
Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin
- Nicole C. Riddle1,9,
- Aki Minoda2,9,
- Peter V. Kharchenko3,9,
- Artyom A. Alekseyenko4,
- Yuri B. Schwartz5,6,
- Michael Y. Tolstorukov3,
- Andrey A. Gorchakov4,
- Jacob D. Jaffe7,
- Cameron Kennedy2,
- Daniela Linder-Basso5,
- Sally E. Peach7,
- Gregory Shanower5,
- Haiyan Zheng8,
- Mitzi I. Kuroda4,
- Vincenzo Pirrotta5,
- Peter J. Park3,
- Sarah C.R. Elgin1,10 and
- Gary H. Karpen2,10
- 1 Department of Biology, Washington University St. Louis, Missouri 63130, USA;
- 2 Department of Molecular and Cell Biology, University of California at Berkeley and Department of Genome Dynamics, Lawrence Berkeley National Lab, Berkeley, California 94720, USA;
- 3 Center for Biomedical Informatics, Harvard Medical School and Informatics Program, Children's Hospital, Boston, Massachusetts 02115, USA;
- 4 Division of Genetics, Department of Medicine, Brigham & Women's Hospital, and Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 5 Department of Molecular Biology & Biochemistry, Rutgers University, Piscataway, New Jersey 08901, USA;
- 6 Department of Molecular Biology, Umea University, 90187 Umea, Sweden;
- 7 Proteomics Group, The Broad Institute, Cambridge, Massachusetts 02139, USA;
- 8 Biological Mass Spectrometry Resource, Center for Advanced Biotechnology and Medicine, University of Dentistry and Medicine of New Jersey, Piscataway, New Jersey 08854, USA
-
↵9 These authors contributed equally to this work.
Abstract
Eukaryotic genomes are packaged in two basic forms, euchromatin and heterochromatin. We have examined the composition and organization of Drosophila melanogaster heterochromatin in different cell types using ChIP-array analysis of histone modifications and chromosomal proteins. As anticipated, the pericentric heterochromatin and chromosome 4 are on average enriched for the “silencing” marks H3K9me2, H3K9me3, HP1a, and SU(VAR)3-9, and are generally depleted for marks associated with active transcription. The locations of the euchromatin–heterochromatin borders identified by these marks are similar in animal tissues and most cell lines, although the amount of heterochromatin is variable in some cell lines. Combinatorial analysis of chromatin patterns reveals distinct profiles for euchromatin, pericentric heterochromatin, and the 4th chromosome. Both silent and active protein-coding genes in heterochromatin display complex patterns of chromosomal proteins and histone modifications; a majority of the active genes exhibit both “activation” marks (e.g., H3K4me3 and H3K36me3) and “silencing” marks (e.g., H3K9me2 and HP1a). The hallmark of active genes in heterochromatic domains appears to be a loss of H3K9 methylation at the transcription start site. We also observe complex epigenomic profiles of intergenic regions, repeated transposable element (TE) sequences, and genes in the heterochromatic extensions. An unexpectedly large fraction of sequences in the euchromatic chromosome arms exhibits a heterochromatic chromatin signature, which differs in size, position, and impact on gene expression among cell types. We conclude that patterns of heterochromatin/euchromatin packaging show greater complexity and plasticity than anticipated. This comprehensive analysis provides a foundation for future studies of gene activity and chromosomal functions that are influenced by or dependent upon heterochromatin.
Two types of chromosomal regions are generally recognized in eukaryotic genomes, heterochromatin and euchromatin. Initially defined based on histological staining patterns in interphase cells (Heitz 1928), these subtypes are now known to represent distinct genomic and nuclear domains distinguished by a variety of properties including DNA sequence composition, gene density, replication timing, nuclear localization, frequency of meiotic recombination, and biochemical composition (for review, see Grewal and Elgin 2007; Eissenberg and Reuter 2009). Genomic studies generally focus on the euchromatin, which contains most of the genes. In addition, analyses of heterochromatin are challenging due to enrichment for repetitive sequences. Thus, although heterochromatin encodes essential structural and regulatory features such as centromeres, telomeres, and meiotic pairing sites (Allshire and Karpen 2008; Peng and Karpen 2008; Hughes et al. 2009), as well as several hundred genes (Smith et al. 2007b), its structure and organization remain poorly characterized.
At the core of chromatin are the histone proteins, which assemble DNA into nucleosomes, providing the basis for higher order chromatin packaging. A variety of post-translational histone modifications are used in combination to define alternative chromatin states (Jenuwein and Allis 2001; Ruthenburg et al. 2007; The modENCODE Consortium 2010; Kharchenko et al. 2011). Despite the complexity and the many organism-specific intricacies observed in the use of histone modifications, some common themes have emerged (Kouzarides 2007). For example, histone hyperacetylation, in general, and methylation of H3 lysine 4 (H3K4), in particular, are correlated with open chromatin conformations and gene expression; these “activation marks” are generally enriched in euchromatic regions. In contrast, heterochromatic regions generally display low levels of histone acetylation and H3K4 methylation, and instead are enriched for “silencing marks” such as H3 lysine 9 (H3K9) methylation (Kouzarides 2007; Eissenberg and Reuter 2009).
Heterochromatic domains are enriched for a number of specialized proteins implicated in epigenetic regulation, including those involved in deposition or recognition of specific histone modifications (Ruthenburg et al. 2007; Marmorstein and Trievel 2009). The first of such proteins identified was heterochromatin protein 1a [HP1a, also known as SU(VAR)205], which shows strong enrichment in the pericentric and telomeric regions of Drosophila melanogaster chromosomes (James and Elgin 1986; James et al. 1989) and binds di- and trimethylated H3K9 (Lachner et al. 2001). Similar enrichment patterns are observed for other proteins, including known histone-modifying enzymes [e.g., the SU(VAR)3-9 histone H3 K9 methyltransferase] and the SU(VAR)3-7 zinc finger protein (Cleard et al. 1997; Schotta et al. 2002). Mutations in such proteins cause defects in heterochromatin formation and associated gene silencing, while overexpression increases heterochromatin establishment, suggesting that these proteins directly participate in heterochromatin assembly and function (Eissenberg and Reuter 2009). The patterns of enrichment and depletion of histone modifications and of the proteins associated with epigenetic regulation can be used to distinguish chromatin domains across the genome and to assess chromatin changes that occur in different cell types.
The modENCODE project was initiated by NIH to provide a complete annotation of the functional elements in the C. elegans and D. melanogaster genomes (Celniker et al. 2009; Gerstein et al. 2010; The modENCODE Consortium 2010). We have used chromatin immunoprecipitation (ChIP) array analysis to define the genome-wide patterns of an extensive list of histone modifications and chromosomal proteins in D. melanogaster using chromatin from different cell culture lines, embryos, larvae, and adult heads. These data have provided an unprecedented opportunity to map the heterochromatin/euchromatin borders at high resolution, to ask whether this border is fixed or variable in different cell types, and to search for “facultative heterochromatin,” i.e., tissue-specific domains present in the euchromatic chromosome arms that display modifications typical of heterochromatin. We find that the organization and composition of Drosophila heterochromatin is surprisingly complex. Both single-copy and repeat-rich regions of the heterochromatin exhibit a mosaic of distinct chromatin signatures. Most striking is the packaging of active genes embedded within heterochromatin, which exhibit both “silencing” and “activation” marks, differentially distributed across different gene features. We observe some variability of heterochromatin/euchromatin border positions, as well as plasticity in the distributions and properties of facultative heterochromatin in different cell types. These findings demonstrate that heterochromatin contains more complex and plastic chromatin patterns than previously appreciated, which must be considered in any future analysis of heterochromatin assembly and function.
Results
Epigenomic borders between euchromatin and heterochromatin differ among cell types
By cytological criteria, about one-third of the D. melanogaster genome is heterochromatic, including pericentric regions, telomeres, and all of the 4th and Y chromosomes (Gatti and Pimpinelli 1992; Pimpinelli et al. 1995); the long arms of the X, 2nd, and 3rd chromosomes are euchromatic. There are three types of heterochromatic sequences in Release 5 of the D. melanogaster genome (Fig. 1A): (1) sequences assembled contiguously with the chromosome arms (“h”; e.g., 2Lh), (2) scaffolds not linked to the euchromatic sequences but mapped to a specific chromosome arm (“Het”; e.g., 2LHet), and (3) unmapped assemblies (arm U) (Hoskins et al. 2007; Smith et al. 2007b). The unique components of these sequences are included in the Affymetrix genome tiling arrays (version 2) used in our studies. Arm U and the Y chromosome sequences, respectively, were excluded from analysis, as they are predominantly repetitive and not well represented.
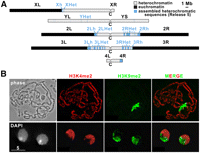
Chromatin marks define heterochromatin and euchromatin. (A) Diagram of the heterochromatic and euchromatic regions in the D. melanogaster genome. For each chromosome, euchromatin is represented in black, heterochromatin in light blue or light gray, and “C” is the centromere. The targets of this analysis, the assembled heterochromatic sequences in Release 5 of the D. melanogaster genome (h and Het regions), are indicated in light blue. (B) Euchromatin and heterochromatin occupy distinct genomic compartments. Polytene chromosomes from third instar larvae of Oregon R (wild type) (top) or interphase nuclei of S2 cells (bottom) were stained with antibodies specific for H3K4me2, which is enriched in euchromatic regions of the genome, and for H3K9me2, which is enriched in heterochromatin. (Left to right) Phase image (polytene) or DAPI staining (S2 cells); H3K4me2 staining (red); H3K9me2 (green); merged image of the signals for H3K4me2 (red) and H3K9me2 (green).
The borders between pericentric heterochromatin and euchromatin in the chromosome arms have been defined previously using cytogenomic methods (Fig. 1A; Hoskins et al. 2002, 2007). Here, we refine “cytogenomic” borders into “epigenomic” borders by mapping marks such as H3K4me2 and H3K9me2 by ChIP-array analysis (Fig. 2). As expected, the pericentric h and Het regions are enriched for classical “silent” marks (e.g., H3K9me2, H3K9me3, and HP1a) and depleted for “active” marks (e.g., H3K4me3) (Fig. 1B shows a cytological result; Fig. 2, the ChIP with microarray hybridization [ChIP-chip] result; additional marks are shown for BG3 cells in Supplemental Fig. 1). (The terms “active” and “silent” marks will be used in quotes, since these marks are not strictly associated with activation or silencing of expression.) It is of note that previous analysis concluded that Drosophila heterochromatin in third instar larvae contains high levels of H3K9me2, but not H3K9me3 (Ebert et al. 2004), which is in contrast to the high enrichment of H3K9me3 observed in mammalian heterochromatin (Peters et al. 2003). Here, using antibodies validated by Western and peptide blots, our analyses by ChIP-array and immunofluorescence show that H3K9me2 and H3K9me3 have, in fact, similar distributions and enrichments in the heterochromatin of Drosophila cells, as well as 3rd instar larvae. Furthermore, quantitative mass spectrometry analysis showed that H3K9me3 is only 1.7-fold less abundant in Drosophila cells compared with human cells (see Methods; Supplemental Fig. 2).
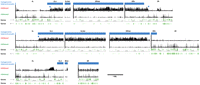
Chromatin marks define the epigenomic border between heterochromatin and euchromatin. Centromere-proximal euchromatin/heterochromatin borders were delineated based on ChIP-array data. Enrichments for H3K9me2 and H3K4me3 in 2–4-h embryos are shown for the centromere-proximal 3 Mb of chromosomes 2, 3, and X, as well as the distal portion of the 4th chromosome (1.35 Mb). The complete Het regions are shown also for chromosomes 2, 3, and X. Log intensity ratio values (y-axis) are plotted for each mark relative to the chromosomal position (x-axis). Boxes below the bar graph demarcate genomic regions with significant enrichment (0.1% false discovery rate [FDR]). Genes are shown in green below the ChIP-array data with their orientations as indicated by the arrows, and the cytogenomically defined heterochromatin is marked by a blue bar. The blue arrowheads indicate the positions of the epigenomic borders for chromosome arms 2L, 2R, and 3L. Patterns for multiple “silent” and “active” marks on chromosome arms 2R and 3L are shown in Supplemental Figure 1.
To define the epigenomic euchromatin–heterochromatin borders, we determined locations of sharp H3K9me2 transitions (Table 1), which were similar to the transitions for the other silent marks (Supplemental Fig. 1). The borders in 2–4-h embryos were located close to the previous cytogenomic borders for chromosome arms 2L, 2R, and 3L (within 300 kb) (Fig. 2; Table 1; Supplemental Fig. 1). However, no transition to H3K9me2 enrichment was observed for chromosome X and arm 3R, suggesting that in early embryos the currently available contiguous sequence for these arms do not reach into the epigenomically defined heterochromatin. No border was observed for chromosome 4 because it shows chromosome-wide H3K9me2 enrichment; note that the assembled sequence does not extend into the pericentric region. The close congruence between previously determined borders and our epigenomic analysis supports the validity of these approaches, justifying an expansion of our study to other cell types.
Heterochromatin–euchromatin border positions in different cell types
We next carried out ChIP-array analysis with 2–4- and 14–16-h embryos, third instar larvae, fly heads, and four cultured cell lines (S2 = embryo, undifferentiated, male; BG3 = peripheral neuron, differentiated, male; Kc = embryo, female; clone 8 = imaginal disc, male). In most cases, the epigenomic borders lie within 300 kb of the cytogenomic borders (Fig. 3; Table 1; Supplemental Fig. 3). One exception is S2 cells, in which the epigenomic borders are shifted distally by at least 700 kb (Fig. 3; Table 1; Supplemental Fig. 3).
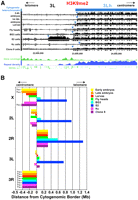
Heterochromatin–euchromatin borders differ among cell types. (A) H3K9me2 log intensity ratio values (y-axis) in the proximal region of chromosome arm 3L (x-axis, sequence coordinates in base pairs) are shown for 2–4-h embryos, 14–16-h embryos, third instar larvae, and adult heads, and for S2, BG3, Kc, and Clone 8 cells. Boxes below the bar graphs demarcate genomic regions with significant enrichment (0.1% FDR). The cytogenomically defined heterochromatin is shown in blue, and the blue arrowheads indicate the positions of the epigenomic border between euchromatin and heterochromatin. The “Repeat Density” track shows the fraction of each 10-kb window that consists of repeated DNAs, based on RepeatMasker (Release 3.28) (http://www.repeatmasker.org). “Gene coverage” plots the number of genes within 50-kb windows, and individual genes are shown below with their orientations as indicated by the arrows. (B) The barplot summarizes the positions of the epigenomic euchromatin–heterochromatin borders on each chromosome arm in the eight cell types examined. On the x-axis, 0 represents the positions of the cytogenomic borders; minus and plus numbers indicate that the epigenomic border was centromere-proximal or -distal to the cytogenomic border, respectively (in Mb). No enrichments for heterochromatic marks were observed for region 3Rh in any cell type, and for the X chromosome in three cell types (?<–), indicating that the borders lie in more proximal regions that are not in the current assemblies. Sequence coordinates of the epigenomic borders are shown in Table 1.
What determines the euchromatin–heterochromatin border is currently unknown. Previous studies have linked such borders to a sharp drop in the density of repetitive elements (Yasuhara and Wakimoto 2008). However, improved repeat identification (Smith et al. 2007a,b) reveals that high H3K9me2 levels are not always associated with high-repeat density (Supplenmental Fig. 3). Thus, repeat density in the arms alone does not determine the extent of heterochromatin or the position of the epigenomic border.
The expansion of pericentric heterochromatin in S2 cells represents a special case, as S2 cells are known to have undergone considerable genomic change compared with the D. melanogaster reference genome. Thus, the expansions may result from an increase in the number of repeats in the pericentric core region (marked by H3K9me2 in all cell types), by the invasion of repetitive elements into the extension regions themselves, or from changes in the dosage of heterochromatin proteins. High-throughput sequencing shows that the total repeat content in S2 cells is higher than in other cell types (data not shown); however, in the absence of a complete S2 cell genome assembly, we cannot distinguish between these and other possibilities.
We conclude that the locations of the epigenomic and cytogenomic borders for most chromosome arms are similar in fly tissues and most cultured cells, as observed previously for a more limited set of marks and cell types (Yasuhara and Wakimoto 2008). However, there is a significant expansion of heterochromatic marks in S2 cells into sequences that are euchromatic in other cell types. The variable euchromatin–heterochromatin borders impact how heterochromatin should be defined with respect to genomic sequence; thus, the epigenomic borders given in Table 1 will be used for the analyses described below.
Heterochromatin contains distinct combinatorial chromatin patterns
Next, we determined the landscapes of multiple histone modifications and chromosomal proteins in BG3 and S2 cells (see Methods) in pericentric heterochromatin and the 4th chromosome, which will be referred to hereafter as “heterochromatin” for simplicity. For the examination of individual modifications and proteins, the enrichment profiles are normalized genome-wide (Fig. 4A; Supplemental Fig. 4A, BG3 cells; Supplemental Fig. 4B, S2 cells). As euchromatin constitutes the majority of the genome sampled (94% in BG3 cells, 92% in S2 cells due to the different epigenomic borders), it shows few deviations from the average pattern, and thus no enrichments or depletions are observed. In contrast, in both BG3 and S2 cells, the pericentric sequences assayed are strongly enriched for “silent” marks, such as HP1a, H3K9me2, H3K9me3, and SU(VAR)3-9, and weakly enriched for the linker histone H1 (Fig. 4A; Supplemental Fig. 4A,B, panels 1,2). These enrichments are accompanied by strong depletion of H3K23ac and reduced levels of many chromatin marks typically associated with transcriptionally active regions of the genome (e.g., RNA polymerase II (Pol II), H4K16ac, H3K18ac, H3K27ac, H3K4me3, and ubiquitinated H2B). The pericentric regions also show an overall depletion for modifications and proteins associated with Polycomb group (PcG)-mediated silencing [e.g., H3K27me3, PC, and E(Z)]. Chromosome 4 resembles pericentric heterochromatin in terms of average patterns, but more detailed analysis shows some significant differences (see below).
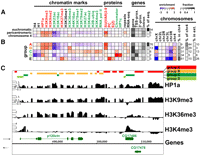
A number of specialized chromatin states characterize the centric heterochromatin and chromosome 4 in BG3 cells. (A) Average levels of enrichment of individual chromatin marks and proteins (panels 1 and 2; green, “active” marks; red, “silent” marks; black, undefined) are shown for euchromatin, pericentric heterochromatin, and chromosome 4. The colors show enrichment (red) or depletion (blue) on a log2 scale after genome-wide normalization (see Methods). There is less depletion of “active” marks on chromosome 4 (e.g., H3K4me2 and H3K27ac) and higher enrichment for H3K36me3, a modification associated with transcript elongation, compared with pericentric heterochromatin. Panel 3 gives the average enrichment for repeats and the RNA-seq signal (Z-score, relative to the array average). The fraction (represented by the gray scale) of the three genome domains associated with genes/gene elements is shown in panel 4 (gene, entire gene; TSS-prox., ±500 bp of the TSS annotated in Flybase; 3'-prox., ±500 bp of the 3'end; intron, within annotated introns). The far-right column indicates the percent of the tiled genome sequence on the oligonucleotide array in each group. See Supplemental Figure 4B for the same analysis of the enrichment patterns in S2 cells. (B) Prevalent combinatorial patterns of chromatin marks within the pericentric heterochromatin (“heterochromatin”) and chromosome 4. Sequences displaying specific combinatorial patterns of “chromatin marks” (panel 1) were first identified by a 15-state K-means PCA cluster analysis (presented in Supplemental Fig. 4A), then combined into five similarity groups (A–E) (see Methods). Other properties, shown in the remaining panels, were then assessed relative to these groups. Each column (panels 1 and 2) indicates average enrichment levels for a given histone modification or protein within the five groups (A–E). The color-coding for each group reflects the predominant patterns of “active” and “silent” marks (see text). Panels 3, 4, and 5 are as described above. The “chromosomes” panel shows the fold over-/under-representation of each group (log2 scale) relative to the amount of heterochromatin in each chromosome arm (h plus Het regions). The next two columns give the percentage of the group found in chromosome 4 (“% in chr4”), and the percentage of chromosome 4 that is accounted for by each group (“% of chr4”). “% in extensions” reports the percentage of each group present in the heterochromatin extensions (Fig. 3B; Table 1). See Supplemental Figure 4, B and C for the same analysis of the chromatin states in S2 cells. (C) An example of the interspersion of different chromatin states in the pericentric region of chromosome 2R. The region shows two transcribed genes (p120ctn and CG17486) within a heterochromatic context. The enrichment profiles of four marks are shown in black (y-axis: log intensity ratio values, x-axis: position on the chromosome), and the groups are illustrated as colored bars on the top. Genes are indicated in green with orientations indicated by the arrows. The upstream promoter regions of each gene are associated with the group D pattern (light-green; low H3K9me2 and me3, depletion of H4 and H1, and moderate HP1a enrichment). The regions immediately downstream from TSSs are associated with group C (dark green) and show enrichment in H3K4me2/3, H2B-ubi, along with low levels of HP1a and even lower levels of H3K9me2/3. The sequences within the body of the genes fall into group B (yellow), with strong enrichment for H3K36me3 along with HP1a and H3K9me2/3. The intergenic regions are associated with the group A pattern (red), showing enrichment only for H3K9me2/3 and HP1a. Group E describes a small group of loci under PC regulation.
We utilized cluster and principal component analysis to examine what specific combinations of modifications and proteins occur throughout heterochromatin (chromatin “states”) (see Methods). A total of 15 clusters were initially determined (the number of clusters was chosen to be sufficiently high to capture most of chromatin variability) (Supplemental Fig. 4A,B). To facilitate the interpretation of the resulting patterns, however, the clusters were then grouped into five states (A–E) based on enrichment similarity (see Fig. 4B for BG3 cells, and Supplemental Fig. 4C for S2 cells). The majority (76%) of the heterochromatin sequences are contained within group A, which displays strong enrichments for “silent” marks and depletion for “active” marks, and is nearly identical to the average heterochromatin pattern. However, 13% of the heterochromatic sequences (group B) are strongly enriched for both “silent” marks and H3K36me3, a modification linked to transcriptional elongation (Carrozza et al. 2005). Furthermore, group C (6% of sequence) is moderately enriched for some “silent” marks, and strongly enriched for many “active” marks. Group D (2% of sequence) is distinguished from group C by the lack of enrichment for several active marks, and group E (3% of sequence) exhibits enrichments for PcG domain markers. Regions associated with these chromatin states are typically interspersed; an example of a 30-kb heterochromatin region of chromosome 2Rh from BG3 cells is shown in Figure 4C.
The right arm of chromosome 4 exhibits characteristics of both heterochromatin and euchromatin (Riddle et al. 2009); it is similar to pericentric heterochromatin in its enrichment for “silent” marks. However, unlike pericentric regions, chromosome 4 does not exhibit overall depletion of “active” marks and shows increased levels of H3K36me3 (see Fig. 4A for BG3 cells; Supplemental Fig. 4B for S2 cells). Consistent with this pattern, chromosome 4 contains a higher proportion of combinatorial states enriched for “active” marks (groups B,C, Fig. 4B; “% of chr4”) and a reduced proportion of states with the most extreme heterochromatic signature (group A, Fig. 4B; Supplemental Fig. 4B, “chromosomes”).
Overall, this analysis reveals that combinatorial patterns of chromatin marks in the assayed heterochromatin are more complex than suggested by the average patterns. As indicated by previous work, active marks can be present within pericentric heterochromatin (Pimpinelli et al. 1995; Johansson et al. 2007b; Yasuhara and Wakimoto 2008). Our analysis significantly extends the number of marks used in prior work and reveals the variability observed in the combinations of “silent” marks such as H3K9m2, H3K9me3, and HP1a with many active marks. These combinatorial patterns are likely to be functionally relevant, since they are closely coordinated with gene structure (see Fig. 4C) and transcriptional activity. Thus, we next analyzed chromatin states associated with genes.
Distinct combinations of marks associated with “silent” and “active” genes in heterochromatin
There are hundreds of protein-coding genes within the pericentric heterochromatin (Smith et al. 2007b), which function in a chromatin environment historically known for silencing euchromatin-derived reporter genes (Cryderman et al. 1998; Konev et al. 2003). We examined the chromatin patterns across these genes to gain a better understanding of their regulation. We defined heterochromatic genes by using the cell-type-specific epigenomic borders described above for pericentric heterochromatin, including chromosome 4 (Figs. 2, 3; Table 1), and excluding 46 genes in chromosome 3Rh now reassigned to euchromatin.
Active heterochromatic genes display H3K9me2, H3K9me3, and HP1a
We first examined euchromatic, pericentric, and chromosome 4–linked genes in BG3 cells, separating transcriptionally active and silent genes on the basis of RNA-seq data (Fig 5B,C, respectively; see Supplemental Fig. 5 for S2 cells). Enrichment profiles for all nonoverlapping genes were analyzed for five gene segments of each gene: 500-bp regions upstream and downstream from the 5' and 3' ends, as well as the remaining internal gene bodies (Fig. 5A). Surprisingly, over half of all heterochromatic genes are expressed in BG3 and S2 cells (51% and 54%, respectively), a percentage similar to that observed for euchromatic genes (51% and 52%, respectively) (Fig. 5C; Supplemental Fig. 5B). As expected, enrichment of HP1a, H3K9me2, and H3K9me3 are seen at transcriptionally silent heterochromatic genes. However, what is striking is that transcriptionally expressed heterochromatic genes, on average, are also enriched for these marks, in addition to the expected “active'”marks (Fig. 5, cf. B and C). The average levels of HP1a, H3K9me2, and H3K9me3 are, in fact, comparable to those of silent heterochromatic genes, and the average levels of “active” marks are also similar to those of active euchromatic genes (Fig. 5C).
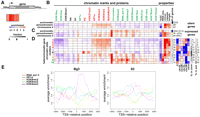
Genes within heterochromatin have specialized properties. (A) The observed chromatin state of each annotated gene was summarized by calculating average enrichment within the 500-bp regions flanking the 5' and 3' ends, the first and last 500 bp within the gene, and the remaining gene body. Each region is represented by the small rectangles (various shades of red in the diagram). Only nonoverlapping genes are considered in this analysis. Levels of modifications and proteins in each gene segment are indicated by shades of red (enrichment) and blue (depletion) in B–D. (B) Average patterns of enrichment for chromatin marks and proteins (log2 scale) for transcriptionally silent genes in BG3 cells. The second panel shows average G/C nucleotide content, repeat content, RNA-seq level, and gene length for each group of genes. The number of genes within each group is indicated in the last column. Transcriptionally inactive genes within heterochromatin and chromosome 4 are highly enriched for H3K9me2/me3, HP1a, and SU(VAR)3-9 over all gene segments, and depleted for most active marks, in comparison to inactive euchromatic genes. (C) Average patterns of chromatin mark enrichment for transcriptionally active genes in BG3 cells. Genes transcribed within the heterochromatic regions show enrichment for “active” marks at comparable levels to expressed euchromatic genes (e.g., H3K36me3, Pol II, H3K4me2/3, and CHRO (a chromodomain protein associated with interband regions on polytene chromosomes; Gortchakov et al. 2005; Rath et al. 2006). However, enrichment levels were noticeably reduced for some active marks (e.g., H4K16ac, H3K18ac, H3K23ac, and H3K79me1/2) compared with active euchromatic genes. Most importantly, the heterochromatic and 4th chromosome genes also contain high levels of HP1a, H3K9me2, and H3K9me3, which are not observed at active euchromatic genes. Expressed heterochromatic genes are, on average, shorter, and contain fewer intronic repeats compared with silent heterochromatic genes (cf. “length” and “repeats” in B and C). (D) Combinatorial chromatin patterns exhibited by heterochromatic genes. Genes were clustered according to their enrichment summary (A) across multiple histone modifications and chromosomal proteins (columns in panel 1; see Methods). Each row shows the average enrichment pattern of the genes within one of the 10 determined clusters. Cluster numbers are color-coded to indicate chromatin states with similar predominant patterns of “active” and “silent” marks. The last three panels show fold enrichment/depletion of each chromosome within the clusters (log2 scale), percentage of cluster regions in chromosome 4 (% in chr4), and percentage of each cluster present in the heterochromatic extensions (“% in ext.”). (E) TSS enrichment patterns at actively transcribed heterochromatic and 4th chromosome genes. The plots show average enrichment profiles for HP1a (blue), H3K9me2 (orange), H3K9me3 (green), and Pol II (red) around TSSs in BG3 cells (left, clusters 7,8 in D and corresponding clusters in S2 cells (right, Supplemental Fig. 5C, clusters 7,8). Genes with divergent promoters (of <2 kb separation) and overlapping genes were excluded, resulting in analysis of a total of 25 genes for BG3 and 32 genes for S2 cells. Average enrichment levels (log2 scale) are plotted on the y-axis relative to the TSS (0) on the x-axis (bp). The results show significant depletion of silencing marks at the TSS.
Interestingly, we also find HP1c associated with expressed heterochromatic genes. HP1c is one of four D. melanogaster paralogs of HP1a. It interacts with the zinc-finger proteins WOC and ROW, and from immunofluoresence localization is considered to be absent from heterochromatin (Smothers and Henikoff 2001; Font-Burgada et al. 2008; Abel et al. 2009). However, we observe HP1c enrichment in the 500 bp immediately upstream of the transcription start sites (TSSs) of active genes in both euchromatin and heterochromatin. This restricted localization, combined with the presence of many more active genes in euchromatin compared with pericentric heterochromatin, could explain the discrepancy between cytological and ChIP localization results. While Western blot analysis indicates our antibody is specific to HP1c (Supplemental Fig. 17), a small amount of signal is detected with this antibody in chromatin prepared from homozygous mutant larvae (<4% of wild-type binding sites; Supplemental Fig. 2E,F). This signal might reflect cross-reactivity to a nontarget protein, or it might reflect the minimal levels of HP1c remaining from the maternally loaded protein. Overall, our data for HP1c suggest a general role in gene regulation, which is consistent with recent work by Kwon and colleagues showing an interaction between HP1c, FACT, and Pol II (Kwon et al. 2010).
Chromatin states of heterochromatic genes reflect chromosomal location as well as expression status
In an attempt to gain further insight into the chromatin organization of heterochromatin, we examined chromatin states at nonoverlapping heterochromatic genes to determine what combinations of modifications and proteins occur specifically at genes (BG3 cells, Fig. 5D; S2 cells, Supplemental Fig. 5C). The genes were grouped into 10 clusters (this number of clusters was chosen based on the ability to interpret the resulting patterns). More than half of the 10 clusters contain transcriptionally silent genes (clusters 1–6), showing the expected enrichment of “silent” marks across these genes. However, one cluster shows lower levels of enrichment for “silent” marks (BG3 and S2, clusters 5), and another almost lacks such marks (S2, cluster 6, Supplemental Fig. 5C). The distributions of these genes in the “silent” gene clusters are not random on the chromosomes (Fig. 5D, panel 4; Supplemental Fig. 5C, panel 4). For example, BG3 clusters 1 and 2 are strongly enriched on chromosome 3RHet and depleted from the X chromosome, whereas clusters 4 and 5 are strongly enriched on the X chromosome. The four genes in BG3 cluster 6, found almost exclusively on chromosome 4, display high levels of PC, H3K27me3, and H3K4me2 surrounding the TSSs; in fact, three of these genes were previously identified as Polycomb targets (toy, zfh2, and fd102C) (Schwartz et al. 2006). Polycomb target genes are very rare in pericentric heterochromatin, but occur at a higher frequency on chromosome 4 in specific cell types. Interestingly, this specific combinatorial chromatin pattern is mostly absent from S2 cells, except for the sv gene on chromosome 4, which was excluded from analysis due to overlap with another gene.
BG3 and S2 clusters 7–10 (Fig. 5D; Supplemental Fig. 5C) predominantly contain expressed genes based on RNA-seq signal, Pol II binding, and enrichment for the elongation-associated modification H3K36me3 within gene bodies. As observed for the overall average of transcribed heterochromatic genes, these clusters contain chromatin marks typical for active euchromatic genes, as well as HP1a, H3K9me2, and H3K9me3. Genes in clusters 7 and 8 display the most extreme mixture of marks that are typically considered to be “active” and “silent,” although the enrichments of these marks across the genes vary; at their 5′ ends, “active” marks are enriched where the levels of “silent” marks are low, and over the gene bodies and 3′ ends, both “silent” and “active” marks are enriched. Strikingly, the levels of these “silent” marks over the gene bodies and 3′ ends are at levels comparable to the silent gene clusters, yet these genes display moderate levels of expression. It is of note that there is a local depletion for “silent” marks near the promoters of these genes (Fig. 5E), as has been described previously for some pericentric genes (Yasuhara and Wakimoto 2008). Depletion of methylated H3K9 in this region is not due solely to nucleosome loss, since levels of active marks such as H3K4me3 are high (Fig. 5D; Supplemental Fig. 5C). Furthermore, this region of depletion does not correspond to the “nucleosome free region” (NFR) observed at active genes in many organisms, which lies upstream of the TSS (Yuan et al. 2005; Mavrich et al. 2008). It is also worth noting that HP1a can be enriched in regions where H3K9me2 and H3K9me3 levels are low or depleted (e.g., clusters 7 and 8, upstream of and downstream from TSSs), suggesting that HP1a localization is not strictly H3K9me2/3-dependent at these sites.
Active gene clusters 9 and 10 (Fig. 5D; Supplemental Fig. 5C) are distinguished by much lower enrichments for silent marks across most gene segments, and much stronger enrichments for “active” marks. BG3 and S2 clusters 9 display the highest levels of Pol II, H3K4me3, and RNA-seq signal. Despite inclusion of the most highly expressed heterochromatic genes, cluster 9 also exhibits HP1a enrichment, especially in the 500 bp upstream of the TSSs, but low H3K9 methylation in all gene segments. Genes in BG3 and S2 clusters 10 are distinguished by the lowest levels of “silent” marks among the active heterochromatic genes, and display high enrichments for “active” marks, yet, on average, are expressed at lower levels. These clusters are preferentially located in X chromosome heterochromatin (Fig. 5D, panel 4), and display high enrichment for H4K16ac, a mark associated with dosage-compensating X-linked genes (Gelbart et al. 2009).
Active genes on chromosome 4 are similar to pericentric genes in average chromatin patterns (Fig. 5C; Supplemental Fig. 5B) and are present in all active gene clusters in the chromatin state analysis (Fig. 5D; Supplemental Fig. 5C, see “% in chr4”). The high level of HP1a over active chromosome 4 genes has been previously observed (Johansson et al. 2007b). However, comparison of the levels of different marks across gene bodies revealed significantly higher average enrichments for H3K9me3 across active 4th chromosome gene bodies compared with active pericentric genes, at a level that exceeds that of 4th chromosome intergenic regions (Fig. 6A,B). The patterns of HP1a and H3K9me3, in fact, closely follow the profile of the elongation-linked modification H3K36me3 across chromosome 4 genes (Fig. 6A,B; correlation analysis in Supplemental Fig. 6).
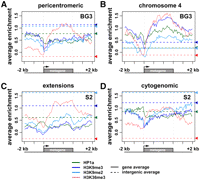
Chromatin patterns vary for expressed genes and intergenic domains located in different heterochromatin regions. The plots show log2 enrichment (y-axis) for H3K36me3 (red), H3K9me2 (light blue), H3K9me3 (dark blue), and HP1a (green) relative to a scaled metagene and 2-kb flanking regions (x-axis). The dashed horizontal lines show average levels of enrichment within intergenic regions for each modification/protein, using the same color key. (A) Average enrichment profiles for expressed pericentric genes in BG3 cells indicate that the levels of HP1a and H3K9me2/3 are higher in intergenic regions compared with gene bodies, whereas H3K36me3 levels are higher over gene bodies than in intergenic regions. Pericentric genes are located in the regions that are centromere-proximal to the BG3 epigenomic borders, including the cytogenomic heterochromatin plus the BG3 extensions (n = 235). (B) Average enrichment profiles for expressed chromosome 4 genes in BG3 cells show significantly higher levels of HP1a, H3K9me2/3, and H3K36me3 enrichment within gene bodies compared with intergenic region averages (n = 58). (C) Average enrichment profiles genes in S2 cells located (C) within the S2-specific extension regions (between the S2 and BG3 epigenomic borders). Profiles for 60 such genes that are expressed in both S2 and BG3 cells are shown. (D) Average enrichment profiles in S2 cells for expressed genes located within the pericentric heterochromatin defined by the cytogenomic borders (excluding 3Rh; n = 117). In S2 cells, the extensions and cytogenomic heterochromatin have comparable levels of enrichment for all four marks within the intergenic regions. However, at active genes, the levels of HP1a and H3K9me2/3 are lower in the extensions than in the cytogenomic regions.
We conclude that both active and silent heterochromatic genes display unusual combinatorial patterns of chromatin marks, which differ from euchromatic genes in levels of enrichment/depletion and in distributions across gene segments. Overall, higher expression levels are correlated with lower enrichments for H3K9me2 across all gene segments, but are not correlated with HP1a levels (compare silent and expressed genes in Fig. 5B,C, and all heterochromatic to heterochromatic expressed genes in Supplemental Fig. 7A,B). Perhaps the most atypical feature of active heterochromatic genes is the presence of modifications and proteins normally associated with gene silencing and the depletion of these marks immediately downstream from TSSs. This is consistent with previously observed reductions in HP1a levels for a subset of heterochromatic genes (de Wit et al 2007). Given the presence of “silent” marks over the gene bodies, the dramatic depletion of these marks specifically at TSSs of active genes may be critical for gene expression.
Genes in S2 cell heterochromatic extensions are not silenced
The S2 “extensions” provide a unique region of study, where the same sets of genes lie in a heterochromatic or a euchromatic environment in different cell types. Comparison of the S2 “extensions” (2.8 Mb) to the relevant regions in BG3 cells reveals a decrease in the average levels of most “active” chromatin marks, in addition to the average increase in “silent” marks (Supplemental Fig. 8, green and orange, respectively). However, 98% of the genes in the S2 extension regions that are expressed in BG3 cells are also expressed in S2 cells. Only five nonoverlapping S2 genes in the extensions show significant changes in expression (clusters 1–3, Supplemental Fig. 9C); of these, only one gene is silenced in the S2 extension (cluster 1), and the other four actually have higher levels of expression in S2 cells compared with BG3 cells. Transcriptionally silent genes in S2 extensions that are also silent in BG3 cells show higher enrichments for “silent” marks on average (Supplemental Fig. 9A, panel 1), although they are lower than those in the cytogenomically defined heterochromatin (Supplemental Fig. 9A, panel 2). In contrast, intergenic regions in the extensions display similar levels of “silent” marks to cytogenomic heterochromatin (Fig. 6C,D, dashed lines).
We conclude that the accumulation of “silent” heterochromatic marks within the S2 extensions is most prominent within intergenic regions. Surprisingly, gene expression is virtually unchanged between BG3 and S2 cells in these “extension” regions, despite acquisition of moderate levels of heterochromatic marks within many of the genes. These observations suggest that genic regions within the S2 extensions, especially expressed genes, are more resistant to establishment or maintenance of heterochromatic patterns than are intergenic regions (see Discussion).
Chromatin patterns in intergenic regions are complex
We conservatively defined intergenic regions as sequences >2 kb away from the nearest annotated gene, which comprise 30% of the pericentric heterochromatin and 7.4% of chromosome 4 sequences on the tiling array. As expected, intergenic regions in heterochromatin are, on average, enriched for HP1a and H3K9me2/3 and depleted for most “active” marks in both BG3 and S2 cells (Supplemental Fig. 10A,C). Combinatorial patterns are similar for all S2 and most BG3 (86%) intergenic regions (Supplemental Fig. 10B,D; BG3 clusters 3, 4, 6, and 8–10, S2 clusters 1–10). However, 14% of the BG3 intergenic regions show chromatin patterns typical of active transcription (Supplemental Fig. 10B; clusters 1, 2, and 7), suggesting the presence of transcribed repeats or currently unannotated genes. Interestingly, intergenic regions have lower levels of “silent” marks and higher enrichments for “active” marks in chromosome 4, compared with pericentric heterochromatin, even though repeat densities are comparable (Fig. 6A,B; Supplemental Figs. 3, 10A,C). Furthermore, within chromosome 4, intergenic regions have a higher density of repeats than active genes, but contain less HP1a and H3K9me2/3 (Fig. 6B). These findings provide further evidence for a link between “silent” mark enrichments and gene activity in some heterochromatic regions (see above and Discussion).
Transposable elements display complex patterns of epigenomic marks
One of the major distinctions between heterochromatin and euchromatin is the density of repeated sequences. At least 80% of the assembled heterochromatic sequences are repeats, predominantly organized as scrambled clusters of transposable elements (Smith et al. 2007b), whereas only 6% of the euchromatin is classified as repetitive (Kaminker et al. 2002). The 4th chromosome domain analyzed here contains ∼30% repetitious DNA (Leung et al. 2010). Previous analyses have found that H3K9me2 is highly enriched at both satellite and rDNA sequences in a SU(VAR)3-9 HMTase-dependent manner (Peng and Karpen 2007).
Many repeat types, in particular tandem repeats, cannot be adequately assessed using tiling arrays due to cross-hybridization and signal intensity issues. We therefore focused on analyzing the chromatin patterns associated with unique transposable element sequences (intact remnants and scrambled clusters; see Methods) in heterochromatin and euchromatin (Fig. 7A; Supplemental Figs. 11, 12). Most TEs that were examined in heterochromatin show strong enrichment in HP1a and H3K9me2/3, and moderate enrichment for H1 (Fig. 7A, panel 2; Supplemental Figs. 11, 12), consistent with the HP1a association reported by Dam-ID for TEs in repeat-rich regions (de Wit et al 2007). However, some TEs in heterochromatin show weak or moderate enrichment for H3K36me3, Chromator (CHRO; a chromo-domain protein), and H3K4me3. Thus, although most TEs in heterochromatin are predominantly associated with silent marks, a few show complex patterns that include active marks.
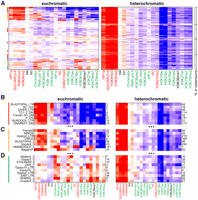
Repetitive elements integrated within heterochromatic regions show similar epigenomic signatures. (A) Average enrichments (red) and depletions (blue) for particular chromatin marks (columns) in BG3 cells are shown for specific repetitive element types (rows) in euchromatic (left) and heterochromatic (right) regions (extended version with repeat names is shown in Supplemental Fig. 11 for BG3 cells, and Supplemental Fig. 12 for S2 cells). The color spectrum for the enrichment level (log2 scale) is the same as in Figure 5. The fraction of the heterochromatic repeats found in the BG3 extension regions is reported in the grayscale column on the right. The heterochromatic instances of all repeat types are marked by strong enrichment in HP1a, SU(VAR)3-9, and H3K9me2/3. In contrast, euchromatic repeat instances are associated with different types of chromatin patterns that vary in the levels of “active” and “silent” marks. Elements with similar patterns are marked by colored vertical bars on the left; red, highly enriched for “silent” marks, depleted for “active” marks; green, low enrichment or depletion for “silent” marks, highly enriched for “active” marks; orange, mixed enrichments for both “active” and “silent” marks. (B) Full-scale view of the top-most portion of the plot, showing repeat types for which euchromatic and heterochromatic instances show similar average chromatin patterns with predominant enrichments for “silent” marks. The RepBase repeat type names are shown on the left, with the number of instances found within each region to the right. In contrast, repeat types with mixed (C) and “active” (D) chromatin patterns in euchromatic regions (left) show predominantly “silent” mark enrichments when located in heterochromatin (right).
In contrast, TEs within euchromatic regions are associated with at least three types of chromatin signatures: (1) enriched for “silent” marks (∼30% of the repeat types; Fig. 7A, marked by red side-bars on the left), some of which are at the same level as those in heterochromatin (Fig. 7B), (2) enriched for both “silent” and “active” marks (Fig. 7C, orange side-bars), and (3) enriched for “active” chromatin marks only (Fig. 7D). Remaining repeats show less HP1a, but no significant enrichment of Pol II (Fig. 7A).
We conclude that the chromatin state of TEs depends on the genomic context, consistent with the previous study that found a TE's likelihood of HP1a association depends on the density of repeats in the region (de Wit et al 2007). Our results suggest that TEs inserted into euchromatic regions avoid chromatin-mediated silencing, in line with observations that Drosophila cells contain many developmentally regulated TE transcripts (Flavell et al. 1980; Lankenau et al. 1994). It is possible that TE remnants are sufficiently evolved so that they are no longer recognized as TEs, and/or that single TEs in a euchromatic environment are unable to maintain stable heterochromatin, as suggested by studies of the 1360 TE (Haynes et al. 2006). As such, the higher overall density of TEs and TE fragments in heterochromatic regions compared with euchromatin may ensure uniform silencing. Future studies focused on providing a more complete catalog of the chromatin states associated with both TEs and other highly repeated sequences should reveal whether these hallmarks of heterochromatin are universally enriched for “silent” marks, or display the kind of complex enrichment patterns that we observed for heterochromatic genes. Furthermore, limitations of the microarray platform did not allow us to take into account TE instances with a high degree of sequence identity, and therefore, further investigations are needed to extend the analysis to a complete set of TEs.
Euchromatin contains domains enriched for H3K9me2
Since the locations of the epigenomic euchromatin–heterochromatin borders differ among cell types, we asked whether there are other cell-type differences in the rest of the genome. Whole-genome classification shows that there are clusters of H3K9me2 enrichment in the euchromatin; some of these domains are present in all of the cell types examined, whereas others are cell-type specific (Supplemental Fig. 13). Focused segmentation analysis identified common H3K9me2 enrichment in euchromatic regions of all examined animal and tissue culture cell types except for Kc cells (Fig. 8A, cluster 1). There are also domains of H3K9me2 enrichment that are unique to adult heads (cluster 6), BG3 cells (cluster 5), S2 cells (cluster 3), and domains shared between S2 and BG3 cells (cluster 2) or S2 and Kc cells (cluster 4).
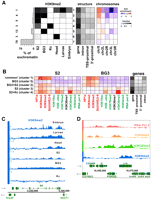
BG3 and S2 cells show novel domains of H3K9me2 enrichment within euchromatic regions. (A) Regions of H3K9me2 enrichment across different cell types. The euchromatic portion of the genome (excluding regions defined as heterochromatin by the border analysis; see Fig. 3A) was subdivided into sets of regions that exhibit a common pattern of H3K9me2 enrichment across different cell types. Each box shows the fraction (grayscale) of the regions belonging to the set (row) that are enriched for H3K9me2 in a particular cell type (column). The histogram on the left shows the fraction of the euchromatic genome in each row (1–7), with exact %s to the left. Regions in row 7 lack H3K9me2 across all examined cell types, whereas row 1 groups regions enriched for H3K9me2 in all examined cell types (except for Kc cells). Rows 2–6 identify other euchromatic regions that display H3K9me2 enrichment in only a subset of cell types (e.g., only BG3 cells (row 5) or S2 cells (row 3), or both (row 2). Panel 2 shows the fraction of sequence within each group associated with different parts of annotated genes (gene, entire gene; TSS-prox. [±500 bp of the TSS annotated in Flybase], 3'-prox. [±500 bp of the 3'end annotated in Flybase], and intron are a subset of the sequences included in the “gene” column). The third panel shows over-/under-representation of each cluster on different chromosome arms, which was calculated by comparing the fraction of sequence of a cluster on a specific chromosome with the fraction of sequence the chromosome contributed to the array. (B) Average enrichment of chromatin marks in the cell-type-specific H3K9me2 enrichment domains. Each row shows average enrichment levels (log2 scale) within regions corresponding to the main patterns seen in A. The specific regions were identified using HMM segmentation (see Methods). Panel 1 shows the average enrichment patterns in S2 cells, panel 2 shows the average enrichment patterns for the same genomic regions in BG3 cells, and panel 3 indicates the fraction of the particular H3K9me2 enrichment domain associated with gene features. While “common,” BG3 and BG3+S2 domains (rows 1–3) are enriched only for heterochromatic marks, the S2-specific and S2+Kc-specific domains (rows 4,5) include actively transcribed genes that in S2 cells are enriched for heterochromatic marks along with marks normally associated with transcription, similar to “mixed” state genes found in heterochromatin (Fig. 4). (C) Browser shot showing an example of a gene from a “common” (row 1) domain, located in the euchromatic arm of chromosome X, and enriched for H3K9me2 across all examined cell types except Kc cells. x-axis, chromosomal position in base pairs (centromere to the left). Genes are indicated in green with their orientations as indicated by the arrows. y-axis, H3K9me2 enrichment levels (log2 scale) for the indicated tissue. (D) A representative region of arm 3R containing an S2-specific domain (row 3), showing a combination of H3K9me2 (blue) and marks associated with active transcription—H3K36me3 (green), H3K4me3 (orange), and Pol II (red). Two sets of genes display a divergent promoter orientation typical of the S2-unique domain genes. X-axis, chromosomal position in base pairs; y-axis, enrichment levels (log2 scale).
There are significant differences between these H3K9me2 domains in their sizes, chromosome distributions, gene activities, and patterns of chromatin marks (Fig. 8; Table 2). The 14 H3K9me2 domains that are found in all but Kc cells (cluster 1) contain 20 functionally diverse genes, which are small and generally specific to the X chromosome, with only two of 14 such regions found on autosomes. These genes are transcriptionally inactive (only 10% are expressed) and show enrichment for all other heterochromatic marks [H3K9me3, SU(VAR)3–9, and HP1a] in both BG3 and S2 cells (Fig. 8B). With the exception of two domains (covering SteXh and the skpC/skpD/skpE gene clusters), each chromosome X-specific domain includes a single, multi-exonic gene, with H3K9me2 enrichment strongly biased toward the 3′ end of the gene (Fig. 8C; Supplemental Fig. 14). The absence of these “common” H3K9me2 domains in Kc cells is particularly interesting, given that the domains are enriched on the X chromosome, and Kc cells alone are derived from female flies (see Discussion).
Properties of euchromatin H3K9me2 domains in different cell types
Although they are enriched in H3K9me2 in S2 cells, the majority of genes in clusters 3 and 4 are actively transcribed in this cell type (59% S2 specific and 73% S2+Kc cells, respectively) and comprise 7.7% of the euchromatic sequence (Table 2). Similar to active heterochromatic genes, these genes are enriched for activation-associated marks (H3K4me3, H3K36me3, and H2B-ubi), as well as multiple “silent” marks (Fig. 8B,D, Lrrk gene). H3K9me2 enrichment is also biased toward the middle and 3′ end of these genes, with a decline at the TSSs, similar to active heterochromatic genes (Supplemental Fig. 7). These mixed patterns are specific to the S2 and Kc cells; the same set of genes is also expressed in BG3 cells with the same pattern of “active” marks as in S2 cells, but without the heterochromatic marks (Fig. 8B, middle panel). In addition, these genes are strongly biased toward divergent promoter orientation (Table 2, 43% for S2-specific P-value = 1.9 × 10−4, 48% for S2 + Kc-specific P-value = 8.2 × 10−5).
Compared with other cell-type-specific H3K9me2 domains, the 180 domains specific to BG3 cells in cluster 5 account for a much higher fraction of the euchromatic arms (12%), are notably larger (78 kb mean size), and have a lower gene density (Table 2). A total of 90% of genes within these domains are transcriptionally silent in BG3 cells and are also silent in embryo and S2 cells, despite the fact that they lack the heterochromatin-like patterns in those cell types. These genes show a strong preference for tandem orientation (18% divergent promoters). This group shows significant over-representation for genes associated with sensory perception functions (P-value = 6 × 10−19) (Table 2). Further analysis of these large BG3-specific domains reveals that all clusters are depleted for marks associated with transcription (e.g., Pol II and H3K4me3) and are enriched for H3K9me2, but differ in the levels of enrichment for HP1a, SU(VAR)3-9, and H3K9me3 (Supplemental Fig. 15).
Since both S2 and BG3 cells are known to have abnormal chromosome counts, and are most likely segmental aneuploids, we looked at the possible correlation between the occurrence of novel H3K9me2-enriched domains and copy number, but no correlation was found (D MacAlpine and P Kharchenko, data not shown). We therefore examined whether these domains are caused by genomic rearrangements. Four different heterochromatin-like domains occurring in BG3 cells were analyzed using PCR and Southern analysis (Supplemental Fig. 16). While one domain was associated with a rearrangement in BG3 cells (Supplemental Fig. 16A), this H3K9me2 domain is present in both S2 and BG3 cells, and no rearrangement was observed in S2 cells. Thus, it is unlikely that genomic alterations are responsible for the bulk of the “heterochromatin-like” domains in BG3 cells.
We conclude that there are extensive H3K9me2 domains present in euchromatic sequences, and that their sizes and impact on gene expression differ among cell types (Table 2). These domains do not appear to arise from genomic rearrangements that juxtapose euchromatic with pericentric regions.
Discussion
Our genome-wide analysis of histone modifications and chromosomal proteins is consistent with and extends previous findings of enrichment for marks known to define heterochromatin, such as HP1a and H3K9me2/3, in pericentric regions and the 4th chromosome (Gatti and Pimpinelli 1992; Yasuhara and Wakimoto 2008; Eissenberg and Reuter 2009; Riddle et al. 2009). Our new data provide a high-resolution map of the epigenomic borders between heterochromatin and euchromatin, revealing intriguing differences among cell types. Our findings also illuminate significant variations in marks classically associated with transcriptional activity and silencing, revealing unexpected complexity in heterochromatic chromatin patterns (summarized in Fig. 9). We also show that repetitive elements, thought to be uniform targets of heterochromatin formation, consistently carry silencing marks within the heterochromatic regions, but vary within euchromatin. Finally, we identify novel, cell-type-specific regions within euchromatic sequences that contain heterochromatin marks. These findings raise important questions regarding how different chromatin states are established and maintained within heterochromatic domains, and about their impact on genomic functions.

Summary of chromatin patterns observed in Drosophila heterochromatin. The predominant enrichment patterns observed for selected histone modifications and proteins are summarized for active and silent genes and intergenic regions, in euchromatin, pericentric heterochromatin (including the S2-specific extensions), and the 4th chromosome. Red, “silent” marks and proteins; green, “active” marks. Heights of color blocks within each row indicate enrichment levels relative to the features shown below, whose combinatorial patterns are reflected in the colors and intensities. For example, the lighter red used for intergenic regions and silent genes in chromosome 4 indicate lower enrichments for “silent” marks compared with pericentric heterochromatin. Gradients across active genes reflect differences in the relative levels of “active” and “silent” marks; red, predominantly “silent” marks; green, predominantly “active” marks; yellow, enrichments for both. Silent genes in euchromatin are shown in gray to indicate the absence of “silent” marks.
Epigenomic patterns demonstrate variable positioning of heterochromatin–euchromatin borders in different cell types.
Overall, there is a gratifying congruence between the heterochromatin–euchromatin borders determined previously by cytogenomic techniques (Hoskins et al. 2002, 2007) and the epigenomic borders determined here (Fig. 2). Future analyses should utilize the more relevant epigenomic borders to define the heterochromatin domains for each cell type studied. As a border was not identified for arm 3R, we now consider the available 3Rh sequences to be euchromatic. Supporting this reassignment is the finding that this region has a lower repeat content and higher gene density compared with other pericentric regions (Smith et al. 2007b; see also Supplemental Fig. 3). Identifying the border in 3R will require assembling sequences and analyzing chromatin patterns in the gap between 3Rh and 3RHet.
In S2 cells, we observed larger extensions of the pericentric heterochromatin compared with other cell types, marked by high enrichment for “silent” marks, mostly restricted to intergenic regions (summarized in Fig. 9). Despite acquiring moderate levels of “silent” marks, genes within the extensions retain chromatin marks typically associated with transcription, and expression levels are surprisingly similar between BG3 and S2 cells, with only one gene becoming silenced. Similarly, genes in euchromatic regions are not uniformly silenced when juxtaposed with heterochromatin by chromosome rearrangements (Rudolph et al. 2007; Vogel et al. 2009). We propose that many active genes are resistant to heterochromatin formation or spreading, despite being embedded in large domains that acquire “silent” marks in specific cell types. Determining whether “heterochromatinization” of extension regions alters other aspects of gene function, such as cell-cycle regulation of transcription or protein levels, requires further analysis.
The variable nature of the border positions in different cell lines (Fig. 3), embryos, and fly tissues (arms 2L and 3L, Supplemental Fig. 3) argues against a mechanism involving strict sequence-based boundary elements, as observed in S. pombe (Scott et al. 2006; Wheeler et al. 2009). We favor the hypothesis that border positions depend on the “epigenetic balance” between euchromatic and heterochromatic chromatin components, as suggested by studies of rearranged chromosomes (Ebert et al. 2004; Rudolph et al. 2007). However, border positions also appear to be influenced by general properties, such as repeat and gene densities. We propose that high-repeat content and/or low gene density contributes to the extent of heterochromatin formation (as in S2 cells), but what is attained will depend on global cell-specific properties, such as heterochromatin protein levels. Heterochromatin also could be restricted in a cell-specific manner in cis by the presence of chromatin states that are incompatible with heterochromatin formation or spreading, such as high gene activity or blocks of Polycomb marks and proteins (as observed for three arms in BG3 cells).
Heterochromatic genes display complex chromatin patterns
Overall, our analysis of combinatorial patterns of chromatin marks revealed that the composition of heterochromatic genes is more complex than suggested by the average patterns for individual marks (e.g., Fig. 4).
Active heterochromatic genes display an unusual distribution of ‘active’ and “silent' marks
Surprisingly, we found that a similar proportion of genes in pericentric heterochromatin and euchromatin are transcriptionally active (∼50%). The levels of most “active” marks are also comparable between euchromatin and heterochromatin at active genes (Fig. 5C; Supplemental Fig. 5B). However, enrichments for some marks at active heterochromatic genes are noticeably reduced (e.g., H4K16ac, H3K18ac, H3K23ac), accompanied by high-average enrichments for H3K9me3, H3K9me2, and HP1a relative to active euchromatic genes (summarized in Fig. 9). Interestingly, HP1a enrichments do not track precisely with H3K9me2/3 levels near TSSs; at most active heterochromatic genes, a prominent peak of HP1a is centered 800 bp upstream of TSSs where H3K9me2/3 levels are low, followed by moderate depletion of HP1a immediately downstream from TSSs. This reduced enrichment for all three silent marks immediately downstream from active heterochromatic gene TSSs corresponds to the peaks of Pol II and H3K4me3 enrichments (Fig. 5D,E) and is not due simply to local nucleosome depletion.
The prominent association of HP1a, H3K9me2/3, and SU(VAR)3-9 across active gene bodies, with high levels of H3K36me3, suggests that once transcription is initiated, Pol II can elongate through regions highly enriched for these supposedly “silent” marks. In fact, enrichments for “silent” marks across active chromosome 4 gene bodies are higher than for pericentric genes, and surprisingly, higher than observed for 4th chromosome intergenic regions (Fig. 6A,B).
Importantly, we have established that the levels of H3K9me3 in D. melanogaster are similar to those seen in mammals. Although the observed overall distributions of H3K9me2 and H3K9me3 are similar, they show some interesting differences as well (summarized in Fig. 9). For example, HP1a is, on average, more highly correlated with H3K9me3 compared with H3K9me2, especially on the 4th chromosome (Supplemental Fig. 6). This finding suggests that in a chromatin context HP1a may have a greater affinity for H3K9me3 nucleosomes versus those containing H3K9me2. Differences in the distributions and levels of H3K9me2 and H3K9me3 are more extreme at specific regions and gene groups, as revealed by the combinatorial cluster analyses. Whether differences between these marks have biological impact on heterochromatic gene expression and other functions is unknown and warrants further analysis.
Do ‘silent’ marks and proteins play both positive and negative roles in regulating heterochromatic gene expression?
Our findings suggest a model for heterochromatic gene transcription that accommodates enrichments for HP1a and other “silent” marks. Interpreting the functional implications of marks such as HP1a on active genes is complicated, as evidenced by previously reported observations of both positive and negative effects on gene expression. Although HP1a clearly is required for heterochromatin-mediated silencing (Eissenberg et al. 1990), it is also essential for the expression of some heterochromatin genes (e.g., light) (Hearn et al. 1991). We propose that the inhibitory functions of HP1a and associated proteins/marks must be eliminated at TSSs to allow transcription initiation, whereas they are required over heterochromatic gene bodies to maintain high levels of expression. According to this model, the main distinction between active and silent heterochromatic genes is initiation through depletion of “silent” marks near TSSs.
For transcription initiation to occur in heterochromatin, Pol II may be recruited first, followed by loss of “silent” marks near TSSs, or Pol II may only be recruited after “silent” marks are removed. The latter model is supported by the observation that most putative binding sites are not actually bound by their cognate transcription factors unless the chromatin is already “open” (MacArthur et al. 2009; Weber et al. 2009). One candidate that may be responsible for the transcription initiation of the heterochromatic genes is HP1c, which is enriched at TSSs of active heterochromatic genes, and coincides with HP1a enrichment around TSSs (see Fig. 5B,C). Given its reported association with transcription factors (Smothers and Henikoff 2001; Font-Burgada et al. 2008), it is possible that HP1c promotes initiation of transcription at heterochromatic genes, perhaps through a physical or functional interaction with HP1a that promotes local chromatin changes. Whether HP1c or other factors such as H3K9 demethylases (Marmorstein and Trievel 2009) act at heterochromatic TSSs to promote initiation must be addressed in future studies.
It is perhaps more surprising to consider factors such as HP1a as positively impacting gene expression, in this case through their association with transcribed gene bodies. While a previous study suggested that HP1a positively impacts the expression of some euchromatic genes (particularly a subset expressed at high levels), through interactions with RNA and RNA processing factors (Piacentini et al. 2003, 2009) we observe that HP1a is not enriched at active euchromatic genes in general (Fig. 5B). Another potential positive link between HP1a and transcription comes from the observation that HP1a can bind to KDM4A and stimulates its H3K36me2/me3 demethylase activity (Lin et al. 2008), which in S. cerevisiae results in histone hypoacetylation and blocks initiation from cryptic promoters in ORFs (Carrozza et al. 2005). Although these latter observations provide an attractive solution as to how “silent” marks could promote transcription, the relevance of this biochemical interaction is not supported by our observations. In particular, HP1a-mediated activation of the KDM4A demethylase predicts reduced levels of H3K36me3 and increased levels of H3K36me1 at sites of HP1a enrichment; however, both HP1a and H3K36me3 are highly enriched across active chromosome 4 and pericentric heterochromatic gene bodies, and H3K36me1 is depleted across all gene segments (Figs. 5, 6). Finally, we observe no enrichment for HP1a at active euchromatic genes, inconsistent with a general role for HP1a recruitment of KDM4A in promoting gene expression.
Clearly, we currently lack a mechanistic understanding of how genes embedded in heterochromatin are regulated and expressed, but we expect that these comprehensive chromatin landscapes will provide a foundation for future advances. Direct experimental dissections are needed to elucidate how the distributions and levels of H3K9 methylation and HP1a impact the initiation, elongation, and RNA processing associated with heterochromatin gene expression.
The 4th chromosome is enriched for specialized heterochromatic domains
The distal 1.35 Mb of chromosome 4R exhibits characteristics of both heterochromatin and euchromatin (Riddle et al. 2009). As is the case for pericentric heterochromatin, the entire 4th chromosome is late replicating (Zhimulev et al. 2003), enriched for HP1a and H3K9me2 (Fig. 1B; Greil et al. 2003; Slawson et al. 2006; Johansson et al. 2007a; Yasuhara and Wakimoto 2008), and shows no meiotic recombination under normal conditions (Sandler and Szauter 1978). However, this region of the 4th chromosome has a gene density comparable to euchromatin on other chromosome arms (Supplemental Fig. 3) and is amplified during polytenization, unlike the underreplicated pericentric heterochromatin. Genes are interspersed with repetitious sequences that make up 30% of this region (Leung et al. 2010), a value that is intermediate between euchromatin (6%) and pericentric regions (>80%) (see Supplemental Fig. 3).
Our studies confirm that chromosome 4 generally resembles pericentric heterochromatin (Fig. 4A; summarized in Fig. 9), but it has higher levels of many “active” chromatin marks that are likely due to its higher gene density (Supplemental Fig. 3). Several combinatorial chromatin states are enriched specifically on chromosome 4 (Supplemental Fig. 4); they are clearly associated with transcribed genes (e.g., BG3 clusters 4–6, 11, and 14, Suppemental Fig. 4A), and one cluster is associated with silent genes (cluster 1).
Sequences associated with Polycomb (PC) and H3K27me3 are rare in pericentric heterochromatin. However, we observed a distinctive chromatin state enriched for H3K27me3 and PC that is associated with seven 4th chromosome genes in BG3 cells (Fig. 4B, group E), some of which were detected in previous studies (Negre et al. 2006; Schwartz et al. 2006; Tolhuis et al. 2006). The presence of PcG marks in BG3 cells but not S2 cells is consistent with the previously reported cell-type-specific differences in PC binding at euchromatic genes (Kwong et al. 2008; Schwartz et al. 2010). Interestingly, the association of 4th chromosome genes with PC and H3K27me3 in BG3 cells correlates with reduced levels of HP1a, whereas the same genes devoid of PcG binding in S2 cells display high levels of HP1a that correspond well with the chromosome-wide averages. This suggests that the chromatin state of the genes repressed by PcG is in some way incompatible with H3K9methyl-mediated silencing and HP1a binding.
We conclude that the chromatin composition of the 4th chromosome is, in general, most similar to pericentric heterochromatin, consistent with previous genetic, biochemical, and developmental studies (Johansson et al. 2007a; Riddle et al. 2009); however, the 4th also has unique domains that distinguish it from both pericentric and euchromatic regions.
A surprisingly large fraction of euchromatin sequences display cell-type-specific heterochromatin features
In mammals, there are well-studied examples of HP1 and H3K9me2 enrichment at single genes in euchromatin, generally associated with silencing as part of a signaling response or process of cell differentiation (Ayyanathan et al. 2003; Cammas et al. 2004). One recent study identified large domains of H3K9me2 present in differentiated mouse ES cells (Wen et al. 2009), though there is some dispute about the significance (Filion and van Steensel 2010). Our genome-wide analysis of H3K9me2 distributions identified a large fraction of D. melanogaster euchromatic sequences (up to 12% or ∼900 genes in BG3 cells) that are enriched for “silent” marks (Fig. 8). Interestingly, the impact of these “silent” mark enrichments in Drosophila on gene expression is not uniform. The basis for these differences in gene expression is unknown. However, it is intriguing that the H3K9me2 domains that contain more expressed genes (clusters 3 and 4) are, on average, smaller than domains with few expressed genes, have a two- to fourfold higher gene density, and are more enriched for genes with divergent promoters (Table 2). These observations suggest that the cell-type-specific establishment, maintenance, or spreading of heterochromatin features in euchromatic sequences may be inhibited by genes or facilitated by intergenic regions, similar to the observed resistance of active genes to “heterochromatinization” in the S2 extensions (see above).
Several hypotheses can be considered to explain the presence of “heterochromatin-like” domains in euchromatin. There are previously described regions of intercalary heterochromatin in all D. melanogaster euchromatic arms (Belyaeva et al. 2008) that could be responsible for the H3K9me2-enriched domains found in all cell types and tissues. Comparing regions of intercalary heterochromatin with the positions of the H3K9me2-enriched domains, we find that only one of 23 regions coincide with intercalary heterochromatin identified by polytene chromosome analysis (Semeshin et al. 2001), and six of 23 regions overlap with intercalary heterochromatin defined by ChIP-chip mapping of the SUUR protein (Belyakin et al. 2005). Our results demonstrate that it is also unlikely that such domains result from chromosome rearrangements or changes in copy number. We note that when chromosome rearrangements in animals have been analyzed by ChIP, there is a gradient of heterochromatin marks spreading from the breakpoint (Rudolph et al. 2007; Vogel et al. 2009), while here we see distinct borders between these domains and flanking regions.
We favor the hypothesis that at least some of these domains represent the establishment of heterochromatic chromatin patterns to accomplish local, cell-type-specific silencing of euchromatic genes. Gene-poor regions appear to be particularly susceptible to acquisition of “silent” marks; perhaps they lack the high levels of transcription required to resist silencing, or these regions contain genes with extensive regulatory regions characteristic of developmental regulators. A novel observation from our study is that the “common” domains of this type are predominantly on the X (12 out of 14 domains). They are absent from the female Kc cell samples and present in other sources that are either entirely male (S2 and BG3 cells) or a mix of male and female cells (heads, larvae, embryos). These H3K9me2 domains on the X chromosome are reminiscent of the previously reported HP1a enrichment on the male X chromosome, shown by DAMid mapping (de Wit et al. 2005). Although de Wit and colleagues observed X chromosome-wide enrichments for HP1a, our ChIP-chip analysis clearly shows distinct domains (that contain mostly single genes), covering 1.1% of the X, which exhibit high levels of multiple heterochromatic marks (H3K9me2, H3K9me3, and HP1a). It is possible that these “common” H3K9me2 domains contain X-linked genes that are silenced by “heterochromatinization” only in male cells, suggesting a strategy for avoiding the effects of dosage compensation mechanisms which up-regulate expression of X-linked genes in these cells (Gelbart and Kuroda 2009). The genes associated with these domains are inactive in female Kc cells as well as male S2 and BG3 cells, suggesting that accomplishing this goal requires an additional layer of gene repression in male cells. Further analyses are required to fully test the hypothesis that Drosophila euchromatic sequences acquire heterochromatic features during developmental determination or differentiation, and to ascertain whether other organisms exhibit large domains of heterochromatic marks in euchromatic sequences.
In conclusion, this analysis has provided a much more detailed picture of the complexity of heterochromatin from an epigenomic perspective. Heterochromatin, sometimes referred to as the “black hole” of the genome, contains a much richer and more complex landscape of histone modifications and chromosomal proteins than previously imagined. The effect is like looking at a pointillist painting by Georges Seurat—before we were standing at a distance, now we are looking close-up and perceiving the complex patterns used to achieve different effects. We expect that this study will provide a solid foundation for future experimental analyses aimed at addressing the relationships among chromatin composition, organization of heterochromatin sequences, and the functions of heterochromatic domains. It will be particularly important to determine how heterochromatic gene expression and silencing are regulated, how the borders between heterochromatin and euchromatin are established and maintained, and what roles the euchromatic H3K9me2 domains may play in developmental regulation of gene expression.
Methods
Detailed materials and methods descriptions can also be found at http://www.modENCODE.org.
Growth conditions
Cell lines were obtained from the Drosophila Genome Resource Center (DGRC) and grown according to DGRC protocols (https://dgrc.cgb.indiana.edu/): S2-DRSC cells (stock #181), Kc-167 cells (DGRC, stock #1), ML-DmBG3-c2 cells (DGRC, stock #68), and Clone 8 cells (DGRC, stock #151).
OR flies (Bloomington stock #25211) were raised in population cages at 25°C with 70% humidity on grape juice-agar medium supplemented with yeast paste (Shaffer et al. 1994). Two to 4 h and 14–16 h embryos and adult flies were collected from these cages, frozen in liquid nitrogen, and stored at −80°C. Fly heads were prepared from frozen flies using sieves (sieve sizes: 710 μm, 600 μm, and 500 μm). Heads were reimmersed in liquid nitrogen and stored at −80°C. Third instar larvae were collected from OR flies grown in bottles (at low density) at 25°C/70% humidity on standard cornmeal-agar medium (Shaffer et al. 1994). Larvae were frozen in liquid nitrogen and stored at −80°C.
Antibody validation
We only used antibodies that were validated as specifically recognizing the modification or protein, based on Western analysis of nuclear extracts, peptide blot analysis (see Supplemental Fig. 17), and in some cases, mass spectrometry analysis and IF analysis of cells (Kharchenko et al. 2011).
Histone antibodies
Commercial histone modification antibodies were tested for cross-reactivity with unmodified recombinant histones and other proteins by Western blotting according to standard protocols (Sambrook and Russell 2001). An antibody was considered validated if (1) there was no significant cross-reactivity with other proteins (<50%), and (2) if there was no significant cross-reactivity with the unmodified histone from the E. coli extract (>10-fold difference in signal intensity).
In addition, histone antibodies were tested by slot/dot blot analysis according to standard protocols (Sambrook and Russell 2001) with modified histone peptides (Diagenode), using amounts ranging from 100 pmol to 3 pmol on nitrocellulose membrane with 0.1-μm pore size.
Despite previous claims that Drosophila lacks the H3K9me3 modification, we observed that antibodies that recognize H3K9me3 colocalize extensively with H3K9me2 (Supplemental Fig. 2A) and HP1a (data not shown) in IF experiments. The specificity of the three H3K9me3 antibodies used in our studies were validated by Western blot analysis (Supplemental Fig. 2B), as well as peptide blot analysis, with no cross-reactivity with H3K9me2, and ∼10-fold higher binding to H3K9me3 versus H3K27me3 peptides (Supplemental Fig. 2C). In addition, we compared the abundance of H3K9me1, H3K9me2, and H3K9me3 in Drosophila S2 and human HeLa cells using quantitative mass spectrometry (Supplemental Fig. 2C). The results demonstrate that H3K9me3 is only ∼1.8-fold more abundant in HeLa than in S2 cells (Supplemental Fig. 2D). We conclude that Drosophila does contain significant amounts of H3K9me3, whose distributions and enrichments are similar but not identical to H3K9me2 (see Discussion).
Other proteins
Protein antibodies were tested for cross-reactivity with nontarget proteins, and specificity was tested on mutant protein extracts or extracts from RNAi knockdowns in S2 cells (RNAi experiments were carried out according to Worby et al. 2001). Western blotting was carried out according to standard protocols (Sambrook and Russell 2001). An antibody was considered validated if (1) there was a band of the correct size detected in the wild-type sample, which lessened in intensity in the knockdown/mutant sample (>50% depletion), and (2) if there was no significant cross-reactivity with other proteins (<50%).
ChIP-chip
Chromatin preparation, immunoprecipitation, and microarray hybridization were done as described in Schwartz et al. (2006) and Kahn et al. (2006) with the following modifications. To prepare chromatin from fly heads and larvae, the nuclei were isolated first and then cross-linked with 1% formaldehyde as a suspension in 1× PBS. Bioruptor (Diagenode) sonicator was used for solubilization and shearing of the chromatin. Cross-linked cultured cells were permeabilized with 1% SDS prior to ultrasound treatment. The ChIP products were amplified with Genome Plex Complete WGA Kit (Sigma) according to the manufacturer's recommendations, with omission of the first chemical DNA fragmentation step.
Data analysis
Enrichment profiles
For M-value normalization, the log-intensity ratio values (M-values) were calculated for all perfect-match (PM) probes as log2(ChIP intensity)–log2(input intensity). The M values were then shifted so that the mean is equal to 0. The smoothed log intensity ratios shown in the example plots were calculated using lowess with a smoothing span corresponding to 500 bp, combining normalized data from two replicate experiments. Enrichment P-values are calculated using a sliding window (1-kbp window size; step size 30 bp). The P-value enrichment score is calculated at each step using a one-sided t-test on the M-values of probes that fall within the window. To capture both significant enrichment and significant depletion, P-values for enrichment test (ePv) and depletion test (dPv) are calculated, and the score is given as -log10(min(ePv,dPv)). The score is then multiplied by −1 if dPv was smaller than ePv. Quality control measures for all ChIP-chip datasets included in our study are provided in Supplemental Table 1.
Chromatin states and gene clustering
To study combinatorial patterns (i.e., Fig. 4; Supplemental Fig. 4A), average enrichment was calculated for 500-bp blocks tiling the genome. The enrichment scores for each mark were shifted to a mean of 0 and scaled to a unit variance. To reduce excessive contribution of intercorrelated marks, the normalized matrix was projected to its principal components. The projected matrix was then used to cluster genome regions based on the similarity of the chromatin state. The problem of determining the number of clusters to use is complex, and no satisfying solution exists given the inherently inexact nature of the question. The number of clusters (15) was chosen because we thought it would sufficiently capture the variability. While the number of clusters was chosen to maximize the number of different patterns detected without creating redundancy, for ease of the reader, these 15 clusters were classified into five groups based on similarity. This classification results in some loss of information, which is why the original 15 cluster plots are provided as supplemental figures. Analogous procedures were used for gene clustering (i.e., Fig. 5), with each of the five cells representing gene enrichment in a particular mark being considered as an independent column. Again, we chose 10 states as a compromise between comprehensiveness and ease of interpretation.
Chromatin context of the repetitive elements
Repetitive elements within each class were identified using RepeatMasker (http://www.repeatmasker.org), excluding microsatellite repeats. Average enrichment was estimated based on the array probes that fell within the relevant repeat instances and 200-bp margins extending on either side of the identified instance boundaries. Complete-linkage hierarchical clustering was used to group repeat types based on the combined enrichment states (Fig. 6).
H3K9me2 enrichment across different cell types
For each cell line, continuous regions of H3K9me2 enrichment were determined using a Viterbi algorithm, based on a three-state hidden Markov model (HMM). The adjusted ChIP/input log intensity ratios were modeled using Gaussian emission probabilities, with means of −0.5, 0, and 0.5 corresponding to the enriched, neutral, and depleted states. All three emission signals utilized a fixed variance of 0.3 and a fixed transition probability of 1e-120 to transition between the states. Following HMM segmentation, a K-means clustering procedure was applied to the enrichment domains to determine patterns of H3K9me2 coverage across cell lines (Fig. 8A). The functional analysis of these patterns (Fig. 8B) utilized a more conservative set of loci matching the overall enrichment pattern shown in Figure 8A; i.e., a position included in the “common” regions (cluster 1) had to be covered by H3K9me2 HMM domains in all cell lines except for Kc (for WIG files of domains, see Supplemental material).
PCR and Southern analysis
PCR and Southern blots were carried out according to standard protocols (Sambrook and Russell 2001). For details, see Supplemental Methods.
Acknowledgments
We thank NIH and the NHGRI modENCODE project (U01HG004258 and R21-DA025720) for their support. We thank Sarah Gadel and Sarah Marchetti (Washington University) for technical assistance, and Dave MacAlpine (Duke University) and Sue Celniker (LBNL) for sharing their modENCODE data prior to publication. We thank the staff of the Bionomics Research and Technology Center of Rutgers University where the microarray processing and scanning were carried out. We also thank Sasha Langley and Serafin Colmenares for insightful comments that improved this manuscript.
Footnotes
-
↵10 Corresponding authors.
E-mail karpen{at}fruitfly.org.
E-mail selgin{at}biology.wustl.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.110098.110.
- Received May 9, 2010.
- Accepted December 8, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








