential to predict nucleosomal positioning in different species. The fact that they position nucleosomes in a species-specific manner (Figs. 6⇑–8), together with the differences among matrices (Fig. 5), suggests that they would probably not perform very well as positioning predictors across species.
The potential of nucleosomal signatures to customize nucleosome positioning in coding and noncoding sequences, together with the design of promoters of variable strength, based on their capacity to position or exclude nucleosomes (Curran et al. 2014), opens up the possibility of incorporating this information into the design of synthetic genomes (Annaluru et al. 2014; Haimovich et al. 2015). On a different scale, it will be worth exploring whether the engineering of exogenous sequences to mimic the endogenous nucleosomal pattern of eukaryotic hosts has the potential to improve the expression, maintenance, or stability of genes and vectors of biotechnological interest.
Methods
Yeast strains and growth conditions
The S. pombe 972 h- and h- leu 1-32 ura4 DS-E (harboring an internal deletion of the ura4 ORF) and S. cerevisiae aw303-1a (MATa ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100) strains were transformed with the appropriate DNA fragments to generate all the mutants used in this work. Cell culture conditions are described in Supplemental Methods.
Synthesis of modified DNA sequences and yeast transformation
All modified coding and noncoding DNA molecules used in this work were synthesized by GeneArt (Life Technologies) and GeneWiz. S. pombe ura4 DS-E cells were transformed by electroporation with modified versions of the ura4 ORF (Figs. 1, 4, 7) flanked by recombination cassettes. Transformant colonies were directly selected in minimal medium. S. pombe 972 h- cells transformed with constructs different from the ura4 gene (Figs. 2, 3, 6, 8) were ligated to the kan (kanamycin resistance) gene, and transformants were selected on rich medium plates containing 100 µg/mL of G-418 antibiotic (except 25 µg/mL in Fig. 8A, where the analyzed wild-type and modified ORF was that of the kan gene). A control strain for each mutant strain was constructed by targeting the kan gene alone to the same loci in S. pombe 972 h- cells. S. cerevisiae aw303-1a cells were transformed by the lithium acetate protocol, and transformants were selected in plates containing 100 µg/mL of G-418. Correct integration in the targeted loci in all transformants was monitored by PCR or by standard DNA sequencing.
Chromatin immunoprecipitation and qPCR
ChIP analysis in Supplemental Figure S5 was performed as described in Supplemental Methods.
Digestion with MNase, indirect end-labeling, and preparation of mononucleosomal DNA
Exponential cultures of S. pombe and S. cerevisiae were processed as described in Supplemental Methods.
Next-generation sequencing and DANPOS analysis
Libraries of mononucleosomal DNA were constructed following the Illumina protocol and were sequenced in an Illumina NextSeq500 platform using the paired-end protocol.
We generated between 18 and 58 million reads per experiment, representing 186- to 582-fold genome coverage. Reads were aligned using Bowtie (Langmead et al. 2009) to the S. pombe genome (ASM294v2.20 assembly 13/08/2013 from PomBase), S. cereviase (SacCer 3), or to genome versions where specific wild-type sequences were replaced by their respective modified versions (Supplemental Fig. S1). Alignment files were processed using the NUCwave algorithm (Quintales et al. 2015b) to generate the nucleosome occupancy maps. Altogether, we generated duplicates of 23 MNase-seq maps corresponding to the mutants described in the text. The DANPOS 2 application was used to calculate the difference in fuzziness between specific regions using the dpos utility with a span of 1 bp and a read extension of 50 bp to make it compatible with NUCwave maps (Chen et al. 2013a).
Generation of the position-specific weight matrix
Nucleosomal signatures were defined by the asymmetrical and palindromic distribution of the four nucleotides along 38154,
46120, 27024, and 34526 mononucleosomal DNA sequences of S. pombe, S. octosporus, S. japonicus and S. cerevisiae, respectively, aligned to their central (dyad) position (Quintales et al. 2015a). The dimension of the position-specific weight matrix (PSWM) is 16 (dinucleotides) × 150 (positions along mononucleosomal
DNA). Values for each position (i) and for each dinucleotide (NN) are calculated according to the expression
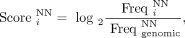 where
where 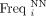 is the frequency of the NN dinucleotide at position i in the group of aligned sequences, and
is the frequency of the NN dinucleotide at position i in the group of aligned sequences, and 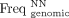 is the average genomic frequency of the same dinucleotide. Each of the 2384 (16 × 149) elements of the matrix represents
the score for each dinucleotide depending on their position along mononucleosomal DNA. Each species generates a different
PSWM depending on their different nucleosomal signatures. The genomic GC content of S. pombe, S. octosporus, S. japonicus and S. cerevisiae is 36.1%, 38.8%, 41.2%, and 39.7%. Despite this relatively similar composition, the distribution of dinucleotides (Fig. 5), mononucleotides, trinucleotides, and A + T profile (Quintales et al. 2015a) is very different among the four species. Maps of nucleosomal occupancy generated using different methods like MNase digestion
or chemical cleavage of DNA at the dyad position are very similar (Brogaard et al. 2012; Moyle-Heyrman et al. 2013; Lieleg et al. 2015; Quintales et al. 2015a, b). This implies that matrices derived from well-positioned nucleosomes from different experiments will probably be very similar
to those described in this work.
is the average genomic frequency of the same dinucleotide. Each of the 2384 (16 × 149) elements of the matrix represents
the score for each dinucleotide depending on their position along mononucleosomal DNA. Each species generates a different
PSWM depending on their different nucleosomal signatures. The genomic GC content of S. pombe, S. octosporus, S. japonicus and S. cerevisiae is 36.1%, 38.8%, 41.2%, and 39.7%. Despite this relatively similar composition, the distribution of dinucleotides (Fig. 5), mononucleotides, trinucleotides, and A + T profile (Quintales et al. 2015a) is very different among the four species. Maps of nucleosomal occupancy generated using different methods like MNase digestion
or chemical cleavage of DNA at the dyad position are very similar (Brogaard et al. 2012; Moyle-Heyrman et al. 2013; Lieleg et al. 2015; Quintales et al. 2015a, b). This implies that matrices derived from well-positioned nucleosomes from different experiments will probably be very similar
to those described in this work.
Design of sequences for nucleosome positioning on synthetic noncoding DNA molecules
To incorporate the information from S. pombe nucleosomal signatures in the noncoding synthetic sequences in Figure 6, we generated six random sequences 153 bp long (147-bp core DNA plus 6-bp linker) (Lantermann et al. 2010) with a 36% average G + C content. In the case of S. cerevisiae, the six sequences were 168 bp long (147-bp core DNA plus 21-bp linker) (Lantermann et al. 2010) and had a 38% G + C content. We subjected these individual sequences to reiterate cycles of random single-point mutation and selected the resulting sequences after each cycle if they had a higher score in the position-specific weight matrix) than in the previous cycle. Since unlimited reiteration would generate six identical mononucleosomal sequences, we repeated the process while the average identity between them was not significantly higher than the 25% average identity between individual genomic mononucleosomal sequences.
Integration of nucleosomal signatures into ORF sequences by codon substitution
To reproduce the nucleosomal pattern of an endogenous ORF in an unrelated ORF of the same size in S. pombe, we identified the midposition of any of the well-positioned endogenous nucleosomes. From this coordinate, we considered that the midposition of flanking nucleosomes in S. pombe would be at a distance equivalent to multiples of 153 bp, as described in the previous section. In a second step, for each codon along mononucleosomal DNA, we selected the synonymous codon with the highest score in the S. pombe PSWM at the corresponding position. The same protocol was applied to S. cerevisiae, but in this case, the distance between nucleosomal dyads was 168 bp and the S. cerevisiae PSWM was used as a reference. We did not modify wild-type codons corresponding to linker sequences in either yeast. Since none of the ORFs replaced in S. pombe (ura4 in Fig. 7 and SPBC16G5.03 in Fig. 8) and in S. cerevisiae (YKL007W in Fig. 8) encompassed an integer number of nucleosomes, we maintained the ATG and STOP codons and modified only the codons included in the ORF. Wild-type codons in the remaining 150 bp of the two mononucleosomal DNAs including the two ends of the ORFs were not modified.
Data access
All genomic sequencing data generated for this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE84910.
Acknowledgments
We thank Emma Keck for helpful revision of the manuscript. This work was funded by grant BFU2014-52143-P from the Spanish Ministerio de Economía y Competitividad (MINECO).
Author contributions: S.G., A.G., and R.S. performed the end-labeling chromatin analyses. S.G., E.V., A.G., and M.S. carried out the MNase-seq experiments. E.V. carried out the bioinformatic and DANPOS analyses of the MNase-seq experiments. L.Q. carried out computational analyses and generated the PSWM. F.A. supervised the general strategy of the work, analyzed data, and wrote the article. All authors contributed to the designing of the experiments, analyzed data, and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.207241.116.
-
Freely available online through the Genome Research Open Access option.
- Received March 18, 2016.
- Accepted September 19, 2016.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








