
The genetic basis for individual differences in mRNA splicing and APOBEC1 editing activity in murine macrophages
Abstract
Alternative splicing and mRNA editing are known to contribute to transcriptome diversity. Although alternative splicing is pervasive and contributes to a variety of pathologies, including cancer, the genetic context for individual differences in isoform usage is still evolving. Similarly, although mRNA editing is ubiquitous and associated with important biological processes such as intracellular viral replication and cancer development, individual variations in mRNA editing and the genetic transmissibility of mRNA editing are equivocal. Here, we have used linkage analysis to show that both mRNA editing and alternative splicing are regulated by the macrophage genetic background and environmental cues. We show that distinct loci, potentially harboring variable splice factors, regulate the splicing of multiple transcripts. Additionally, we show that individual genetic variability at the Apobec1 locus results in differential rates of C-to-U(T) editing in murine macrophages; with mouse strains expressing mostly a truncated alternative transcript isoform of Apobec1 exhibiting lower rates of editing. As a proof of concept, we have used linkage analysis to identify 36 high-confidence novel edited sites. These results provide a novel and complementary method that can be used to identify C-to-U editing sites in individuals segregating at specific loci and show that, beyond DNA sequence and structural changes, differential isoform usage and mRNA editing can contribute to intra-species genomic and phenotypic diversity.
Splicing is an obligatory step in the processing of both coding and noncoding RNA precursors (pre-RNA) to mature RNA, and is executed by a ribonucleoprotein megaparticle known as the spliceosome. Even though the sequential assembly of the spliceosome (Kornblihtt et al. 2013) can occur around any splice site that consists of consensus sequences recognized by the spliceosomal components, splice sites that conform better to a consensus sequence are strongly recognized and favored by the spliceosome complex. Besides the consensus sequences, the selection of optimal splice sites is regulated, in part, by a minimal set of conserved cis-acting GU and AG sequences at the 5′ and 3′ splice sites, respectively (Wang et al. 2008), and sequence elements known as intronic/exonic splicing enhancers and silencers (Wang and Burge 2008; Lee et al. 2012). As such, genetic variation at the splice site, in the consensus sequences, or in the intronic/exonic enhancers and repressors may lead to alternative splicing (AS), resulting in alternative transcripts and protein isoforms from a single gene. AS is known to affect biological processes such as cell death, pluripotency (Irimia and Blencowe 2012; Han et al. 2013), and tumor progression (Venables et al. 2009), and can also contribute to phenotypic diversity within species. Indeed, independent studies have recently revealed that AS may supersede variable gene expression in determining human phenotypic differences (Heinzen et al. 2008). For example, individual differences in the splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) affect the efficacy of statins (Medina and Krauss 2009), while induced exon skipping in the dystrophin gene is being explored for Duchenne muscle dystrophy therapy (Mitrpant et al. 2009). Therefore, the identification of genetic variants that modulate splicing may result in a better understanding of the genetic basis for individual differences in susceptibility to disease.
In addition to AS, biological systems have evolved additional mechanisms that alter the fidelity of the information coded in DNA and RNAs, thereby increasing genome diversity. One such mechanism is DNA and RNA editing (Gray 2012). Since its discovery in trypanosomatids (Benne et al. 1986), editing has been found in Drosophila (Rodriguez et al. 2012), humans (Fritz et al. 2013), and mice (Rosenberg et al. 2011). Editing is generally catalyzed by cytidine deaminase or adenosine deaminase enzymes (Hamilton et al. 2010). Broadly, the cytidine deaminase family of enzymes includes the activation-induced cytidine deaminase (AID or AICDA) and the apolipoprotein B mRNA editing enzyme, catalytic polypeptides (APOBECs). Even though related, AICDA has been shown to primarily deaminate cytidine to thymidine in DNA and is important in antibody diversification processes (Fritz et al. 2013), while APOBECs can edit both DNA and RNA, converting cytidine to uridine in RNA or to thymidine in DNA. The APOBEC deaminase family includes APOBEC1, APOBEC2, APOBEC3, and APOBEC4, with APOBEC3 reported to have various isoforms in humans (A3A, A3B, A3C, A3D, A3F, A3G, and A3H). No biological function for APOBEC2 and 4 has been reported, while APOBEC1 and 3 have been implicated in cancer development (Fritz et al. 2013; Roberts et al. 2013) and inhibition of viral replication (Niewiadomska and Yu 2009; Krisko et al. 2013). However, the most common form of editing in higher eukaryotes is catalyzed by the adenosine deaminase acting on RNA (ADAR) enzyme and involves the deamination of adenosine to inosine (Athanasiadis et al. 2004; Kim et al. 2004; Levanon et al. 2004; Li et al. 2011), which is then recognized as guanosine by the RNA translation machinery. The mammalian genome encodes three Adar genes (Adar, Adarb1, and Adarb2), whose proteins share a common variable N-terminal mRNA binding and a C-terminal catalytic deaminase domain (Melcher et al. 1996; Valente and Nishikura 2005). Even though both ADAR and ADARB1 are catalytically active (Nishikura 2010), ADAR is the best characterized, while the function of ADARB2 is unclear (Chen et al. 2000). Like AICDA and APOBEC, ADAR-catalyzed editing has been implicated in a variety of biological processes, including AS (Solomon et al. 2013), viral replication (Doria et al. 2009), and immune development (Hartner et al. 2009). Similarly to mRNA splicing, the determination of which cytidine or adenosine to deaminate is guided by consensus sequences, proximal to the edited nucleotide, which are then recognized by the mRNA binding motifs in the editing enzymes (Anant and Davidson 2000; Rosenberg et al. 2011; Bahn et al. 2012). Therefore, like AS, genetic variation in the editing mooring sequence or in the mRNA binding site of the editing enzyme may affect the rate of editing between and within species.
Even though mRNA splicing and editing have been implicated in a variety of biological processes and potentially contribute to phenotypic diversity within species, individual differences in, and the genetic transmissibility of, mRNA splicing and editing are still ambiguous. Individually or together, AS and mRNA editing contribute to define macrophage biology and are important in the response to various stimuli. For instance, macrophage activation with endotoxin or interferon gamma (IFNG) has previously been shown to result in increased ADAR editing activity (Rabinovici et al. 2001), while APOBEC3A expression has been implicated in the differential resistance of monocytes and macrophages to HIV-1 (Peng et al. 2007). Here, we hypothesized that the macrophage genetic background and environmental milieu modulate the rate of mRNA splicing and editing. To test this, we investigated isoform usage and the rate of mRNA editing in bone marrow–derived macrophages (BMDMs) obtained from the laboratory inbred A/J and C57BL/6J mice and from AXB and BXA recombinant inbred (RI) mouse strains before and after stimulation with a variety of stimuli, including IFNG and the intracellular pathogen Toxoplasma gondii. We show that both isoform usage and the rate of mRNA editing are quantitative traits influenced by the macrophage genetic background and environment.
Results
Macrophage genetic background and environmental milieu modulate isoform usage
The widely used laboratory inbred A/J (AJ) and C57BL/6J (B6) mice display variable susceptibility to a variety of infectious agents—such as Staphylococcus aureus, T. gondii, and Trypanosoma cruzi (McLeod et al. 1989; Parekh et al. 1998; Ahn et al. 2010; Silva et al. 2013)—and inflammatory pathologies such as atherosclerosis, obesity, and type 2 diabetes (Paigen et al. 1985; Surwit et al. 1988; Surwit et al. 1995). These phenotypic differences often replicate in the AXB/BXA RI mice, derived from an initial reciprocal cross of AJ and B6 mice followed by multiple rounds (≥20) of inbreeding (Nesbitt and Skamene 1984; Marshall et al. 1992). Importantly, the RI mice have stable genomes that are homozygous for virtually every polymorphic parental allele and have been useful for mapping AJ and B6 genetic differences (McLeod et al. 1989; Matesic et al. 1999; Boyle and Gill 2001). Therefore, to investigate how the genetic background and stimulation conditions modulate isoform usage in macrophages, we performed high-throughput RNA-sequencing (RNA-seq) on BMDMs obtained from female AJ, B6, and 26 AXB/BXA mice (28 samples in total) before and after stimulation with IFNG/tumor necrosis factor alpha (TNF), inflammatory cytokines important in the immune response against most intracellular pathogens, or CpG, a synthetic double-stranded DNA that activates TLR9, or infection with T. gondii, an obligate intracellular apicomplexan parasite. Using Illumina HiSeq 2000, we generated at least 100 million paired-end reads from each of the 28 samples in each macrophage condition, except for the IFNG/TNF-stimulated macrophages that were sequenced on a single end.
Initially, we aligned the RNA-seq reads to the mouse reference genome (mm9, NCBI build 37.2 downloaded from Illumina iGenomes; http://cufflinks.cbcb.umd.edu/igenomes.html) using TopHat (Trapnell et al. 2009), which automatically integrates Bowtie (Langmead et al. 2009). However, because sequence polymorphisms between AJ and B6 can skew read mapping toward the reference allele, we made a synthetic reference genome in which all the polymorphic nucleotides between AJ and B6 were converted to a neutral nucleotide (Degner et al. 2009). This way, if a polymorphism initially affected the mapping of reads obtained from AJ alleles to the reference (B6) genome, it would now affect reads from the B6 alleles as well, thereby eliminating read mapping bias. On average, 70% of reads in the individual samples uniquely mapped to the synthetic genome, which was on average 1% less than the number of reads mapped to the iGenome. Henceforth, unless otherwise stated, all the RNA-seq data used herein are based on alignment to the synthetic genome.
Because the expression of individual isoforms may be linked to the expression level of the parent transcript and not splicing per se, overall isoform expression levels may not be a good measure for differential isoform usage. Therefore, to measure isoform usage, we computed the ratio of reads supporting each splicing event, described as percent-spliced-in (PSI), using the mixture-of-isoforms (MISO) program (Katz et al. 2010) and a compendium of exon-centric and 3′ UTR alternative events (Wang et al. 2008; Hoque et al. 2013). MISO uses a Bayesian logic to model the generative process by which RNA-seq reads are produced from isoforms and generates confidence intervals (CIs) for estimates of exon and isoform abundance and a Bayes factor (BF), which can be used to estimate differential isoform usage between samples (for a detailed description of MISO, see Katz et al. 2010). Thus, treating each macrophage condition (nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and Toxoplasma-infected) as an independent cohort, we calculated the PSI values for each splicing event in each of the 28 samples. Of the 16,974 annotated exon-centric and 3′ UTR alternative events (Wang et al. 2008; Hoque et al. 2013), excluding alternative first and last exons, 9954, 9198, 10,604, and 9963 events had a PSI ≥ 0.2 (i.e., supported by at least 20% of the total reads originating from the constitutive and alternative exons) in at least five mice in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and Toxoplasma-infected macrophages, respectively. Out of the 9036 nonredundant genes with annotated exon-centric and 3′ UTR alternative events, 6646 genes had PSI ≥ 0.2, in more than five mice, in at least one macrophage condition.
Because the RI mice are homozygous at each polymorphic parental allele, and assuming there is no epistasis, we reasoned that if the macrophage genetic background modulates mRNA splicing, then isoform usage should vary in the RI mice and their progenitors. To investigate this, we used B6 as a reference and calculated the BF for each splicing event in each sample using MISO. To identify isoforms that are differentially used among mouse strains, we required that an event must have a BF ≥ 5 (i.e., the isoform is five times more likely to be differentially used) in at least five mice. The BF calculations and the filtering steps were performed separately for each macrophage condition. The requirement for five samples was due to the observation that at each of the 934 unique AXB/BXA genetic markers (Sampson et al. 1998; Williams et al. 2001), at least four RI mice carry the AJ or the B6 allele. In the end, we observed 2376, 961, 3067, and 787 events, from 1870, 821, 2309, and 678 nonredundant transcripts, in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and Toxoplasma-infected BMDMs, respectively, for which the relative isoform usage was determined by the macrophage genetic background (Supplemental Table 1A–D). This translates to a total of 4677 unique alternative events from all the macrophage conditions, of which 1719 were present in at least two macrophage conditions (Supplemental Table 1E). Included in this category were interleukin 3 receptor alpha (Il3ra), Apobec3, and C-type lectin domain family 7, member a (Clec7a) (also known as dectin-1) transcripts (Fig. 1), which independent studies have shown to be differentially spliced and to contribute to phenotypic diversity between several mouse strains, including AJ and B6 (Ichihara et al. 1995; Jimenez-A et al. 2008; Li et al. 2012).
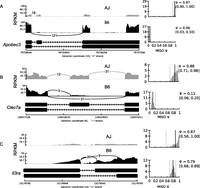
Isoform usage varies between laboratory mouse strains. We observed differential isoform usage (reflected by the different PSI values) of Apobec3 (A), Clec7a (B), and Il3ra (C) between the different mouse strains. Shown are the percent-splice-in (PSI, ψ) for the longest isoform for each gene, and the lower and upper 95% confidence intervals (CIs) (in parentheses). Also shown is the total number of reads supporting each splice junction. Due to the low, almost absent, expression of the alternative Il3ra isoform in the AJ, which is consistent with a previous observation (Ichihara et al. 1995), there are no reads supporting the alternative splice junction, which is also the reason for the large CI. The alternative isoforms for both Apobec3 and Clec7a are due to a skipped exon, while the Ilr3a alternative isoform is due to an alternative 3′ splice site (alternative acceptor).
Besides genetic variants, splicing can also be modulated by the physiological state of the cell, e.g., cancerous versus healthy cells or stimulated versus nonstimulated cells. If a splicing event is modulated by cellular homeostasis, irrespective of the macrophage genetic background, then we do not expect its isoform usage to vary among the mouse strains. Therefore, to investigate stimulus-induced differential isoform usage, we calculated the BF for each splicing event between matched mouse strains in the different macrophage conditions, e.g., to investigate differential usage of event “A” in the nonstimulated versus infected BMDM, we calculated the BF for event “A” in mouse strain “X” in the nonstimulated condition relative to event “A” in mouse strain “X” in the infected condition. In total, we observed 594 differentially used (BF ≥ 5 in at least five samples for each pairwise comparison) isoforms from all the possible pairwise macrophage condition comparisons (Supplemental Table 1F). For example, even though Samhd1 AS, in which the 14th exon is skipped, did not vary between the mouse strains, the IFNG/TNF-stimulated macrophages express more of the primary isoform, while Toxoplasma-infected macrophages express mostly the alternative isoform (Fig. 2). SAMHD1 is known to restrict intracellular HIV-1 virus replication by depleting cellular dNTP levels. Interestingly, the primary isoform is known to be stable and able to degrade dNTPs, while the alternative isoform is unstable and metabolically inactive (Welbourn et al. 2012).
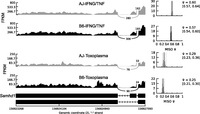
Stimulus-specific differential splicing. Even though isoform usage for Samhd1 was the same between the strains, we observed stimulus-specific differential isoform usage for Samhd1 between IFNG/TNF-stimulated and infected BMDM. The Samhd1 event is due to the skipping of the 14th exon, and the alternative isoform is known to be metabolically unstable and catalytically (dNTPase activity) inactive. Shown are the PSI (ψ) for the longest isoform for each strain, and the lower and upper 95% CIs (in parentheses).
Thus, both the genetic background and the environmental milieu determine isoform usage in murine macrophages.
Distinct loci modulate isoform usage in murine macrophages
The results described above implicate the macrophage genetic background in mRNA splicing without revealing the dynamics of isoform usage in murine macrophages. Therefore, to identify the genetic factors that modulate differential isoform usage, we used the PSI values obtained from the RI mice as quantitative traits, and the corresponding genetic map (containing 934 informative genetic markers) in a genome-wide scan to identify the genomic loci that modulate mRNA splicing (splicing quantitative trait loci [sQTL]) using R/qtl (Broman et al. 2003). To correct the QTL P-value of individual isoforms for multiple testing at the 934 genetic markers, we used 1000 permutation tests linked across phenotypes, i.e., permutation on the rows of the phenotype matrix relative to the row of the genotype matrix (Burrage et al. 2010) in R/qtl. Next, to correct for testing of multiple isoforms, we calculated the corresponding false-discovery rate (FDR) using the qvalue package (Storey and Tibshirani 2003) and used FDR ≤ 10% to select significant sQTL. As in the previous section, all the linkage analysis and subsequent filtering steps were performed separately for each macrophage condition. In the end, we identified 205, 285, 395, and 268 significant sQTL in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and infected macrophages, respectively (Supplemental Table 1G–J). Therefore, in agreement with previous studies (Ichihara et al. 1995; Jimenez-A et al. 2008), we have shown, using Il3ra, Apobec3, and Clec7a as examples, that isoform usage is modulated by the mouse genetic background (Fig. 3A). Similarly, although not significantly associated with a genomic region, when averaged based on the genotype at the suggestive sQTL, the PSI for Samhd1, which is modulated entirely by the macrophage stimulation condition, was significantly variable between the relevant conditions (Fig. 3B). However, due to the mosaic introgression of AJ and B6 alleles in the RI genome, the complex regulation of mRNA splicing, and the few number of mice available in this RI line, linkage was not observed for all the splicing events modulated by the macrophage genetic background.
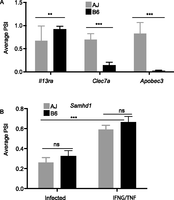
mRNA splicing is genetic. (A) The dependence of Apobec3, Clec7a, and Il3ra splicing on the macrophage genetic background is genetically transmissible and is revealed when the PSI values are averaged based on the genotype at the relevant splicing QTL (sQTL). (B) Conversely, the differential splicing of Samhd1 is stimulus-induced and is independent of the macrophage genetic background. Averaging the Samhd1 PSI values in the RI mice based on the genotype at the Samhd1 suggestive sQTL confirms that the splicing of Samhd1 is only variable between the stimuli and not the mouse strains. The AJ allele (AA) is presented as gray bars and the B6 allele (BB) as black bars (mean ± SD, [***] P < 0.001, Student's t-test)
AS can be due to variations at the splice site (cis) or to polymorphic or differentially expressed splice factors, enhancers, and repressors (trans) (Wang and Burge 2008; Lee et al. 2012; Zaphiropoulos 2012). Therefore, based on the distance from the corresponding splice site, the sQTL can be categorized as cis or trans. Thus, we computed sQTL distance from the relevant splice site, and using a genomic window of 10 Mb, we designated each sQTL as cis (≤10 Mb from splice site) or trans (>10 Mb from splice site) (Table 1; Supplemental Table 1G–J). Generally, cis-sQTL are expected to be due to structural or sequence variations in the vicinity of the splice site (Lee et al. 2012). Therefore, we inspected the significant sQTL for single nucleotide polymorphisms (SNPs) proximal to the splice site (≤250 bp from splice site) and observed that, unlike the trans-sQTL, significantly (Student's t-test, P < 0.05) more cis-sQTL had SNPs close to the splice site (Table 1; Supplemental Table 1G–J). A single splice factor, enhancer or repressor, can modulate the splicing of several events. In linkage analysis, we expect the sQTL for such events to colocalize to the same genomic region (trans-sQTL hotspots or trans-band), proximal to the common splicing regulator. Indeed we observed such trans-sQTL hotspots in the current study. For example, we observed a trans-band on chromosome 5 for the significant events in the IFNG/TNF-stimulated macrophages (Fig. 4). However, it is plausible that sQTL can colocalize by chance alone. Therefore, to eliminate this possibility, we used Bonferroni-corrected P-values and Poisson distribution to compute the number of trans-sQTL that need to colocalize in a 10-Mb genomic window to constitute a trans-band and found six, seven, six, and five for the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and infected macrophages, respectively. Subsequently, using these cutoffs, we identified three, four, four, and three trans-sQTL hotspots in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and infected macrophages, respectively (Supplemental Table 1G–J) that were functionally enriched for a variety of biological processes, including T-cell differentiation and RNA processing (Table 2). Overall, chromosome 5 contained most of the trans-bands, and a single region (80–118 Mb) not only contained the highest number of trans-sQTL in the IFNG/TNF-stimulated macrophages (66 trans-sQTL), but also was replicated in all the macrophage conditions. About 45 putative splice factors are physically located in this region of chromosome 5, out of which 31, 26, 30, 28 were expressed (average FPKM ≥ 5) in the nonstimulated, IFNG/TNF-stimulated, infected, and CpG-stimulated macrophages, respectively. Since all the macrophage conditions have a large trans-sQTL in this region, 21, 66, 13, and 28 in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and infected macrophages, respectively, either a differentially expressed or a polymorphic factor modulates the splicing of the transcripts with sQTL in this region.
Number of significant splicing QTL (sQTL) in the four macrophage conditions
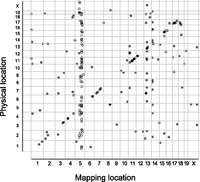
Linkage analysis of splicing events in RI mice reveals distinct trans splicing QTL (trans-sQTL). Shown is a representative of a trans-sQTL enrichment (trans-sQTL hotspot) on chromosome 5 in IFNG/TNF-stimulated macrophages. The linkage analysis was based on PSI values across 26 recombinant inbred AXB/BXA mice. QTL significance was estimated at adjusted P < 0.05 after 1000 permutation tests in R/qtl and FDR ≤ 10%. Each dot represents an individual event, and the circles indicate events within a 10-Mb window.
Large trans-splicing QTL hotspots (trans-bands), their functional enrichment, and putative regulators
A variation at the Apobec1 locus causes differential C-to-U mRNA editing
AS and mRNA editing are intricately linked (Lee et al. 1998; Laurencikiene et al. 2006; Solomon et al. 2013), and the differentially spliced genes, described above, included Apobec1, which encodes an important editing enzyme. Therefore, we postulated that the rate of editing might also be variable between the mouse strains. To test this, we used the RNA-seq data from all the 28 mouse strains to identify single nucleotide variants against the RefSeq genome (mm9, NCBI build37) using SAMtools/VarScan (Li 2011), followed by a filtering step, which removed all the known AJ and B6 SNPs (for details, see Methods; for an overview, see Supplemental Fig. 1). Because editing is relatively conserved within species (Danecek et al. 2012; Ramaswami et al. 2013), and to reduce variant artifacts due to sequencing errors, we required a putative edited nucleotide to be observed in at least seven of the 28 samples and to be supported by at least five independent RNA-seq reads. Additionally, since the RI mice are homozygous for almost all polymorphic AJ and B6 alleles, we excluded variants with more than two possible alleles. As in the previous sections, the macrophage conditions were treated as independent cohorts for these analyses, resulting in 6716, 3702, 17,898, and 6772 variants, of which 4176, 3631, 8623, and 3992 were either A-to-G and C-to-T or the possible reverse-strand T-to-C (Danecek et al. 2012) and G-to-A variants in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and Toxoplasma-infected macrophages, respectively. From all the four macrophage conditions, we observed 13929 nonredundant A-to-G or C-to-T transitions, of which 3886 were observed in at least two conditions (Supplemental Table 2A), i.e., reproduced in 14 independent samples considering the initial filter required an edited nucleotide to be present in at least seven samples in each macrophage condition. Probably due to the pervasive ADAR editing activity, 61%, 63%, 57%, and 61% of the variants in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and Toxoplasma-infected macrophages, respectively, were A-to-G or T-to-C, with most genes edited at multiple proximal sites (Danecek et al. 2012).
Previously, using RNA and DNA sequencing of wild-type and Apobec1 knockout mice, C-to-U editing was found to occur at the 3′ UTR of many transcripts (Rosenberg et al. 2011). Therefore, we computed the number of edited reads as a percentage of the total reads overlapping these previously confirmed 3′ UTR edited sites for a subset of genes expressed in murine macrophages—B2m, App, Tmbim6, and Sh3bgrl—in each of the 28 samples. Using the genotype at the Apobec1 sQTL to differentiate the mouse strains, we observed significantly (Student's t-test P < 0.05) lower rates of editing in macrophages from strains carrying the B6 Apobec1 allele compared to the AJ allele (Fig. 5). Investigation of Apobec1 isoform usage revealed that regardless of the stimulus, the B6 macrophages, relative to AJ, predominantly express the truncated Apobec1 isoform (Fig. 6A,B), suggesting that isoform usage may be the source of C-to-T editing variability among the mouse strains. However, we observed that the alternative Apobec1 isoform, which is due to an alternative acceptor site, did not result in a different protein isoform. Additionally, even though not statistically significant, the QTL modulating Apobec1 transcript expression (eQTL) colocalized with its sQTL on chromosome 6 (119.9 Mb NCBI mm9), proximal to its physical location (∼122 Mb NCBI mm9). Furthermore, Apobec1 expression was significantly lower (P < 0.05) in the BMDM from mice carrying the B6 allele at the Apobec1 sQTL, relative to AJ allele, regardless of the stimulus. Except for the IFNG/TNF-stimulated BMDM, which had lower Apobec1 expression levels, the expression level for Apobec1 was not significantly variable among the macrophage conditions (Fig. 6C). Similar to isoform usage and expression levels, APOBEC1 editing activity did not significantly vary with the macrophage microenvironment. An exception to this was the CpG-stimulated macrophages that, regardless of the mouse strain, exhibited lower rates of editing, even though these macrophages did not have the lowest Apobec1 FPKM or PSI values (Fig. 6B,C). Finally, currently there is no known nonsynonymous sequence or structural variation in Apobec1 between AJ and the B6 mice. As such, it is not clear if the differential isoform usage or expression of Apobec1 modulates its editing activity. However, if differential Apobec1 expression was regulating its editing activity, then we can expect the rate of editing to be lower in the IFNG/TNF-stimulated BMDM. Yet this is not the case; for example, even though the average expression of Apobec1 in IFNG/TNF-stimulated RI macrophages was significantly (Student's t-test P < 0.05) lower than in the nonstimulated macrophages, the rate of editing in these macrophages was not significantly different (Fig. 6D). Nevertheless, if we disregard the genotype at the Apobec1 sQTL, we found the rate of editing to correlate significantly (P < 0.05) with the Apobec1 PSI and FPKM, the latter being similar to an observation previously made in humans (Roberts et al. 2013). Therefore, through Apobec1 expression or isoform usage or both, a variation at the Apobec1 locus modulates the variable rate of C-to-U editing, although the fact that Apobec1 PSI is significantly mapped to the genome while the FPKM is not makes isoform usage a strong candidate.
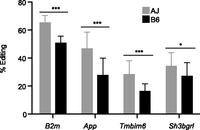
The rate of APOBEC1-mediated editing is dependent on the mouse genetic background. We observed strain-specific variable rates of editing at previously confirmed APOBEC1-edited sites in a set of genes expressed in macrophages. Using the genotype at the Apobec1 sQTL to distinguish the RI mice followed by averaging the rate of editing in mice with the same genotype (∼13 for each genotype), we show that the rate of editing at these previously identified sites varies between macrophages with the AJ (gray) and B6 (black) genotypes. All the genes, except App, were reported to have a single APOBEC1 edited site. For App, only one site supported by the most number of reads was considered (mean ± SD, [***] P < 0.001, [*] P < 0.05, Student's t-test).
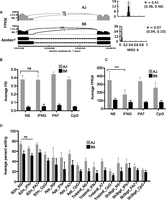
Apobec1 isoform usage and editing activity is variable between mouse strains. (A) Isoform usage for Apobec1 varies between the mouse strains, independent of the stimulus, which is confirmed by averaging the PSI values in the RI mice based on the genotype at the Apobec1 sQTL (B). (C) Like the isoform usage, the Apobec1 mRNA level is variable between the mouse strains, independent of the stimulus. (D) The rate of editing does not significantly vary between macrophage conditions, except for CpG-stimulated macrophages, which generally have lower rates of editing. The average PSI or FPKM values for RI lines having the AJ Apobec1 sQTL allele is represented by the gray bars and B6 allele by the black bars (mean ± SD, [ns] not significant, [***] P < 0.05, Student's t-test).
Previously, C-to-T editing was observed in human B cells even though APOBEC1 and its complementation factor (A1CF) transcripts were not detected in these cells (Li et al. 2011), and non A-to-G (or non T-to-C) transitions observed in different mouse strains were considered artifacts (Danecek et al. 2012). Therefore, to further validate and confirm the genetic transmissibility of the C-to-T editing observed here, we reasoned that if Apobec1 editing activity is genetic and determined by a variant at its locus, as suggested above, then in linkage analysis, the rate of editing at individual sites should map to the Apobec1 locus on chromosome 6 (∼122 Mb NCBI build37). This hypothesis is anchored on the observation that C-to-T transitions do not occur in Apobec1 knockout mice (Rosenberg et al. 2011) and the observation of C-to-T transitions, in the present study, despite the nondetectable expression of A1cf (FPKM = 0), both of which suggest a dominant role for Apobec1 in C-to-T transitions. Therefore, as proof-of-concept, we used the rate of editing at the previously confirmed APOBEC1 edited sites (Rosenberg et al. 2011), in the same subset of genes described above, to identify the genomic loci that modulate the rate of C-to-U mRNA editing (editing QTL or edQTL) in murine macrophages, using R/qtl. As expected, the rate of editing at the previously identified APOBEC1 edited sites (Rosenberg et al. 2011), in genes expressed in macrophages (FPKM ≥ 100), mapped significantly to the Apobec1 locus and overlapped perfectly with the Apobec1 sQTL (Fig. 7; Supplemental Table 2B–E). Next, we extended this concept to include all the edited sites, described above, separately for all macrophage conditions. Because we observed in the preliminary linkage analysis that the rate of editing in genes with average FPKM < 100 did not significantly map to the Apobec1 sQTL, we introduced a parsimonious filtering step which required that the edited transcript must be highly expressed (FPKM ≥ 100). At FDR ≤ 10%, we observed 63, 119, 138, and 116 edQTL in the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and infected macrophages, respectively (Supplemental Table 2B–E). Additionally, because the genotype at Apobec1 sQTL determines the rate of editing, to identify novel C-to-T edQTL we restricted the APOBEC1 edited sites to those with edQTL ≤ 10 Mb from the Apobec1 sQTL. All the variants that significantly mapped within 10 Mb at either side of the Apobec1 sQTL locus (chr6: 109.9–129.9 Mb) were either C-to-T or G-to-A (Supplemental Table 2B–E). This observation was true even when we dropped the average FPKM ≥ 100 requirement. In the end, from all the macrophage conditions, we identified a total of 62 C-to-T edited sites that significantly mapped to the Apobec1 sQTL, of which 51 (36 nonredundant) were not reported by Rosenberg et al. (2011) and hence were considered novel sites (Supplemental Table 2F). Although not intentionally filtered for, we found that 92% (47/51) of the novel edited sites were located at the 3′ UTR, the preferred site for APOBEC1 editing activity, and all the edited sites were flanked on both sides by either an adenosine or thymidine nucleotide (Rosenberg et al. 2011). Using Sanger sequencing, we confirmed editing of three randomly selected novel sites. In addition, using the same three novel sites and one previously confirmed site, we confirmed that the rate of editing is variable in the classical laboratory AJ and B6 mice (Fig. 8). Thus, linkage analysis can be complementary to other methods in identifying novel sites in genes edited by APOBEC1.
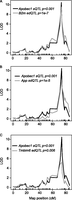
APOBEC1-mediated mRNA editing is genetic. Using linkage analysis and the rate of editing at previously confirmed sites on the 3′ UTR of B2m (A), App (B), and Tmbim6 (C) in murine macrophages, we show that APOBEC1-mediated editing is genetic and linked to the same genetic marker as the Apobec1 sQTL on chromosome 6. The Apobec1 sQTL is shown in black, and the edQTL for each gene is indicated in gray. While significant sQTL were identified at adjusted P < 0.05 after 1000 permutations in R/qtl, we show on the plots the actual adjusted P-values for each edQTL and the Apobec1 sQTL.
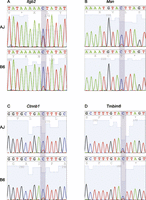
Validation of novel sites and the strain variability of C-to-T editing. Representative examples of Sanger sequencing chromatograms for editing of Itgb2, at chr10:77028356 (A); Msn, at chrX:93363499 (B); and Ctnnb1, at chr9:120869190 (C) in AJ and B6 macrophages. (D) Also shown is a previously confirmed edited site in Tmbim6 at chr15:99239051, which we used, together with the novel sites, to show the variability (chromatogram height for the nonreference base) of APOBEC1 editing activity in AJ and B6 macrophages.
Discussion
AS and mRNA editing, pervasive in eukaryotic transcripts, are known to contribute to a variety of mammalian phenotypes (Caceres and Kornblihtt 2002; Dhir and Buratti 2010; Gee et al. 2011; Burns et al. 2013). However, despite the unequivocal characterization of the cellular factors that mediate these events, the genetic features that delimit individual differences for these tightly regulated mRNA biogenesis processes are largely understudied. In the current study, we used the fraction of reads supporting constitutive and alternative exons to establish the genetic basis for AS in murine macrophages. Further, we used the genetic variation at the Apobec1 locus to demonstrate the variability and genetic basis of C-to-T editing in classical inbred laboratory mice. We have ensured reproducibility by performing this study, separately, on four different macrophage conditions, each containing 26 RI mouse strains and their two progenitors (a total of 112 samples), and using a stringent filtering step requiring an event to be observable in at least five (for isoform usage) or seven (for mRNA editing) mouse strains.
AS has been linked to various human phenotypes (Lee et al. 2012; Braunschweig et al. 2013). In the present study, we detected significant linkage between murine genetic variants and AS events for various genes, including Apobec3, Il3ra, Clec7a, and Apobec1, congruent with previous studies that demonstrated association between individual AS events and SNPs in the human genome (Cartegni et al. 2002; Narla et al. 2005; Lee et al. 2012). AS can result in nonfunctional protein isoforms (Lango Allen et al. 2010), which may alter cellular physiological states (David et al. 2010; Gao and Cooper 2013). However, investigating differential isoform usage in genetically and phenotypically divergent individuals is still uncommon. Therefore, the use of AJ and B6 mice and the corresponding AXB/BXA RI progenies, known to diverge for more than 30 disease phenotypes (Mu et al. 1993), is likely to illuminate the influence of AS in murine phenotypic differences. For example, differential splicing of Il3ra, Apobec3, and Clec7a is reported to contribute to a variety of phenotypes (Jimenez-A et al. 2008; Li et al. 2012; Stier and Spindler 2012), and we have demonstrated that isoform usage for these genes is genetically transmissible. Additionally, the observation that Toxoplasma, which is an opportunistic pathogen in HIV-infected patients, induces the expression of an unstable and nonfunctional isoform of SAMHD1, known to restrict HIV-1 replication by limiting cellular levels of dNTP (Welbourn et al. 2012), may have implication in macrophage response to Toxoplasma or exacerbate HIV replication in coinfected HIV patients. This possibility is reinforced by the fact that while SAMHD1 depletes intracellular dNTPs (Welbourn et al. 2012), Toxoplasma is dependent on host cellular nucleotides for optimal growth (Yu et al. 2009). Further experiments are thus needed to test these hypotheses. Besides genetic variations, epigenetic modifications, such as DNA methylation, are known to regulate mRNA splicing (Malousi and Kouidou 2012; Gelfman et al. 2013). It is thus conceivable that by limiting analysis to genetic variants in the current study, we have excluded splicing events modulated through epigenetic memory. In addition, due to the limited number of mice available in this RI line, it is possible that we have overlooked splicing events that are regulated by complex genetic interactions. Consequently, a comprehensive study, involving large cohorts and incorporating epigenetics, may reveal additional loci modulating splicing events in murine macrophages.
Like AS, mRNA editing is known to contribute to a variety of mammalian phenotypes (Jiang et al. 2013). However, to the best of our knowledge, the genetic basis and variability of mRNA editing in genetically diverse individuals is currently unknown. Here, we have shown that APOBEC1-editing activity is both variable and genetic in murine macrophages. While the consensus method for validating edited nucleotides has been the use of Sanger sequencing of both DNA and RNA, a method that relies solely on RNA-seq data has recently been published (Ramaswami et al. 2013). We have applied this method, together with other rigorous filters, to confirm known edited sites and identify novel sites. Although the use of linkage analysis to confirm edited sites is novel, it requires complete penetrance or dominance, preferably related to the editing enzyme. Editosomes composed of several genetic variants with small effects may confound linkage analysis, thereby hindering accurate validation of edited sites. This probably explains the lack of genetic coherence in the present study for ADAR editing activity. For instance, the editing activity of ADAR does not correlate with its expression (Jacobs et al. 2009; Wahlstedt et al. 2009), but with, among other factors, AS, self-editing, and sumoylation (Rueter et al. 1999; Desterro et al. 2005). Additionally, ADAR editing activity requires enhancers (Garncarz et al. 2013), which may complicate linkage analysis. Consistent with this complex regulation of ADAR editing activity, we did not observe a clear pattern of variability for the rate of A-to-G editing in the mouse strains, which is similar to an observation made in 15 mouse strains (Danecek et al. 2012), including the two progenitors used in the present study. Even though we observed significant edQTL for some A-to-G and T-to-C transitions, these did not colocalize to the Adar genomic locus (chr3: 89.53–89.55) or any other loci.
The colocalization of Apobec1 sQTL and eQTL proximal to its physical location has complicated the identification of the molecular basis for its differential editing activity. Compounding this further, is the observation that the alternative Apobec1 isoform, identified here, does not result in an alternative protein isoform. Even though a previous study reported a correlation between APOBEC expression and its editing activity in humans (Roberts et al. 2013), in the present study it appears that either or both Apobec1 isoform usage and expression modulate its editing activity. We can, however, conclude that a genetic variation at the Apobec1 locus, which we place upstream of both its splicing and expression, modulates its editing activity. It is worth noting that APOBEC1 editing activity studies in humans and mice have revealed some broad differences. For example, C-to-T editing occurs in humans even in the absence of APOBEC1 expression (Li et al. 2011; Roberts et al. 2013), while in mice the absence of Apobec1 abrogates C-to-T transitions (Rosenberg et al. 2011). Also worth noting is that, while this previous study (Roberts et al. 2013) investigated APOBEC editing activity in general, which includes all the different APOBEC gene variants in humans, in the present study, we looked specifically at edited sites with significant edQTL at the Apobec1 locus. It may well be that Apobec1 expression determines its editing activity and that there is a minimum expression threshold that once reached, i.e., in the IFNG/TNF-stimulated BMDM, maximal editing activity is achieved. However, we speculate that while the alternative splice acceptor site does not alter the protein sequence, it can alter the mRNA translation efficiency. A detailed investigation of both Apobec1 mRNA transcription and translation efficiency may provide further insight into how the different Apobec1 isoforms correlate with its editing activity.
Unlike for AICDA, APOBEC3, and ADAR, the biological significance for APOBEC1 editing activity is still equivocal. Although APOBEC1 was initially reported to edit C-to-U in the exon of Apob, it is now known that it binds mRNA at AU-rich templates (Navaratnam et al. 1995) and, from the present study and a previous study (Rosenberg et al. 2011), edits predominantly the 3′ UTR. However, the biological relevance of this predominant 3′ UTR editing is not clear. However, due to the observation that both APOBEC1 and miRNA bind the AU-rich templates at the 3′ UTR and that several miRNA-binding seeds overlap the APOBEC1 edited sites (Rosenberg et al. 2011), we can speculate on the importance of 3′ UTR editing on mRNA biogenesis. Because the 3′ UTR can influence post-transcriptional regulation of gene expression, by modulating mRNA localization, translation, and stability (Winter et al. 2008), one hypothesis is that 3′ UTR editing is biologically designed to selectively regulate these processes. To this end, although APOBEC1 has been shown to modulate intracellular viral replication mostly through the editing of viral DNA/RNA, it is possible that 3′ UTR editing of host mRNA indirectly affects viral pathogenesis. Particularly since some viruses, such as human cytomegalovirus, produce miRNA that target specific isoforms of host mRNAs at the 3′ UTR, thereby modulating various immune processes such as antigen processing and presentation (Kim et al. 2011), editing of these viral miRNA binding sites might inhibit viral miRNA binding. Additionally, some of the edited genes, including B2m and Itgb2, are known to function in immune processes. As such, the stability, localization, and translation efficiency of these transcripts may influence immune processes (Cogen and Moore 2009; Yee and Hamerman 2013). Collectively, the lowering of plasma LDL levels by the ectopic expression of Apobec1 (Teng et al. 1994; Greeve et al. 1996); the localization of plasma cholesterol level QTL to the Apobec1 locus (Mehrabian et al. 2005); and the observation that App, an APOBEC1-edited gene, regulates insulin secretion from pancreatic cells (Tu et al. 2012) suggests that APOBEC1 may be the causative gene for common physiological traits such as atherosclerosis and diet induced obesity.
Methods
RNA sequencing
BMDMs were plated in six-well plates (3 × 106 cells/well) overnight before stimulation or infection. For stimulation, IFNG (100 ng/mL)/TNF (100 ng/mL) or CpG (50 ng/mL) was added to each well for 18 h; while for the infected sample, a type II strain of T. gondii (Pru) was added to the confluent BMDM at an MOI of 1.3 for 8 h. Total RNA was isolated (Qiagen RNeasy Plus kit), and the integrity, size, and concentration of the RNA were checked (Agilent 2100 Bioanalyzer). The mRNA was then purified by polyA-tail enrichment (Dynabeads mRNA purification kit; Invitrogen), fragmented into 200–400 bp, and reverse transcribed into cDNA followed by sequencing adapter ligation at each end. Libraries were barcoded, multiplexed into four samples per sequencing lane in the Illumina HiSeq 2000, and sequenced from both ends (except for IFNG/TNF-stimulated samples, which were subjected to single-end sequencing), resulting in 40-bp reads after discarding the barcodes. Our preliminary RNA-seq experiments with infected BMDMs have shown that with four samples per lane, we still obtain enough read coverage for reliable gene expression analysis.
Alignment of reads to the genome
Reads were initially mapped to the mouse reference genome (mm9, NCBI build 37) and the Toxoplasma (ME49 v8.0) genome using Bowtie (2.0.2) (Langmead et al. 2009) and TopHat (v2.0.4) (Trapnell et al. 2009) in the n-alignment mode. Because the reference genome to which we mapped the RNA-seq reads is based on the C57BL/6J genomic sequence, and because of the known polymorphisms between the AJ and C57BL/6J, we suspected that sequence variations might introduce read-mapping biases. To mitigate this potential bias toward the reference allele, we created a copy of the mouse reference genome in which all the known single nucleotide polymorphisms (SNPs) between AJ and B6 (ftp://ftp-mouse.sanger.ac.uk/current_snps/) were converted to a third (neutral) nucleotide that is different from both the reference and AJ allele (Degner et al. 2009). However, this did not substantially change the average proportion of uniquely mapped reads in all the samples. In the end we used the mapping data generated from the synthetic genome.
Isoform usage
PSI values were calculated in MISO v.0.4.6 (Katz et al. 2010) in the single-end mode using the splicing event annotation files for major classes of alternative RNA processing downloaded from the MISO web page (http://genes.mit.edu/burgelab/miso/), based on the mm9 (NCBI buld37) genome assembly (parameters:–read-len 40, –overhang 8). PSI values were obtained in each sample for each macrophage condition. The different macrophage conditions were processed separately. BF was calculated in MISO for each splicing event and sample relative to the B6 sample in the corresponding macrophage condition; i.e., to get differential isoform usage of gene A in the nonstimulated condition, we obtained the BF for A in each of the 27 samples relative to the B6 sample in the nonstimulated macrophages. To estimate stimulus specific isoform usage, we calculated the BF for each event in the first condition relative to the corresponding sample in the second condition, for all the possible pairwise macrophage conditions; i.e., to measure differential isoform usage in B6 for the nonstimulated versus infected samples, we calculated the BF for gene A in B6 in the nonstimulated macrophages relative to gene A in B6 in the infected macrophages. Finally, we used custom Perl scripts to generate summary tables at 95% CI width. Differences between samples were deemed significant at |delta PSI| ≥ 0.2 between conditions, and BF ≥ 5.
Identification of edited transcripts
To identify edited mRNA, we processed the four macrophage conditions separately and followed a sequential procedure, which involved first retrieving sequence variation among RNA-seq reads using SAMtools 0.1.18 (r982:295) (Li et al. 2009) mpileup function across all strains, with parameters –f and –B using the reference C57BL/6J genome (mm9). The resulting mpileup files were post-processed using VarScan v2.2.11 (Koboldt et al. 2012) with the following parameters: mpileup2snp –min-coverage 5, –min-reads2 5, –min-avg-qual 15, –min-var-freq 0.01, –P-value 1, –variants 1. Only variants called in at least seven strains, called heterozygous in at least seven independent strains, and with a minor allele frequency of at least 10% across all strains, were retained. We used custom Perl scripts to filter the resulting positions against known sequence variation between C57BL/6J and A/J strains (ftp://ftp-mouse.sanger.ac.uk/current_snps/) and annotated against RefSeq sequence using BEDTools v.2.16.1 (Quinlan and Hall 2010). Finally, we used linkage analysis to confirm the novel edited sites, by treating the rate of editing as a quantitative trait and including for further analysis only genes that mapped to the of Apobec1 sQTL. This was validated by the location of editing QTLs for known APOBEC1 edited sites (Rosenberg et al. 2011)
Quantitative trait locus (QTL) mapping
To map QTLs, we obtained 934 informative AXB/BXA genetic markers from http://www.genenetwork.org. We then used the PSI values or the rate of editing in the AXB/BXA mice to perform genome-wide scans with genotype as main effect in R/qtl. Adjusted P-values at each QTL were calculated using 1000 permutations of the phenotype data as previously described (Churchill and Doerge 1994). Next, we calculated the corresponding qvalues for the genome-wide adjusted P-values in the qvalue package using the bootstrap method (Storey and Tibshirani 2003). QTLs were considered significant at FDR ≤ 0.1. To determine if the number of splicing events mapping in trans to a single genomic window, e.g., the chromosome 5 locus in the IFNG/TNF-stimulated macrophages, can be characterized as trans-sQTL hotspot, we used the Poisson distribution to compute the number of trans-sQTL that need to colocalize in a 10-Mb genomic window to constitute a trans-band and found six, seven, six, and five for the nonstimulated, IFNG/TNF-stimulated, CpG-stimulated, and infected macrophages, respectively. To do this, we divided the mouse mapped genome length, excluding the Y chromosome (since all the mice used here were female), into equal 10-Mb windows (258 windows). For each macrophage condition, using the observed number of trans-sQTL, we then calculated the number of trans-sQTLs that have to map to the same 10-Mb window to reach significance. First we calculated, for each condition, a Bonferroni-corrected P-value by dividing 0.05 (the genome-wide adjusted P < 0.05 used to identify significant sQTL) by the number of possible windows. We then used the Poisson distribution to calculate the minimum number of events that have to map to a single window to reach significance, using this Bonferroni-corrected P-value.
Sanger sequencing
To validate the novel C-to-T edited sites, we randomly selected three nonredundant novel edited sites, in three different genes, together with one previously confirmed edited site for Sanger sequencing. We made cDNA from the relevant mRNA samples using oligo-dT. Next, using primers designed to anneal at least 100 bp from either side of the edited nucleotide, we amplified the edited site using polymerase chain reaction (PCR). The PCR products were purified (QIAquick PCR Purification kit, Qiagen) and commercially sequenced (http://www.genewiz.com).
Data access
The RNA-seq data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE47540.
Acknowledgments
We thank the MIT BioMicroCenter for Illumina library preparation and sequencing. This work was supported by a NERCE developmental grant (U54-AI057159), National Institutes of Health (R01-AI080621), and by PEW Scholar in the Biomedical Sciences grants to J.P.J.S. M.A.H. is a Wellcome Trust-MIT postdoctoral fellow and K.D.C.J. was supported by a Cancer Research Institute Postdoctoral fellowship.
Footnotes
-
↵1 Corresponding author
E-mail jsaeij{at}mit.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.166033.113.
- Received August 31, 2013.
- Accepted November 13, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








