
Polycomb preferentially targets stalled promoters of coding and noncoding transcripts
- Daniel Enderle1,4,
- Christian Beisel1,4,
- Michael B. Stadler2,
- Moritz Gerstung1,
- Prashanth Athri1,5 and
- Renato Paro1,3,6
- 1 Department of Biosystems Science and Engineering, ETH Zurich, CH-4058 Basel, Switzerland;
- 2 Friedrich Miescher Institute for Biomedical Research, CH-4058 Basel, Switzerland;
- 3 Faculty of Science, University of Basel, CH-4056 Basel, Switzerland
-
↵4 These authors contributed equally to this work.
Abstract
The Polycomb group (PcG) and Trithorax group (TrxG) of proteins are required for stable and heritable maintenance of repressed and active gene expression states. Their antagonistic function on gene control, repression for PcG and activity for TrxG, is mediated by binding to chromatin and subsequent epigenetic modification of target loci. Despite our broad knowledge about composition and enzymatic activities of the protein complexes involved, our understanding still lacks important mechanistic detail and a comprehensive view on target genes. In this study we use an extensive data set of ChIP-seq, RNA-seq, and genome-wide detection of transcription start sites (TSSs) to identify and analyze thousands of binding sites for the PcG proteins and Trithorax from a Drosophila S2 cell line. In addition of finding a preference for stalled promoter regions of annotated genes, we uncover many intergenic PcG binding sites coinciding with nonannotated TSSs. Interestingly, this set includes previously unknown promoters for primary transcripts of microRNA genes, thereby expanding the scope of Polycomb control to noncoding RNAs essential for development, apoptosis, and growth.
Development of the adult animal from pluripotent embryonic tissue to highly differentiated cells is realized through lineage-specific gene expression patterns. In order to keep cells committed to a fate, the Polycomb group (PcG) and Trithorax group (TrxG) proteins ensure proper maintenance of transcriptional programs throughout development (Ringrose and Paro 2004; Schuettengruber et al. 2007; Schwartz and Pirrotta 2007; Müller and Verrijzer 2009). Polycomb proteins were first identified in Drosophila melanogaster but are widely conserved across metazoa, where they form specific chromatin complexes binding and repressing their corresponding target genes (Whitcomb et al. 2007). The Polycomb repressive complex 2 (PRC2) is known to trimethylate lysine 27 of histone H3 (H3K27me3) through the subunit Enhancer of Zeste (Cao et al. 2002; Czermin et al. 2002; Müller et al. 2002), while the Polycomb protein itself is part of the Polycomb repressive complex 1 (PRC1) and provides binding specificity to H3K27me3 via its chromodomain (Shao et al. 1999; Fischle et al. 2003; Min et al. 2003). In addition, PRC1 harbors activity for H2A ubiquitination at lysine 119 (Wang et al. 2004; Cao et al. 2005), shown to control transcription elongation of target genes (Stock et al. 2007; Zhou et al. 2008). Several other functions and enzymatic activities have been associated with PcG proteins (Gambetta et al. 2009; Eskeland et al. 2010), emphasizing the diverse mechanisms for transcriptional repression employed by this group. In contrast to PcG proteins, TrxG proteins, with Trithorax (TRX) as the best-characterized member, maintain the active state of gene expression. TRX is an H3K4-specific methyltransferase that undergoes single proteolytic cleavage, resulting in a heterodimer containing two parts termed TRX-N and TRX-C (Kuzin et al. 1994; Hsieh et al. 2003). It was found that TRX-C and PRC1 proteins co-occupy target sequences at inactive HOX genes and that TRX-C additionally binds to promoters of active genes (Beisel et al. 2007; Schwartz et al. 2010). Specific cis-regulatory PcG protein binding sites, termed PcG response elements (PREs) (Simon et al. 1993), were initially identified to regulate the fly HOX genes. But recent genome-wide studies in several organisms expanded the set of target genes substantially, pointing toward a broader role for PcG function (Oktaba et al. 2008; Schuettengruber et al. 2009; Schwartz et al. 2010).
A genome-wide map of chromatin factors binding patterns not only can shed light on the identity of the specific genes affected but may also provide insights into the chromatin structure underlying PcG silencing. We therefore utilized a comprehensive set of chromatin immunoprecipitations (ChIPs) followed by next-generation sequencing (ChIP-seq) to analyze binding profiles of PRC1 components and TRX-C at considerable detail. In contrast to previous studies, we identified thousands of high-confidence PcG binding sites in a single cell line, preferentially locating to core promoter regions of known genes. An annotation-agnostic approach to detect transcription start sites (TSSs) genome-wide allowed us to identify intergenic PcG binding sites at many currently nonannotated core promoters. Interestingly, our findings include promoters of previously unknown primary miRNA transcripts (pri-miRNAs), thereby unraveling a new additional layer of PcG control in Drosophila.
Results
Overview of ChIP-seq and RNA-seq data sets
To gain insight into the structure and function of PcG protein regulated chromatin in D. melanogaster, we used ChIP-seq to acquire high-resolution genome-wide maps of PRC1 components, TRX-C, and H3K4me3 in S2 cells. This cell line is also used by the modENCODE project, thus facilitating future comparative studies. Chromatin from S2 cells was immunoprecipitated using antibodies against Pc, Ph, Psc, TRX-C, or H3K4me3. Isolated DNA was sequenced using the Illumina Genome Analyzer platform, and the sequence reads were aligned to the Drosophila genome. In total, we created five genome-wide maps comprising 6.5–22.5 million aligned reads. In parallel, we isolated RNA from S2 cells and generated global gene expression profiles by RNA-seq to correlate protein binding with transcriptional activity. Furthermore, we surveyed the Drosophila genome for nonannotated TSSs using a newly adapted protocol for Illumina sequencing (termed 5′-MACE) with RNA isolated from S2 cells and embryos. Sequence read statistics are summarized in Supplemental Table 1. All data sets have been submitted to the NCBI Gene Expression Omnibus (GEO) database (accession no. GSE24521), and files containing data in summarized form have been added as supplemental material for visualization in the UCSC Genome Browser (Supplemental Files 1–3).
PcG proteins and TRX-C are enriched in promoter regions
To provide an overview of the genomic regions enriched in our ChIP experiments, we aligned all quality filtered sequence reads to the Drosophila genome and calculated the enrichment of reads in promoters (defined as all bases within 500 bp of a known RefSeq TSS), exons, introns, and intergenic regions (for a detailed definition of the genomic regions, see Methods section) (Fig. 1A). As indicated in previous studies, functionally active chromatin elements containing high-molecular-weight protein complexes have high sensitivity to sonication (Reneker and Brotherton 1991; Schwartz et al. 2004). Consistently, in our analysis sonicated DNA fragments present in the input chromatin fractions used for the ChIPs are enriched in promoters and exons (Fig. 1A). Subsequent analytical steps in this work accounted for this observation by calculating the ChIP enrichment over the input chromatin read count taken as background signal. In Drosophila, PcG proteins have been found to bind large genomic regions encompassing several kilobases, including several genes, or to bind in a considerable distance to gene promoters at PREs (Schwartz et al. 2006, 2010; Tolhuis et al. 2006; Schuettengruber et al. 2009). On the contrary PhoRC, a PcG complex with DNA binding activity, has been mapped to a region within 500 bp of TSSs (Oktaba et al. 2008). In agreement with the latter, all ChIP-seq experiments in the present work show significant enrichment of PcG proteins in promoters binding preferentially in a 500-bp window upstream of TSSs (Fig. 1A,B). Concomitantly, TRX-C binds in the same region, whereas H3K4me3, a mark for transcriptional activity, is positioned directly downstream from the TSS (Fig. 1A,B).
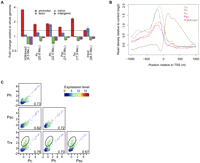
PcG proteins and TRX-C are enriched in promoter regions. (A) Enrichment of read alignments (number in parentheses) in gene promoters (±500 bp around TSS), exons, introns, and intergenic regions. All ChIPs show enrichment in the gene promoters compared with the input control (dotted line). For samples with two biological replicates (Pc, Ph, Psc, TRX, and Input), the bars indicate the mean enrichments, and the whiskers correspond to minimal and maximal enrichments. (B) Composite enrichment profiles relative to the chromatin input controls for all ChIPed proteins in 2-kb windows around nonoverlapping RefSeq TSS (n = 8977). (C) Pairwise scatter-plots of TSS enrichments between Pc, Ph, Psc, and TRX-C samples. Each dot represents one promoter, whereas the color of the dot indicates the expression level of the corresponding transcript, from blue (not expressed) to red (highly expressed). The Pearson correlation coefficient is depicted in the lower right corner of each scatter plot. Promoters with high TRX-C and low or absent PcG protein signals are encircled; note that the corresponding transcripts show generally a higher expression level compared to the ones located on the diagonal.
Next, we compared the binding of the ChIPed proteins globally at gene promoters. We calculated the enrichment signals within ±500-bp nonoverlapping windows of 8977 RefSeq TSSs and performed a pairwise correlation of all four proteins at these promoter regions. All four proteins show high pairwise correlation coefficients in the range from 0.67–0.82, indicating that PRC1 (in our study defined by the simultaneous binding of Pc, Ph, and Psc) and TRX-C co-occupy many gene promoters (Fig. 1C). Additionally, TRX-C seems to bind to a population of TSSs where the PRC1 protein signals are low or absent (Fig. 1C). Previous studies based on ChIP with microarray hybridization (ChIP-chip) indicate that TRX-C co-occupies PRC1-bound sites and binds to active promoters independent of PRC1 (Beisel et al. 2007; Schwartz et al. 2010). In order to compare protein binding and transcriptional activity, we performed a global gene expression analysis based on RNA-seq. TRX has been genetically defined as an anti-silencing factor counteracting PcG protein-mediated repression (Klymenko and Müller 2004). Indeed, the transcripts solely bound by TRX-C are expressed at higher levels compared with the ones co-occupied by PRC1 proteins (Fig. 1C). We separated transcripts into those with a promoter occupied by PRC1 and those with only TRX-C present and analyzed their expression level (Supplemental Table 2). TRX-C binding does not necessarily coincide with active transcription, and also, PRC1-bound genes can be repressed or actively transcribed (Fig. 2A; Supplemental Fig. 2). An analysis including the quantitative nature of ChIP-seq, however, indicates that PRC1 binding correlates with gene repression; genes with lower expression levels tend to have higher ChIP-seq enrichments for Pc, Ph, and Psc (Fig. 2B).
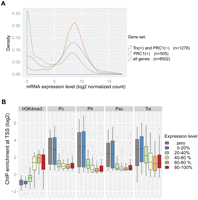
Expression levels of PcG and TrxG target genes. (A) Distributions of mRNA expression levels over all analyzed genes (green), genes only bound by TRX (orange), or PRC1-bound genes (violet). Numbers of mRNAs in the three different classes are indicated. (B) Genes were classified into nonexpressed (log2 mRNA expression level below 5.0, gray) and five groups of increasing expression, each containing the same number of genes (blue to red). ChIP-seq enrichments at the TSS are shown as box plots for each expression level group, with the horizontal line indicating the median, the lower and upper box limits the first and third quartiles, and the lines below and above the box extending to the minimum and maximum enrichments, respectively.
PcG proteins preferentially bind stalled promoters
In Drosophila and mammals, PRC1 has been proposed to mediate gene repression either by regulating the initiation phase of RNA polymerase II (Pol II) or, at a later step, by stalling Pol II elongation (Dellino et al. 2004; Stock et al. 2007). Promoters occupied by stalled Pol II are characterized by the presence of small promoter-proximal RNAs. This class of RNAs has been quantitatively measured by Illumina sequencing in Drosophila S2 cells (Nechaev et al. 2010), enabling us to test in a genome-wide fashion if PRC1 is bound to promoters associated with stalled Pol II. Indeed, PRC1-bound promoters produce more small promoter-proximal RNAs than do non-PRC1-bound promoters (Fig. 3A). Moreover, we found PRC1 preferentially occupying promoters associated with small RNAs (Fig. 3B). We also compared PRC1 binding with the Pol II stalling index of each promoter, which corroborated our view that PRC1 preferentially binds to promoters associated with stalled Pol II (Fig. 3C; Nechaev et al. 2010).
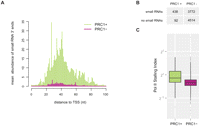
PRC1 preferentially binds stalled promoters in S2 cells. (A) PRC1-bound promoter exhibits a higher abundance of promoter-proximal short RNA 3′-ends indicative of increased Pol II stalling (Nechaev et al. 2010). (B) PRC1 largely binds promoter producing small RNAs (≥1 read from 5′- and 3′-end libraries). (C) Pol II preferentially remains stalled at PRC1-bound promoter as calculated by the ratio of promoter-proximal occupancy versus gene body. The two populations are significantly different (P-value < 2.2 × 10−16, two-sample Kolmogorov-Smirnov test).
PcG proteins target an unexpectedly large set of gene promoters
To gain an unbiased insight into the regulatory capacity of the PcG/TrxG system (up to this point we concentrated our analysis on a nonoverlapping set of 8977 TSS regions), we employed MACS for peak detection and mapped 2826, 4402, 2108, and 5240 binding sites for Pc, Ph, Psc, and TRX-C proteins, respectively (Zhang et al. 2008). In order to increase the stringency for defining PcG/TrxG-regulated genomic sites, we determined the genomic regions that showed overlapping peaks from any three of the four analyzed proteins. Since TRX-C showed a high pairwise correlation with any of the three PcG proteins of the PRC1 complex in the promoter enrichment analysis (Fig. 1C), we included TRX-C for the determination of PcG targets. In total, we determined 2274 overlapping regions, which we defined as PcG binding sites (Supplemental Table 3). At 854 PcG binding sites, MACS detected all four proteins. With further visual inspection of a subset of the other 1420 sites, we would propose that all 2274 PcG binding sites are co-occupied by all four proteins, with one of the proteins falling below the enrichment cut-off we applied in addition to MACS (see Methods), demonstrating a close interplay of PRC1 and TRX-C.
Driven by our sequence read enrichment analysis (Fig. 1A,B), we analyzed the distribution of the PcG binding sites and found that ∼50% are localized within a ±500-bp window of TSSs (Supplemental Table 3). We consider the corresponding genes as a high-confidence target set of the PcG/TrxG system, significantly increasing the number of PcG target genes compared with previous genome-wide mapping studies (for more details, see Supplemental Table 4; Supplemental Fig. 4; Oktaba et al. 2008; Schuettengruber et al. 2009; Schwartz et al. 2010). Given the high number of PcG target genes we identified, we performed a gene enrichment analysis indicating transcription factors decisive for animal development as primary targets for the PcG/TrxG system (Table 1).
Functional categories enriched in PcG targets
PcG proteins bind to many nonannotated TSSs
So far our findings revealed an unexpectedly large proportion (∼50%) of PcG binding sites associated with gene promoters. In the light of these data, we were speculating how many of the other PcG binding sites located in exons, introns, and intergenic regions may represent TSSs that have not yet been annotated (Fig. 4C, right half of the plot). Supporting evidence for such correlations came from our studies of ncRNA transcription within the homeotic bithorax complex (BX-C), where we found that the well-characterized intergenic PcG binding sites Fab-7 and Fab-8 carry TSSs for long noncoding transcripts (Supplemental Text 1). Indeed, noncoding transcription is a well-known feature of the BX-C (Schmitt et al. 2005; Lempradl and Ringrose 2008), and promoter activity of PREs is consistent with known features such as nuclease hypersensitivity and elevated histone replacement (Mito et al. 2007; Deal et al. 2010). In order to test our hypothesis that isolated PcG binding sites are also located at promoters, we engaged in experimentally identifying TSSs in a genome-wide fashion. We turned toward methods based on 5′ rapid amplification of cDNA ends (5′-RACE) (Maruyama and Sugano 1994) that indirectly detect transcription start events by selecting for the 7-methylguanosin modification at the very 5′-end of capped RNAs. The TSSs detected do not occur isolated but are arranged in regions generally referred to as a TSS cluster with the core promoter region loosely defined as the genomic region surrounding it (Carninci et al. 2006; Sandelin et al. 2007). To this purpose, we developed our own specific protocol, related to previous methods (Olivarius et al. 2009; Tsuchihara et al. 2009), which we dubbed 5′-MACE (massive amplification of cDNA ends). Shortly, we combined RNA oligo-capping with reverse transcription using tagged random hexamers to generate a cDNA library of 5′-ends suitable for massive parallel sequencing using Illumina's Genome Analyzer platform (Supplemental Fig. 3C). Sequence reads from a 5′-MACE library should thus directly correspond to the very first nucleotides transcribed during the initiation event. As expected, the vast majority of reads obtained from S2 cells can be mapped uniquely to known promoter regions (Supplemental Fig. 3A), where they faithfully denote the TSS of the current genome annotation (Fig. 4A) and reflect the mRNA expression level of the corresponding gene (Pearson correlation = 0.76) (Supplemental Fig. 3B). As 5′-MACE samples from S2 cells can only serve to detect core promoters active in that particular cell type, we also generated a library from 0- to 16-h-old embryos as a sample representing TSSs active during embryonic development. Similar strategies to discover promoters in mixed tissues have already been successful in Drosophila (Ahsan et al. 2009; Ni et al. 2010). Using MACS peak detection, we are able to identify a total of 9626 TSS clusters in the pooled data set. The position of TSS clusters relative to the annotated RefSeq TSSs revealed a major population representing known TSSs (close to the annotated TSS) and another one indicating a nonannotated set of TSSs (Fig. 4B). Using a conservative cut-off of ±500 bp, we can identify about 2000 novel TSSs in our data set (Fig. 4B, red mark). We went on to utilize this information in finding PcG peaks near to novel TSSs. By plotting all the observed PcG binding sites in S2 cells according to their relative distance toward the TSSs, we can indeed reveal an additional population previously not recognized to be located at the TSSs (Fig. 4C, lower right quarter). In total, we were able to uncover another ∼10% of PcG binding sites at the newly detected TSSs, showing that the majority of PcG binding sites in S2 cells are present at core promoter regions.
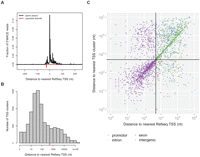
PcG proteins bind nonannotated TSSs. (A) 5′-MACE detects annotated transcription starts shown by metagene analysis of the read distribution. (B) 5′-MACE detects a TSS cluster far from known promoter regions. The red line marks a distance of ±500 bp. (C) PcG binding sites in S2 cells scattered according to their distance to the RefSeq TSS and TSS cluster found by 5′-MACE. The crosshair marks a distance of ±500 bp, and the newly identified PcG-bound promoter can be found in the lower right quarter.
PcG proteins bind promoters of primary microRNA transcripts
We questioned whether the nonannotated TSSs bound by PcG proteins merely consist of alternative promoters for already known genes or might contain novel ncRNA targets under PcG control. Intriguingly, we noticed PcG binding sites in the intergenic regions surrounding loci coding for microRNAs, one example of which is miR-278 (Fig. 5). TSSs at PcG binding sites are sometimes found in the same intergenic region but over 40 kb away from the miRNA sequence. miRNAs are transcribed as precursors, known as primary miRNAs (pri-miRNAs) that can be many kilobases in size and are rapidly processed to mature miRNAs complicating their annotation (Yi et al. 2003; Kadener et al. 2009). We used RT-PCR with primers located at the TSSs and the mature miRNA to amplify the potential transcript and sequenced the cloned cDNAs to unravel the gene structure of the pri-miRNA locus. To this end, we were able to confirm nine out of 11 PcG binding sites tested to be genuine promoters of pri-miRNA transcripts (Table 2; Supplemental Text 2; Fig. 5). This includes the transcription unit of pri-mir-iab8, which we further confirmed by 5′- and 3′-RACE experiments (Fab8-RA) (Supplemental Text 1; see Fig. 7, below). Interestingly, these transcripts contain miRNAs well known for their role in early and late development like mir-iab8, mir-184, and mir-279 (Cayirlioglu et al. 2008; Tyler et al. 2008; Iovino et al. 2009) as well as for regulation of growth and apoptosis, namely, bantam, mir-8, mir-14, and mir-278 (Hipfner et al. 2002; Brennecke et al. 2003; Xu et al. 2003; Nairz et al. 2006; Teleman et al. 2006; Karres et al. 2007; Varghese and Cohen 2007; Hyun et al. 2009). Although mir-282 and mir-275 lack thorough functional evidence, several EP insertions at the mir-282 locus were identified in screens for wing development and circadian rhythm (Molnar et al. 2006; Bejarano et al. 2008; Dubruille et al. 2009) while the presumptive core promoter of pri-mir-275 maps exactly to the cuckold mutation (chr2L: 7,423,896–7,423,995) (Castrillon et al. 1993), strongly suggesting that the observed behavioral defect for cuc1 is caused by a deregulation of the pri-mir-275 transcript. In Drosophila, <50% of all miRNA loci overlap with protein-coding transcripts with the majority remaining as intergenic. To get an overview of PcG control on annotated and nonannotated miRNA genes, we conducted a visual inspection of all miRNA loci using the UCSC Genome Browser (see Methods). Out of the 157 miRNAs listed in miRBase, we found a surprisingly large number of 41 miRNAs to be targeted by PcG proteins (Fig. 6; Supplemental Table 5). Twenty-six out of those miRNAs are located within intergenic regions, including the nine miRNA genes newly annotated in this study. In total, our results show Drosophila miRNAs to be targeted by PcG proteins to an unprecedented extent, revealing pri-miRNAs as a new class of PcG target genes.
Intergenic pri-miRNA transcripts targeted by PcG proteins
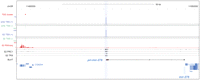
Intergenic PcG binding site acts as promoter for a pri-miRNA. Screenshot from the UCSC Genome Browser including data from ChIP-seq, RNA-seq, and 5′-MACE detecting TSS clusters. The pri-mir-278 transcript (BLAT alignment, black) emanates from a PcG-bound promoter located over 40 kb upstream of the mature miRNA.
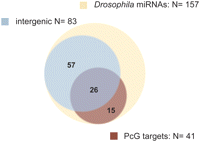
PcG proteins bind the promoter of primary miRNA transcripts. Venn diagram showing 41 Drosophila miRNAs that qualify as PcG targets (see Methods), including many loci lacking information about the primary transcription unit.
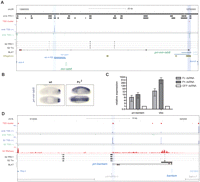
PcG proteins regulate transcription of pri-mir-iab8 and pri-bantam. (A) Screenshot from the UCSC Genome Browser covering the intergenic region between Abd-B and abd-A. TSS clusters at core promoter of pri-mir-iab8 (BLAT result, black) and iab4-RB locate to embryonic PcG binding sites (“emb PRC1”) (Schuettengruber et al. 2009). Fab-8 region (chr3R:12,740,929–12,748,923) (Barges et al. 2000), IAB8 enhancer (chr3R:12,744,584–12,749,967) (Zhou et al. 1999), and transcription factor binding sites (ORegAnno) (Griffith et al. 2008) as indicated. (B) In situ hybridization on whole-mount Drosophila embryos show a derepression of pri-mir-iab8 in homozygous Pc[3] mutant embryos. (C) S2 cells treated with dsRNAs against PRC1 components show derepression of the PcG target genes Ubx and pri-bantam. (D) Screenshot covering the intergenic region between Reg-2 and CG12030. The core promoter of the ∼20-kb pri-bantam transcript is bound by PRC1 and TRX proteins in S2 cells. Three primary transcripts found are shown as BLAT result and boundaries of known lesions and insertions are indicated (Hipfner et al. 2002).
Polycomb group proteins regulate transcription of miRNA genes
Our analysis showed that PcG proteins bind several miRNA promoters. To assess whether this association indicated a functional relationship, we studied the transcriptional control of two developmentally interesting miRNAs. The mature miR-iab8 is encoded in a cis-regulatory region of the BX-C called iab4, building a sense/antisense pair together with miR-iab4 (Fig. 7; Stark et al. 2008). Despite its location, it was termed miR-iab8 since the primary transcript was genetically predicted to reach to the iab8 region >60 kb distant from the mature miRNA (Bender 2008). Indeed, we were able to map pri-mir-iab8 as a long, spliced transcript starting within the IAB8 and terminating at a consensus polyadenylation site 1 kb proximal to the abd-A promoter (Fig. 7A; data not shown). 5′-MACE identifies two TSS clusters between abd-A and Abd-B, one of which is the core promoter of pri-mir-iab4, supporting the notion that the previously cloned iab4 transcript represents the most frequently used TSS (CR31271) (Cumberledge et al. 1990). The other TSS cluster confirms the 5′-end of pri-mir-iab8 adjacent to the Fab-8 PRE region at a site of early transcription factor binding (ORegAnno and BLAT annotation; Barges et al. 2000; Griffith et al. 2008). Consistently, the “IAB8 enhancer” region, including the promoter of pri-mir-iab8, is sufficient to direct parasegment-specific transgene expression (Fig. 7A; Zhou et al. 1999). In order to assess if Polycomb controls the transcription of pri-mir-iab8, we performed in situ hybridization against whole-mount Drosophila wild-type (wt) and Polycomb mutant (Pc3) embryos (Fig. 7B). The wild-type expression pattern in the central nervous system of PS15 to PS13 is similar to previous observations with several probes along this locus, suggesting that they also reflect pri-mir-iab8 expression (Sánchez-Herrero and Akam 1989; Bae et al. 2002; Rank et al. 2002; Stark et al. 2008). This expression is strongly dependent on Polycomb, as null mutant embryos show derepression along the complete anterior-posterior axis (Fig. 7B). A similar control by PcG can be found at the bantam locus, which is located in a transcriptional active genomic stretch of 40 kb without annotated genes. Although the mature bantam miRNA is much smaller, the original ban1 deletion spans 21 kb, containing additional sequences necessary for bantam function (Hipfner et al. 2002; Brennecke et al. 2003). We detected two strong TSSs in this region, one of which is located at the insertion site of the previously described EP3219 line and bound by PcG proteins (Fig. 7D; Hipfner et al. 2002). This promoter seems to connect pri-bantam transcription to the hippo pathway via Hth and Yki (Peng et al. 2009) and produces a long primary transcript processed by drosha (Kadener et al. 2009). We were able to confirm this promoter to be connected to miR-bantam through a cDNA from a spliced transcript, although the RNA-seq profile rather suggests the unspliced transcript to be the predominant form (Fig. 7D, Supplemental Text 2). To confirm the regulation of the bantam locus by the PcG system, we treated S2 cells with dsRNA targeting the PRC1 components Pc or Ph. Indeed, the knock down of both PcG proteins resulted in a robust increase of pri-bantam transcription (Fig. 7C). Altogether, we were able to identify the gene structure of pri-miRNAs and demonstrated that withdrawal of PcG proteins results in the derepression of these novel PcG targets.
Discussion
In this study we used a sensitive ChIP-seq approach on three components of the PRC1 complex and TRX to map thousands of highly resolved chromatin binding sites, revealing that these important chromatin factors preferentially target stalled promoters of coding and noncoding transcripts. In detail, the following main conclusions can be drawn from our analysis: (1) By comparing the chromatin binding profiles with other genome-wide data sets comprising RNA-seq, Pol II stalling, and the mapping of nonannotated TSSs, we found that PcG proteins are highly enriched at gene promoter regions. Chromatin binding profiles of PcG complexes have been mapped in several vertebrate and invertebrate species. While research in the mammalian system has already been concentrated on PcG-bound promoter regions, differing observations among PRC1 components in Drosophila led to the picture of rather large Polycomb-bound domains. Here we report a distribution similar to that observed in mammals, indicating that promoter-bound PRC1 might be a broadly conserved feature of the PcG system. (2) The distinct colocalization at TSSs allowed us to determine an unprecedented rich set of target genes. Using a conservative setting, we can assign about 1000 PcG target genes corresponding to ∼7% of all Drosophila genes, a number similar to the mammalian system (Ku et al. 2008). (3) The use of a homogenous cell population coupled with quantitative chromatin and transcription profiling based on high-throughput sequencing enabled us for the first time to directly correlate protein binding with gene activity on a genome-wide scale in Drosophila. Surprisingly, PcG binding seems to be largely compatible with high as well as low gene expression. Nevertheless, we were able to demonstrate a significant positive correlation between lower levels of mRNA abundance and PcG ChIP signal strength. This supports a model in which PcG proteins are used not only to completely repress their target genes but also to fine-tune their expression level. This may be achieved via modulating the rate of RNA polymerase II (Pol II) elongation, previously recognized as a function characteristic of PRC1 in mammals (Stock et al. 2007). (4) For the first time, using a genome-wide approach, we observe a positive correlation of PRC1 binding with stalled Pol II. In addition, we find stalled genes to share common functional classes with PcG targets in S2 cells (cf. Table 1; Muse et al. 2007), including HOX genes as classical targets of PcG function (Chopra et al. 2009b). Together this further suggests that regulation of Pol II promoter-escape is a conserved feature of PcG silencing. It remains to be addressed whether the small RNAs involved are a mere by-product of PRC1 function in Pol II stalling or, as suggested previously by experiments on human cell lines (Kanhere et al. 2010), are actively involved in recruiting PcG complexes. Also, functional experiments are needed to exclude the possibility of stalled promoters retaining PRC1 to consolidate repression. (5) We can assign isolated PcG binding sites experimentally to nonannotated TSSs, opening the discussion if all PREs might act as promoters of coding/noncoding RNAs. As our results repeatedly indicated that PRC1 binds and functions in the vicinity of promoters, we were interested to further investigate the ∼50% of PcG binding sites that were not associated with annotated promoters. Indeed, using an annotation-agnostic approach for TSS identification, we were able to reveal many more PcG binding sites associated with core promoter regions. This finding stands out against the current model of PcG silencing in which PcG proteins bind to dedicated cis-regulatory elements and control the transcription of the target genes through looping to the promoter region (Schwartz and Pirrotta 2007; Mateos-Langerak and Cavalli 2008). Both views might be compatible in some cases, for example, at the well-described cis-regulatory elements Fab-7 and Fab-8. Strong PcG protein binding as well as stalled polymerase at their promoter regions may account for the dual function of these elements as insulator and PREs (Mihaly et al. 1997; Chopra et al. 2009a). As we are likely to underestimate the total number of TSSs in Drosophila, we still expect many of the intergenic PcG binding sites to be alternative TSSs of known genes or even novel transcription units. In support of the latter case, we were able to identify new pri-miRNA transcripts coding for known intergenic miRNAs. (6) We find primary miRNAs to constitute a new class of PcG targets, further expanding the regulatory capacity of the PcG/TrxG of epigenetic regulators. Many pri-miRNAs are highly regulated (Newman and Hammond 2010; Ryan et al. 2010), very long, and inherently instable (Yi et al. 2003; Kadener et al. 2009), and so, previous research suffered from little information on the position of corresponding TSSs and regulatory proteins. Our approach allowed us to reveal a surprisingly large number of pri-miRNAs as candidates for PcG regulation, and the identified associated functions blend well into the current picture of a typical PcG target gene. Notably, knockout of the miRNA biogenesis core components, ago-1 and dcr-1, leads to severe segment polarity defects (Meyer et al. 2006), suggesting an important role for miRNAs in Drosophila embryonic development, akin to the one observed in mouse (Bernstein et al. 2003; Spruce et al. 2010). In addition, HOX loci are known to host at least three miRNA genes (Lagos-Quintana et al. 2001; Bender 2008; Stark et al. 2008; Tyler et al. 2008), one of which was previously shown to be regulated by Polycomb (Ronshaugen et al. 2005). Among the suspected targets are the HOX genes themselves (Stark et al. 2008; Tyler et al. 2008; Thomsen et al. 2010), thus providing an opportunity for direct regulatory feedback. Another interesting aspect is the novel connection to important growth-regulating miRNAs such as bantam and mir-8, in line with known PcG targets in cell cycle control (Martinez et al. 2006; Oktaba et al. 2008). The complete region of the ban1 deletion has been shown as a strong aneuploid segment in S2 cells (Zhang et al. 2010b), a feature reminiscent of human cancer cell lines (Weaver and Cleveland 2006). Although a repression of pro-proliferative miRNA loci is consistent with a general role of PcG proteins as tumor suppressors (Classen et al. 2009; Martinez and Cavalli 2010), growth effects of PcG proteins seem to be rather diverse and context-dependent when examined in detail (Beuchle et al. 2001; Saj et al. 2010). Accordingly, PcG proteins in mammals also show antagonistic proliferative functions (Lessard et al. 1999; Zhang et al. 2010a), indicating a complex regulatory relationship to growth control in both organisms. In stark contrast to bantam, mir-8 seems to be functionally conserved and controls growth via the PI3K pathway in flies as in humans (Hyun et al. 2009)—possibly engaging in a regulatory feedback, as also demonstrated for mir-214 (Juan et al. 2009; Iliopoulos et al. 2010). Furthermore, examples for PcG-controlled miRNAs begin to emerge in mammals (Marson et al. 2008; Wang et al. 2008; Juan et al. 2009), so it would be interesting to see if miRNAs regulating growth and development are a conserved feature set among PcG targets. In an even broader perspective, various miRNAs, just as PcG proteins, are involved in stem cell maintenance and cancer progression (Calin and Croce 2006; Sparmann and van Lohuizen 2006; Marson et al. 2008; Mills 2010). We are confident that further research into the interconnection of miRNA control by the PcG silencing systems will uncover interesting new details on the regulatory gene networks controlling development and homeostasis.
Methods
S2 cell culture
D. melanogaster S2-DRSC cells (obtained from the Drosophila Genomics Resource Center) were cultured in Schneider's Drosophila medium (Invitrogen) supplemented with 10% FCS (Hyclone).
ChIP and RNA isolation
Chromatin fixation and immunoprecipitation were performed essentially as described by Orlando et al. (1997). Cells (1 × 109) were fixed in 200 mL of medium with 1% formaldehyde for 10 min at room temperature. Cross-linked cells were sonicated to produce chromatin fragments of an average size of 200–400 bp. Soluble chromatin was separated from insoluble material by centrifugation. The supernatant containing chromatin of 5 × 107 cells was used for immunoprecipitation. Psc and Ph antibodies were described by Strutt and Paro (1997), and Pc and TRX-C antibodies were described by Beisel et al. (2007). Anti-H3K4me3 was purchased from Millipore. RNA was isolated using TRIzol reagent (Invitrogen) following the manufacturer's instructions.
Preparation of ChIP-seq and mRNA-seq libraries
Sequencing libraries were prepared with the Illumina mRNA-Seq 8-Sample Prep kit and ChIP-Seq DNA Sample Prep kit according to Illumina's instructions. After adapter ligation, library fragments of ∼250 bp were isolated from an agarose gel. The DNA was PCR amplified with Illumina primers for 15 (RNA-seq) and 18 (ChIP-seq) cycles, purified, and loaded on an Illumina flow cell for cluster generation. Libraries were sequenced on the Genome Analyzer II and Genome Analyzer IIx following the manufacturer's protocols.
Preparation of 5′-MACE libraries
Ten micrograms of TRIzol-extracted, TurboDNAse-digested total RNA was dephosphorylated with CIAP (20 U, Invitrogen), decapped with TAP (1 U, Epicentre), and ligated to 0.42 μg of RNAoligo (35.5 pmol) with T4RNA ligase (10 U, Epicentre). The RNAoligo sequence corresponds to the sequence of Illumina's Genomic DNA PCR Primer 1.1 with the addition of three adenines at the 3′-end (5′-ACACUCUUUCCCUACACGACGCUCUUCCGAUCUAAA-3′). Column-based purifications (RNeasy MinElute, QIAGEN) were used between steps to clean reactions from enzymes and excess RNAoligo. In order to create a pool of first-strand cDNAs with a suitable size distribution, half of ligation product was reverse-transcribed with Supercript III (Invitrogen) and 1 μL of tagged random-hexamers (2 μM, 5′-CAAGCAGAAGACGGCATACGAGCTCTTCCGATCTNNNNNN-3′). To create second-strand cDNA for size-selection on a 1.5% agarose gel, half of the first-strand cDNA was subjected to a limited PCR amplification (eight cycles) using Phusion polymerase (NEB) and Genomic DNA PCR Primer 1.1/2.1. cDNAs running at 350 ± 50 bp were gel-extracted (QIAquick, QIAGEN), and half of the eluate was used in PCR enrichment (18 cycles) for cluster generation according to Illumina's standard protocols. The final cDNA library corresponds to capped transcripts present in ∼1.25 μg of total RNA, and sequencing reads after the initial three adenines match the first nucleotides transcribed. The pooled 5′-MACE reads originate from libraries of Drosophila S2-DRSC cells and 0- to 16-h-old Drosophila embryos, respectively.
Genomic coordinates
The April 2006 D. melanogaster genome assembly (dm3, BDGP Release 5) provided by the Berkeley Drosophila Genome Project (BDGP) (http://www.fruitfly.org/) was used as a basis for all analyses. Annotation of known RefSeq transcripts was obtained from UCSC (http://hgdownload.cse.ucsc.edu/goldenPath/dm3/database/refGene.txt.gz from February 7, 2010). Four types of genomic regions were defined as follows: “promoter” contains all bases within 500 bp of a known RefSeq TSS; “exon” are all nonpromoter bases that overlap exons of RefSeq transcripts; “intron” are all nonpromoter/nonexon bases that are flanked by two exons of a single transcript; and all remaining bases were assigned to the “intergenic” region type. A nonredundant, nonoverlapping set of TSS regions (n = 8977) was generated starting with 1000-bp windows centered at RefSeq TSSs (n = 14,388) and removing all overlapping windows and all windows on chromosomes chrU and chrUextra.
Evaluation of primary microRNAs
All miRNA information has been downloaded from miRBase (release 14: Sept 2009) and analyzed using the UCSC Genome Browser (D. melanogaster April 2006; BDGP R5/dm3 assembly). The genome annotation was provided by FlyBase (v5.12, October 2008) (Kent 2002; Kent et al. 2002; Griffiths-Jones et al. 2008; Tweedie et al. 2009; Rhead et al. 2010). We scored a miRNA as a potential PcG target, if the promoter of the corresponding transcript lies within ±500 bp of a PcG binding site. We also included promoters found within embryonic Pc/H3K27 domains (Schuettengruber et al. 2009). If the miRNA locus was not associated with a transcript yet, we considered all PcG binding sites or embryonic domains within the intergenic region upstream of the miRNA.
Molecular cloning and analysis of primary microRNAs
For identification of primary microRNAs, 2 μg of TRIzol-extracted, TurboDNAse-digested RNA was reverse transcribed using Superscript III and random hexamers. Ten percent of the reaction was subjected to PCRs along with a control cDNA reaction lacking reverse transcriptase. For amplification we used Phusion polymerase (Finnzyme) and primer pairs spanning the region between 5′-MACE signal and corresponding mature miRNA locus. PCR products were gel-extracted using QIAEX II (QIAGEN), subcloned into pCR-II TOPO vectors, and Sanger-sequenced using standard primers. Exceptions are pri-bantam and pri-mir-iab8, which were amplified using Fermenta's Maxima RT with nested PCR or identified in a 5′- and 3′-RACE using the Invitrogen's GENERACER kit, respectively. For functional analysis, S2 cells were subjected to a 7-d incubation with 3.5 μg of T7 RNA polymerase-derived dsRNA in a 24-well plate format. Relative quantification (2−ΔΔCt) with qRT-PCR was performed and analyzed on a LightCycler480 system using SYBR Green Master Mix from Roche.
Acknowledgments
We thank I. Nissen for sample processing for Illumina sequencing and M. Kohler for data processing. Illumina sequencing was carried out at the Laboratory of Quantitative Genomics, D-BSSE, ETH Zurich. Drosophila S2-DRSC cells were received through the Drosophila Genomics Resource Center. R.P. was funded by grants from the DFG, the EU-NoE “Epigenome,” and the ETH Zurich.
Footnotes
-
↵6 Corresponding author.
E-mail renato.paro{at}bsse.ethz.ch; fax 41-61-387-39-96.
-
[Supplemental material is available for this article. The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE24521.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.114348.110.
- Received October 8, 2010.
- Accepted November 22, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








