
Phylogeny of the Serpin Superfamily: Implications of Patterns of Amino Acid Conservation for Structure and Function
- 1Department of Biochemistry and Molecular Biology, Monash University, Clayton Campus, Melbourne, Victoria 3168, Australia; 2Wellcome Trust Centre for the Study of Molecular Mechanisms in Disease, Cambridge Institute for Medical Research, University of Cambridge Clinical School, Cambridge CB2 2XY, United Kingdom
Abstract
We present a comprehensive alignment and phylogenetic analysis of the serpins, a superfamily of proteins with known members in higher animals, nematodes, insects, plants, and viruses. We analyze, compare, and classify 219 proteins representative of eight major and eight minor subfamilies, using a novel technique of consensus analysis. Patterns of sequence conservation characterize the family as a whole, with a clear relationship to the mechanism of function. Variations of these patterns within phylogenetically distinct groups can be correlated with the divergence of structure and function. The goals of this work are to provide a carefully curated alignment of serpin sequences, to describe patterns of conservation and divergence, and to derive a phylogenetic tree expressing the relationships among the members of this family. We extend earlier studies by Huber and Carrell as well as by Marshall, after whose publication the serpin family has grown functionally, taxonomically, and structurally. We used gene and protein sequence data, crystal structures, and chromosomal location where available. The results illuminate structure–function relationships in serpins, suggesting roles for conserved residues in the mechanism of conformational change. The phylogeny provides a rational evolutionary framework to classify serpins and enables identification of conserved amino acids. Patterns of conservation also provide an initial point of comparison for genes identified by the various genome projects. New homologs emerging from sequencing projects can either take their place within the current classification or, if necessary, extend it.
The serpins are a superfamily of proteins, typically 350–400 amino acids in length, with a diverse set of functions including, but not limited to, inhibition of serine proteinases in the vertebrate blood coagulation cascade (Huber and Carrell 1989; Marshall 1993). Serpins are of clinical interest because mutations cause a number of disease states—for example, blood clotting disorders, emphysema, cirrhosis, and dementia—many of which are consequences of polymerization (see Carrell and Lomas 1997). Serpins are also of interest in the context of general protein structure and folding studies because of their dramatic conformational changes and the existence of metastable states.
Several hundred serpins can be identified in higher eukaryotes and viruses. However, despite their appearance in animals and plants, no ancestral homolog from prokaryotes or fungi has yet appeared. One of the findings we report here is our failure, despite extensive database mining, to identify one.
Not all serpins function as proteinase inhibitors. Those that do most commonly inhibit chymotrypsin-like serine proteinases, but some are “cross-class” inhibitors of other types of proteinases. For example, the viral serpin crmA inhibits interleukin-1β–converting enzyme (Komiyama et al. 1994), and Squamous Cell Carcinoma Antigen-1 (SCCA-1) inhibits cysteinyl proteinases of the papain family (Schick et al. 1998). Non-inhibitory serpins perform diverse functions, including roles as chaperones (the 47-kD heat shock protein [HSP47]; Clarke et al. 1991) and hormone transport proteins (e.g., cortisol-binding globulin [CBG]; Hammond et al. 1987) (see Table1)
Role of Members of the Serpin Superfamily
Figure 1A shows the structure of native α1-antitrypsin (Elliott et al. 1996) and defines the nomenclature of the secondary structural elements. Typically, serpins contain three β-sheets and nine α-helices. The reactive center loop (RCL), shown in magenta in Figure 1, is crucial for the function of inhibitory serpins undergoing large structural changes that alter the folding topology of the molecule (Fig. 1B). In α1-antitrypsin, the RCL comprises residues P17–P4′, in the notation of Schechter and Berger (1967), and contains the scissile bond between residues P1 and P1′, cleaved by the target proteinase.
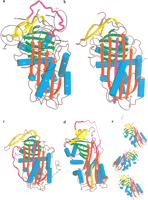
(A) The structure of native α1-antitrypsin. (B) Cleaved α1-antitrypsin. (C) Latent antithrombin. (D) δ-Antichymotrypsin. Part of the F-helix is unwound and inserted into the bottom of the A β-sheet (orange). (E) Polymer of cleaved antitrypsin. Residues P5–P4′ in the RCL, part of which (P5–P1) are making the β-strand linkage, are shown in light green. In all parts of Figure 1, the A β-sheet is in red, the B β-sheet in green, the C β-sheet in yellow, and the reactive center loop (RCL) in magenta. The helices are represented by cylinders colored cyan. Elements of secondary structure are labeled as follows: (hA, hB, etc.) A-helix, B-helix, etc.; (s1A, s2A, etc.) strand 1 of the A β-sheet, strand 2 of the A β-sheet, etc. The important breach, shutter, gate, and hinge regions are indicated by broken circles.
Five conformational states—native, cleaved, latent, δ, and polymeric—appear in serpin crystal structures (Fig. 1A–E). They differ primarily in the structure of the RCL (see Whisstock et al. 1998). In the native state (Fig. 1A), the RCL is exposed and, for inhibitory serpins, accessible for interaction with a proteinase. Upon cleavage of the scissile bond, the reactive center loop forms an additional strand inserted into the A β-sheet, with concomitant conformational changes elsewhere in the molecule (Fig. 1B) (Stein and Chothia 1991; Whisstock et al. 2000a). Cleavage is typically associated with an increase in stability. The native to cleaved change is called the “stressed to relaxed” (S→R) transition (Carrell and Owen 1985). A substate of the native conformation is seen in the X-ray crystal structure of antithrombin, in which the RCL is partially inserted into the A β-sheet (Carrell et al. 1994; Schreuder et al. 1994; Whisstock et al. 2000b).
The latent state is an uncleaved state in which the RCL is inserted into the A β-sheet, as in the cleaved form; this is an alternative R state (Fig. 1C). The latent state was first seen in the crystal structure of Plasminogen Activator Inhibitor-1 (PAI-1; Mottonen et al. 1992). The transition in PAI-1 from the native, active form to the latent, non-inhibitory conformation provides a fine level of functional control, limiting the active lifetime of PAI-1 to a few hours (Levin and Santell 1987). The latent state also occurs in the crystal structure of antithrombin (Carrell et al. 1994; Skinner et al. 1997) (Fig. 1C), and there is evidence for its existence in α1-antitrypsin (Lomas et al. 1995) and α1-antichymotrypsin (Gooptu et al. 2000).
Two additional conformational states have recently been structurally characterized. δ-Antichymotrypsin (which contains the mutation Leu55→Pro) presents an intermediate conformation between the native and latent state (Gooptu et al. 2000) (Fig. 1D). The X-ray crystal structure of cleaved α1-antitrypsin polymers (Fig. 1E) confirms the loop–sheet mechanism of polymerization (Lomas et al. 1992; Huntington et al. 1999; Dunstone et al. 2000).
The S→R transition is integral to the function of inhibitory serpins. The mechanism of inhibition involves the formation of a stable complex between the proteinase and the cleaved form of the inhibitor, analogous to an enzyme-product complex. Some non-inhibitory serpins, such as CBG, use the S→R transition to control ligand release: the native state of CBG has higher affinity for cortisol than does the cleaved form (Pemberton et al. 1988). Note the difference between this mechanism and that of hemoglobin: once cleaved, CBG releases its ligand, and it cannot be re-used; hemoglobin has had to develop a complex allosteric mechanism to achieve reversible release of ligands. Some other serpins (e.g., ovalbumin) do not undergo an S→R transition under normal physiological conditions (Wright et al. 1990).
Several regions are important in controlling and modulating serpin conformational changes (Fig. 1A):
- 1.
- The hinge, the P15–P9 portion of the RCL (Hopkins et al. 1993). The hinge provides mobility essential for the conformational change of the RCL in the S→R transition.
- 2.
- The breach, located at the top of the A β-sheet, the point of initial insertion of the RCL into the A β-sheet (Whisstock et al. 2000a).
- 3.
- The shutter, near the center of A β-sheet (Stein and Carrell 1995). The breach and shutter are two important regions that facilitate sheet opening and accept the conserved hinge of the RCL as it inserts (Whisstock et al. 2000a).
- 4.
- The gate, including strands s3C and s4C, primarily characterized by studies of the transition of active PAI-1 to latency (Mottonen et al. 1992; Stein and Carrell 1995). To insert fully into the A β-sheet without cleavage, the RCL has to pass around the β-turn linking strands s3C and s4C.
Inhibitory serpins can generally be recognized by a consensus pattern in their sequences in the hinge (Hopkins et al. 1993): P17 P16 P15 P14 P12-P9 E E/K/R GT/S (A/G/S)4
P15 is usually glycine, P14 threonine or serine, and positions P12–P9 are occupied by residues with short side-chains, such as alanine, glycine, or serine. These residues are thought to permit efficient and rapid insertion of the RCL into the A β-sheet. The corresponding regions of non-inhibitory serpins deviate from the consensus. Mutations of hinge-region residues often convert inhibitory serpins into substrates.
An unfortunate consequence of conformational lability is the possibility of polymer formation by insertion of the RCL of one molecule into the A β-sheet of another (Fig. 1E) (Mast et al. 1991;Lomas et al. 1992; Huntington et al. 1999; Dunstone et al. 2000). Numerous mutants, including many in the shutter region, have been identified that enhance the propensity for polymerization, leading to dysfunction and disease (for review, see Stein and Carrell 1995).
RESULTS
Alignment Tables
The full alignment of 219 sequences can be found at the following web site (www.med.monash.edu.au/biochem/research/projects/serpins/alignment.html) or is available upon request. The insert included in this issue shows an alignment of 42 representative sequences from the different classes. The secondary structure shown above the sequences is that common to cleaved human α1-antitrypsin, human antithrombin, and ovalbumin.
Variability and Patterns of Sequence Conservation
The insert includes a Kabat variability plot of the 219 aligned sequences (the variability at any position = number of different amino acids observed ÷ frequency of the most common amino acid; Wu and Kabat 1970). The variability is mapped onto the structures of cleaved α1-antitrypsin in Figure2A.
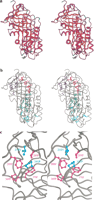
Amino acid conservation in the serpin superfamily. (A) Kabat variability in residues appearing at each site, mapped onto the structure of cleaved α1-antitrypsin. The color scheme ranges from red (low variability) to blue (high variability). Residues corresponding to positions in which >20% of sequences contain gaps are shown in green. The figure was produced using MOLSCRIPT (Kraulis 1991). (B) Cleaved α1-antitrypsin indicating residues conserved in >70% of sequences in ball and stick representation. Residues are colored according to the functional region of the serpin in which they are found: (blue) gate; (red) breach; (green) shutter. Residues outside these regions are in cyan. (C) Packing of conserved residues within the gate region. Phe208, Pro289, Pro369, and Phe370 are almost invariant (conserved in >95% of sequences) and are colored magenta. Two other highly conserved residues—Val218 and Pro391—are colored cyan.
Certain sites show high residue conservation (see Table2). Many others show conservation of physicochemical class. Those conserved in >70% of the serpin sequences are shown in Figure 2B, mapped onto the structure of cleaved α1-antitrypsin. There are 50 conserved residues. In the structure of cleaved α1-antitrypsin, 42 of the residues at these positions are buried (accessible surface area ≤20 Å2) and eight are exposed (in cleaved α1-antitrypsin, these are Asn158, Gly167, Lys191, Thr203, Lys290, Thr307, Phc312, and Pro369). A notable strip of conserved residues extends down the A β-sheet, as a continuous band within, above, and below strands s3A and s5A, along the path of the insertion of the RCL into the A β-sheet. The transition to the latent form requires additional substantial conformational change in the gate region (see Fig. 1A), which also contains a cluster of highly conserved positions (Fig. 2C). Alternatively, the conserved sites appear in the interfaces of the A β-sheet and the α helices that pack against it, and in the interfaces between the A and B β-sheets and the B and C β-sheets.
Residue Conservation: Position of Amino Acids Strictly Conserved in >70% of Sequences
Core of the Structure
The conservation patterns suggest that the serpin scaffold is intolerant of the deletion of all but peripheral elements of secondary structure. Apart from viral serpins and putative gene products, the sequences suggest that all major elements of secondary structure are conserved.
Viral serpins show more extensive changes. The D-helix is predicted to be severely truncated in the viral serpin-2 (SPI-2-like) cluster and the myxoma virus SERP-1 (Lomas et al. 1993). All but four of the sequences in the viral serpin-1/2 clade also have a deletion in the N terminus, which would be predicted to shorten the A-helix by two to three turns. These predictions have recently been confirmed by the X-ray crystal structure of cleaved crmA (Renatus et al. 2000), which revealed a truncated A- and E-helix and deletion of the D-helix.
The most dramatic deletion in a functional serpin is predicted to occur in the myxoma virus SERP-3, which must demonstrate significant perturbation of the region between the B- and F-helices (J.-L. Guerin, J. Gelfi, C. Camus, M. Delverdier, J.C. Whisstock, M.-F. Amardeihl, R. Py, S. Bertagnoli, and F. Messud-Petit, unpubl.). However, the large extent of the deletion and the low sequence similarity to serpins of known structure make it difficult to predict which elements of secondary structure between the B- and F-helices survive.
Most serpins show significant insertions and deletions within the loops joining elements of secondary structure. The RCL and the loop joining the C- and D-helices vary extensively in length. The reasons for the variation in RCL length in inhibitory serpins are not fully understood. Antithrombin utilizes its relatively long RCL (three residues greater than that of α1-antitrypsin) to achieve partial insertion in the native form. However, the X-ray crystal structure of serpin 1K from Manduca sexta (Li et al. 1999) reveals that the RCL, which is two residues longer than that of α1-antitrypsin, is not inserted into the A β-sheet. Presumably in the inhibitory serpins, loop length has evolved in each case for optimal interaction with the target proteinase.
The most striking variation in loop length in serpins is between the C- and D-helices, particularly in the intracellular serpins. PAI-2 has a 33-residue insertion relative to α1-antitrypsin in this region, which has been shown to be important for its intracellular activity (Dickinson et al. 1998). Similarly, the chromatin-condensing myeloid and erythroid nuclear termination stage-specific serpin (MENT) has a 24-residue extension between the C- and D- helices that contains an AT-hook motif, which suggests that it plays a role in DNA binding (Grigoryev et al. 1999).
Phylogenetic Analysis
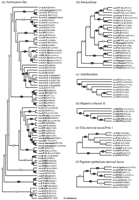
Figure 3 shows the large-scale phylogenetic tree, including the topology and edge lengths, computed from the sequence comparisons. The set of sequences is thereby divided into 16 classes (Table 3). In most cases, the nonvertebrate serpins group according to species. Vertebrate serpins span a number of distinct clusters, in many cases coupled with others of different function; for instance, CBG is closely related to α1-antitrypsin. The data for mammals suggest that intracellular serpins (clade b) were ancestral to the majority of the extracellular ones (the groups typified by heparin cofactor II, α1-antitrypsin, HSP47, and pigment epithelium-derived factor). Figure4A–P shows the boughs of the tree in detail. We also calculated phylogenetic trees using the preexisting alignment available from Pfam. These trees (not shown) were in broad agreement with those reported here; however, several important differences were apparent, including the grouping of the angiotensinogen-like serpins and the uterine serpins as separate clades (rather than including them in the antitrypsin clade a).
Multifurcating phylogenetic tree indicating the overall relationship between members of the serpin superfamily. The tree is a combination of the majority consensus maximum parsimony trees seen in Figure 4, with groups of serpins of similar type (e.g., antithrombin) represented by a single identifier, where possible. The branch lengths reflect maximum likelihood distances introduced using the method of Fitch and Margoliash (1967), as implemented in FITCH (Felsenstein 1996). Conventional bootstrap values from the maximum parsimony trees appear as ovals, rectangles indicate those subtrees whose members were identified using the comparison method, and hexagons indicate those identified by the strict consensus method. The 10 orphans are at the bottom of the tree. Clade identifiers (a, b,c, etc.) are in parentheses and correspond with subgroups identified in Figure 4, Table 3, and the text.
Partitioning into Clades
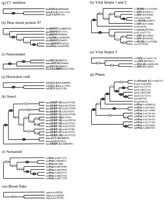
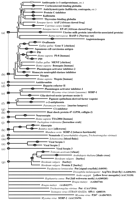
Sequences identified by either the strict consensus method or the comparison method were assembled into majority consensus maximum parsimony bootstrap trees. Bootstrap numbers appear on the branches; filled circles indicate relationships deemed statistically significant (Felsenstein 1985). Sequences are identified by species and name abbreviations, followed by the GenPept accession number in brackets. Species abbreviations: (aae) Aedes aegypti; (asy) Apodemus sylvaticus; (ath)Arabidopsis thaliana; (afa) Avena fatua; (bmo) Bombyx mori; (bta) Bos taurus; (bma) Brugia malayi; (cel)Caenorhabditis elegans; (cca) Callosciurus caniceps; (cpo) Cavia porcellus; (cco)Coturnix coturnix japonica; (cvi) cowpox virus; (cgr) Cricetulus griseus; (ccar)Cyprinus carpio; (dre) Danio rerio; (dvi) Didelphis virginiana; (dme)Drosophila melanogaster; (evi) Ectromelia virus; (eca) Equus caballus; (fru)Fugu rubripes; (gga) Gallus gallus; (hsa) Homo sapiens; (hvu) Hordeum vulgare; (hcu) Hyphantria cunea; (mmu)Macaca mulatta; (mse) Manduca sexta; (mga) Meleagris gallopavo; (mun)Meriones unguiculatus; (mau) Mesocricetus auratus; (mca) Mus caroli; (mmu)Mus musculus; (msa) Mus saxicola; (mvis) Mustela vison; (mvi) myxoma virus; (ocu) Oryctolagus cuniculus; (oar) Ovis aries; (ple) Pacifastacus leniusculus; (pha) Papio hamadryas anubis; (pma)Petromyzon marinus; (rvi) rabbitpox virus; (rno) Rattus norvegicus; (ssci) Saimiri sciureus; (sha) Schistosoma haematobium; (sja) Schistosoma japonicum; (sma)Schistosoma mansoni; (str) Spermophilus tridecemlineatus; (ssc) Sus scrofa; (svi) swinepox virus; (ttr) Tachypleus tridentatus; (tsi) Tamias sibricus; (tvi) Trichostrongylus vitrinus; (tae)Triticum aestivum; (vvi) vaccinia virus; (vavi) variola virus; (xla), Xenopus laevis. Serpin name abbreviations: (A2AP) α2-antiplasmin; (A1AT, AAT) α1-antiproteinase inhibitor or α1-antitrypsin; (AAP) α1-antiproteinase; (ACT) antichymotrypsin; (ANGT) angiotensinogen; (AP) antiproteinase; (API) α1-proteinase inhibitor; (ANT) antithrombin; (C1-I) C1 inhibitor; (CBG) cortisol-binding globulin; (CP-9) carp serine proteinase inhibitor; (EB22/3) antichymotrypsin-like protein; (EP45) estrogen-regulated protein 45 kD; (FXIIA-I) factor XIIA inhibitor; (GDN) glia-derived nexin or proteinase nexin-1; (GP50) HSP-47-like protein; (HEPII) heparin cofactor II; (HP-55) 55-kD hibernation protein; (HSP47) 47-kD heat shock protein; (KAL) kallistatin; (LICI) limulus intracellular coagulation inhibitor; (MC-7) contrapsin-related protein; (MENT) myeloid and erythroid nuclear termination stage-specific protein; (MNEI) monocyte/neutrophil elastase inhibitor; (NEUS) neuroserpin; (OVAL) ovalbumin; (PAI-1, PAI-2, etc.) plasminogen activator inhibitor; (PCI) protein C inhibitor; (PEDF) pigment epithelium-derived factor; (PI-6, PI-8, PI-9, etc.) proteinase inhibitor; (PP-60) 60-kD pregnancy protein; (Put) putative; (RASP-1) Regeneration-Associated Serpin Protein-1; (SCCA) Squamous Cell Carcinoma Antigen; (SERP) serpin; (SPI-1, SPI-2, etc.) serine proteinase inhibitor; (TBG, THBG) thyroxine-binding globulin; (UFAP, UABP) uteroferrin-associated protein; (UTMP) uterine milk protein.
Plants, Nematodes, Insects, and the Horseshoe Crab
The plant serpins (clade p) form a coherent and discrete evolutionary unit. The lack of orthology between plants and animals suggests that at the plant–animal divergence there was only a single serpin gene. With the exception of several “orphans,” the nematode (clade l) and insect (clade k) serpins also cluster into discrete clades. Our analysis suggests a close link between the horseshoe crab anticoagulant serpins (clade j) and the insect, glia-derived nexin (GDN)/PAI-1, and intracellular serpins (see Table4). A link between the horseshoe crab and the insect serpins is consistent with the taxonomic data, as both species share a common ancestor in the Protostomia branch of the Coelomata (Fig. 5).
Relationships between Minor Clades c, i, and jand the Major Subgroups
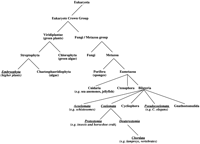
Simplified taxonomic tree constructed using the taxonomy data available at the NCBI. Those taxa in which serpins have been identified are underlined in italics.
The relationships seen in the phylogenetic trees are in agreement with the chromosomal data from the Arabidopsis thaliana andCaenorhabditis elegans genomes (Table5). In the former case, a single gene on chromosome I appears to have given rise to one on chromosome I and several on chromosome II. In C. elegans, a progression of the serpin gene from locus V-20.61→V0.88→V0.68 is apparent.
Chromosomal Location
Viral Serpins
To date, viral serpins have been identified only in the poxviridae. Serpins from the Orthopoxvirus branch (cowpox, ectromelia, vaccinia, variola, and rabbitpox) cluster in two clades: clade n, containing viral serpin-1 (SPI-1-like) and viral serpin-2 (SPI-2-like) serpins, and clade o, the viral serpin-3 (SPI-3-like) serpins. The data suggest that the viral serpins-1 and -2 are closely related, probably arising from a single gene by duplication, and possibly independent of viral serpin-3. The relationships among serpins from other branches of the poxviridae family are more unclear: serpins from myxoma virus (Leporipoxvirus) and swinepox virus (Suipoxvirus) are, with one exception, orphans. Our data suggest that myxoma SERP-1 may be a captured version of the PAI-1/GDN clade e, with which it associates.
Chordata—The Intracellular Serpins
Serpins in higher eukaryotes can be divided into two broad groups: the intracellular serpins or ov-serpins (Remold-O'Donnell 1993) and the extracellular serpins.
The ov-serpins form a well-defined clade (b) and are ancestral to the extracellular serpins. Their most distantly diverged member, megsin, has been shown to potentiate megakaryocyte maturation from bone marrow cells (Tsujimoto et al. 1997). Modification of cellular behavior is a theme evident throughout the subfamily: PAI-2 is able to inhibit tumor necrosis factor-α (TNF)–induced apoptosis (Dickinson et al. 1998), and MENT is involved in chromatin condensation (Grigoryev and Woodcock 1998; Grigoryev et al. 1999). Some ov-serpins also perform intracellular inhibitory roles, for example, PI-6 inhibits cathepsin G (Scott et al. 1999b). The functions of many intracellular serpins are still unknown. However, with the exception of the ovalbumin (which is non-inhibitory), all the ov-serpins contain the conserved hinge region residues essential for inhibitory activity. The exception, ovalbumin, is a major constituent of egg white and is thought to function primarily as a storage protein. However, a recent study by Sugimoto et al. (1999) demonstrates that ovalbumin undergoes conformational rearrangement during chick embryo development.
Chordata—The Extracellular Serpins
The extracellular serpins can be divided into eight clades, the largest of which, clade a, contains the α1-antitrypsin-like serpins. Serpins in this group are involved in a diverse range of processes (see Table 1), most commonly the inhibition of serine proteinases (e.g., kallistatin, Regeneration-Associated Protein-1 [RASP-1], α1-antitrypsin, and α1-antichymotrypsin). However, some are non-inhibitory, including the hormone transport serpins CBG and thyroxine-binding globulin (TBG), the peptide hormone delivery agent angiotensinogen, and the uterine serpins UTMP (uterine milk protein) and UFAP (uteroferrin-associated protein). The uterine serpins are highly diverged and contain a non-inhibitory hinge region. Their function remains obscure; however, a recent study by McFarlane et al. (1999) described binding of ovine UTMP to the growth factor activin, suggesting that it may play a role in sequestering this important factor in the pregnant uterus.
Clade f contains pigment epithelium-derived factor (PEDF) and α2-antiplasmin. PEDF is thought to be a neurotrophic factor. A sea lamprey serpin appears to share ancestry with these mammalian proteins.
Heparin cofactor II forms a separate clade (d), as do the C1 esterase inhibitors (clade g) and HSP47 (clade h). HSP47 serpins are non-inhibitory and function as molecular chaperones involved in the folding of procollagens.
GDN, PAI-1, and the myxoma SERP-1 form a separate clade (e). Reinforcing a potential ancestral link, all three forms of serpin have an interesting substitution in the shutter region, with the consensus His at position 334 on strand s5A replaced with Gln (Fig.6).
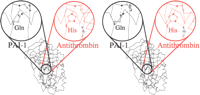
PAI-1 (black) has a Gln at a position 334 in the shutter that makes a hydrogen bond to P10 Ser in the reactive center loop (RCL). The consensus residue (e.g., in antithrombin [red]) at position 334 is a His that makes a hydrogen bond to P8 Thr (blue) in the RCL.
The clustering of antithrombin (clade c) and the neuroserpin (clade i) near the insect/intracellular/PAI-1 portion of the tree (Table 4) suggests that these groups may have diverged relatively early and that antithrombin or neuroserpin may link intracellular and extracellular serpins.
Orphans
Ten orphans failed to group with any other clade, including the accessory gland protein (Acp76a) from Drosophila melanogaster(Coleman et al. 1995) and the Aedes aegypti factor Xa inhibitor (Stark and James 1998). The latter serpin appears to have evolved a novel mechanism of proteinase inhibiton, because it does not possess the consensus sequence for inhibitory serpins in the hinge region and functions as an effective reversible, noncompetitive factor Xa inhibitor.
Chromosomal Location
The phylogenetic clustering agrees with existing chromosomal data and divides taxa effectively into species-based clusters. Table 5 shows the chromosomal location of those serpins for which the information is available.
DISCUSSION
Residue Conservation in the Serpin Superfamily
Conserved residues within the serpin core map to mobile regions that mediate the change in conformation during the S→R transition or the switch to latency. Analysis of known serpin mutations with enhanced lability suggests that the majority of highly conserved positions are directly involved in the mechanism of serpin conformational change or else are located in regions that are known to be important in mediating structural changes (see Table 2; Fig. 2).
The many highly conserved residues in the breach and shutter regions (at the top and in the middle of the A β-sheet) reflect the requirement for RCL insertion during the S→R transition. The breach and shutter regions act as pivot points around which domains rotate to open the A β-sheet (Whisstock et al. 2000a,b).
The gate region also contains a number of highly conserved residues. This region is known to be involved in the transition to latency (Mottonen et al. 1992; Tucker et al. 1995). However, most serpins do not normally form the latent state in vivo, except for PAI-1 and antithrombin (Levin and Santell 1987; Beauchamp et al. 1998) and various dysfunctional serpin variants linked with disease (e.g., Bruce et al. 1994; Gooptu et al. 2000). Thus, the residue conservation seen in the gate may be linked to maintenance of the native form rather than to promotion of the transition to the latent state.
The retention of most of the conserved residues in ovalbumin, which does not undergo the S→R transition under normal physiological conditions, even after cleavage, is somewhat puzzling. However, (1) many of the conserved residues are part of the hydrophobic core of the protein and may be important for maintaining the serpin fold (see the following section), and (2) ovalbumin is closely related to inhibitory serpins and may simply not have diverged very far. Indeed, even angiotensinogen, an extensively diverged non-inhibitory serpin, retains a significant proportion of conserved residues.
Several studies have linked the process of conformational change to the folding pathway of serpins. For example, Yu et al. (1995) showed that the in vivo polymerization of Z-antitrypsin is a result of the formation of a misfolded intermediate that has a propensity to polymerize. Furthermore, studies by James and Bottomley (1998) andDafforn et al. (1999) have shown that α1-antitrypsin is able to adopt a polymerogenic intermediate during guanidine hydrochloride-mediated unfolding. Serpins undergo a change in topology during the S→R transition, and this conformational change can be regarded as a limited “refolding” of the molecule. Thus, serpin folding and serpin conformational change appear to be intimately linked, and it seems reasonable that serpin mutants that fail to fold efficiently might exhibit enhanced lability as a symptom of misfolding. An alternative explanation for the degree of conservation seen in non-inhibitory serpins, such as ovalbumin and angiotensinogen, may be that changes to the conserved core of the serpin molecule could lead to misfolding and dysfunction. Thus, selective pressure will favor changes in nonconserved residues that still allow the serpin to fold efficiently into the native state yet bring about the desired change in function.
Phylogeny of the Serpins
With the exception of the viral serpins, all known serpins appear in organisms of the eukaryote crown group taxon. However, there are important gaps in their distribution (see Fig. 5). Numerous serpins have been identified in the higher plants. However, we failed to identify any putative serpins in Chlorophyta (green algae) or fungi, despite the availability of several complete fungal genomes.
Animal serpins are found exclusively in bilaterian organisms, including the Coelomata (containing the vertebrates), the Pseudocoelomata (e.g.,C. elegans) and the Acoelomata (e.g., schistosomes). Serpins are present in two subtaxa of Coelomata: Deuterostomia (including vertebrates) and the Protostomia (including insects and the horseshoe crab). We found no serpins in Cycliophora or Gnathostomulida, or in the other two taxa within the Eumetazoa: the Cnidaria (including sea anemones and jellyfish) and the Ctenophora, probably because of the paucity of sequence data for these organisms. Perhaps theMetridium senile genome project will extend the serpin superfamily to the Cnidaria.
Known serpins appear confined to multicellular organisms and viruses that infect them. Either prokaryotes and unicellular eukaryotes such as yeasts or algae do not contain serpins or the serpins in these organisms are relatives too distant to be identified using available techniques. The phylogenetic clustering agrees with existing chromosomal data (Table 5) and divides taxa effectively into species-based clusters.
Functionally, most serpins identified to date are involved in regulating processes or cascades that have arisen as a result of being multicellular. We note that the conventional serine proteinases as inhibitory targets are absent from yeasts, algae, and prokaryotes; with one exception (a chymotrypsin-like serine proteinase in pollen [Bagarozzi et al. 1996]), they also appear to be absent from higher plants. In animals, extracellular serpins are involved in processes such as blood coagulation (transport/defense) and hormone delivery (communication). Unicellular organisms have no obvious requirement for the known functions of extracellular serpins. Even intracellular serpins have functions related to multicellular processes, such as granule-mediated apoptosis (Bird 1998; Bird et al. 1998).
In a previous study, we noted that nematode serpins share greatest sequence identity with the intracellular serpins (Whisstock et al. 1999). Database searches performed in this study reveal that insect serpins also are most similar to serpins from the intracellular clade. These results suggest that the intracellular serpins have not evolved as far from their ancestors as have the extracellular serpins.
What then is the evolutionary origin of serpins? The appearance of serpins in animals and plants suggests that, unless there was lateral gene transfer, serpins must have appeared before the animal–plant divergence, ∼1.5 billion years ago (Wang et al. 1999). The ancestor of known serpins may not have survived in any genome of a living species, or it may be so different that we cannot recognize it, or it may appear in a genome to be determined in the future.
Conclusions
We have presented an analysis of relationships among the known serpins, integrating genomic, functional, and structural information. Our classification provides a reference for placement of newly discovered serpins.
All known serpins form a coherent family containing a core of residues alignable in the sequences and amounting to approximately two-thirds of the structure. Patterns of conservation are clearly correlated with mechanism of function common to inhibitory serpins and a few others. Conserved residues flank the pathway of conformational change of the RCL.
The search for an ancestor in fungi or prokaryotes continues.
METHODS
Coordinates
The coordinates of uncleaved α1-antitrypsin (PDB entry2PSI; Elliott et al. 1998), cleaved α1-antitrypsin (7API;Loebermann et al. 1984), native and latent antithrombin (2ANT; Skinner et al. 1997), native antithrombin plus heparin pentasaccharide (1AZX;Jin et al. 1997), uncleaved ovalbumin (1OVA; Stein et al. 1990), δ-antichymotrypsin (1QMN; Gooptu et al. 2000), and native serpin 1K (1SEK; Li et al. 1999) were obtained from the Protein Data Bank (www.rcsb.org; Berman et al. 2000). The coordinates of PAI-1 (Mottonen et al. 1992) were kindly provided by Dr. E.J. Goldsmith.
Database Searching
A PSI-BLAST (Altschul et al. 1997) search of the nonredundant protein database at the NCBI (version of 4 September 1999) identified 433 amino acid sequences with significant similarity (E < 106 [Park et al. 1998]) to the probe sequence, human α1-antitrypsin (SwissProt ID A1AT_HUMAN). We used the BLOSUM62 matrix, gap initiation penalty 10, gap extension 2, and expect value for inclusion in subsequent rounds 0.001. Convergence was achieved at the fifth iteration. Additional PSI-BLASTsearches using the sequences of angiotensinogen, antithrombin, maspin, serpin K, and barley protein Z as probes failed to identify additional homologs. We rejected incomplete sequences shorter than 200 residues and all but one of any set of sequences with ≥98% identity, retaining 219 out of 433 sequences. To confirm our results, we performed further searches using profile hidden Markov model (HMM) tools available at ANGIS (http://www.angis.org.au;http://www.bionavigator.com; Littlejohn et al. 1996). The 219 sequences were aligned (see the following section), and the programHMMER (Durbin et al. 1998) was used to build and calibrate an HMM. The program HMMSEARCH was used to search the GenPept database; however, no additional potential serpin sequences were identified.
Multiple Sequence Alignment
We based our sequence alignment on a structural alignment of three distantly related serpins—uncleaved α1-antitrypsin, native antithrombin plus heparin pentasaccharide, and uncleaved ovalbumin—generated with Quanta (MSI Inc.). Residues falling within sheets and helices in all three structures were given increased gap insertion/extension penalties to guide a profile alignment of the serpin sequences by using CLUSTALW1.7 (Higgins et al. 1996). The resulting multiple sequence alignment was manually refined using SeaView (Galtier 1996). Alignments of the C. elegans sequences were adjusted according to Whisstock et al. (1999). For five highly diverged sequences (GenBank accession nos. AAC58237, AAB96393,CAB04611, AAA82351, and AAB67053), we substituted the original pairwise alignment reported by PSI-BLAST.
Two regions were deemed nonalignable (and are not included in our statistical analysis of residue conservation): (1) the very poorly conserved leader sequences and signal peptides at the N terminus are not included in our alignment table; (2) the residues in the RCL C-terminal to the scissile bond, where most serpins vary in RCL length, are right-adjusted and appear in the alignment table in lowercase. Residues between the N terminus of the RCL and the scissile bond, P17–P1′, are shown in accordance with the assumption, true of inhibitory serpins, that there are no insertions or deletions in this region. Our sequence alignment differs considerably from precalculated serpin alignments that do not take account of secondary structure conservation, such as that available from Pfam (www.sanger.ac.uk/Pfam/;Bateman et al. 1999). The serpin alignment available from SMART (smart.embl-heidelberg.de; Schultz et al. 1998) is in general agreement with that presented here; however, our alignment considers twice as many serpins.
Construction of Phylogenetic Trees
Distance Tree
Sites (columns in the alignment) that contained gaps in >20% of the sequences were removed, and a consensus distance tree (1000 bootstrap trials; Jones, Taylor, and Thornton matrix model of substitution) was generated using the MOLPHY package (Adachi and Hasegawa 1996) and the SEQBOOT and CONSENSE programs of the PHYLIP package (Felsenstein 1996). The tree was rooted at barley protein Z.
Reduced Partition Consensus Profiles
Subsets of taxa found in all bootstrap trees were identified and replaced with single operational taxonomic units (OTU). The trees, reduced from 219 to 77 taxa, were input into REDCON 2.0 (Wilkinson 1996) for generation of strict reduced partition consensus profiles (Wilkinson 1994).
Tree Construction
The neighbor-joining method (Saitou and Nei 1987) with maximum-likelihood distances failed to identify many groups of non-orthologous serpins with satisfactory bootstrap confidence levels. We therefore developed a new technique—which we call the comparison method—making use of the tendency of related sequences to cluster in consistent ways in the ensemble of generated trees. The process is summarized in Figure 7A (available as an online supplement athttp://www.genome.org). This technique resembles, to some extent, the majority-rule reduced partition consensus method of Wilkinson (1996) in that subsets of taxa are combined and poorly resolved associations are excluded. However, our technique tolerates greater variation in taxon clustering and hence is more sensitive to general trends in the data. We were able to identify statistically significant clustering of species within the bootstrap trees (see Table 3). This clustering is supported by the chromosomal localization of the intracellular serpins (Bartuski et al. 1997; Sun et al. 1998; Scott et al. 1999a) and the α1-antitrypsin-like serpins (Rollini and Fournier 1997) (Table 5). Novel associations revealed include the following:
- 1.
- GDN, PAI-1, and myxoma SERP-1;
- 2.
- RASP-1, angiotensinogen, UTMP, TBG, and the cluster of human serpins at 14q32.1 (such as CBG and α1-antitrypsin; see Table 5);
- 3.
- α2-Antiplasmin, PEDF, and sea lamprey serpin;
- 4.
- M. sexta SERP-1 and SERP-2 and Bombyx moriantitrypsin and antichymotrypsin I.
Clade Interrelationships
A second, related technique—tree division (see Fig. 7B, available as an online supplement at http://www.genome.org)—was used to divide each bootstrap tree into subtrees. Nonrandom partitioning into a defined portion of each tree was observed for antithrombin, neuroserpin, and the horseshoe crab coagulation inhibitors. All three associated ≥95% of the time with either the intracellular, GDN/PAI-1, or insect serpin clades; this link suggests that they share a closer ancestor among themselves than with other vertebrate serpins (Table 4).
Maximum Parsimony Trees within Classes
Maximum parsimony (first applied to molecular sequences by Eck and Dayhoff [1966]) in conjunction with bootstrap resampling (Felsenstein 1985) was used to determine the topology within the clades distinguished by the comparison method. Both DNA and protein sequences were used. The nucleotide sequence for each serpin was aligned codon by codon against the corresponding protein sequence. The nucleotide and amino acid alignments were then used to construct maximum parsimony bootstrap consensus trees (1000 bootstrap trials) for each subgroup, using the PROTPARS and DNAPARS programs of the PHYLIP package (Felsenstein 1996). The protein and DNA majority consensus tree in each case was combined into a mosaic tree, with branches selected on the basis of (1) completeness, that is, the availability of sequence data, and (2) the highest total bootstrap value.

Representative alignment of Sequences of Known Serpins.
Regions of secondary structure seen in 1OVA, 2PSI and 1AZX are displayed; cylinders represent helices and arrows represent sheets. The variability (Wu & Kabat 1970) is shown by the jagged line above the sequences. Sequence numbering is according to α1–antitrypsin. Residues are colored according to strict conservation (across all 219 serpin sequences): The darker the shading, the more highly conserved. The following graduations are used: 0–20% (white), 20%–30%, 30%–40%, 40%–50%, 50%–60% and 60%–70%. Residues conserved in >70% of sequences are in dark red and are listed in Table 5. Species abbreviations: ath, Arabidopsis thaliana; bma, Brugia malayi; bmo, Bombyx mori; dme, Drosophila melanogaster; gga, Gallus gallus; hsa, Homo sapiens; hvu, Hordeum vulgare; mvi, Myxoma virus; oar, Ovis aries; pma, Petromyzon marinus; sma, Schistosoma mansoni; svi, Swinepox virus; ttr, Tachypleus tridentatus; tae, Triticum aestivum; vavi, Variola virus. Serpin name abbreviations: A2AP, α2–antiplasmin; AAT, α1–antiproteinase inhibitor or α1–antitrypsin; ACT, antichymotrypsin; ANGT, angiotensinogen; ANT, antithrombin; C1-I, C1 inhibitor; CBG, cortisol-binding globulin; GDN, glia derived nexin or poteinase nexin-1; HEPII, Heparin Cofactor II; HSP47, 47 kDa heat shock protein; KAL, kallistatin; LICI, limulus intracellular coagulation inhibitor; MNEI, monocyte/neutrophil elastase inhibitor; NEUS, neuroserpin; OVAL, ovalbumin; PAI-1, PAI-2 etc., Plasminogen Activator Inhibitor-1 -2 etc; PCI, protein C inhibitor; PEDF, pigment epithelium derived factor; PI-6, PI-8, PI-9 etc., proteinase inhibitor; Put, putative; SCCA, squamous cell carcinoma antigen; SERP, serpin; SPI-1, SPI-2 etc., serine proteinase inhibitor; THBG, thyroxine binding globulin; UTMP, uterine milk protein.
Acknowledgments
We thank Dr. E. Goldsmith for the coordinates of PAI-1. We thank the Wellcome Trust, the Australian Research Council (Grant A10017123), the National Heart Foundation of Australia (Grant G98M0118), and the National Health and Medical Research Council of Australia (Grant 997144) for support. A.M.L. thanks Monash University for its hospitality to him as a Walter Cottman Fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL James.Whisstock{at}med.monash.edu.au; FAX 61 3 9905 4699.
-
Article published online before print: Genome Res., 10.1101/gr.147800.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.147800.
-
- Received May 17, 2000.
- Accepted September 12, 2000.
- Cold Spring Harbor Laboratory Press








