
Cell type–specific gene regulatory atlas prioritizes drug targets and repurposable medicines in Alzheimer's disease
- Yunxiao Ren1,2,
- Ming Hu3,4,
- Yang E. Li5,
- Andrew A. Pieper6,7,8,9,10,11,
- Jeffrey Cummings12 and
- Feixiong Cheng1,2,4,13
- 1Cleveland Clinic Genome Center, Cleveland Clinic Research, Cleveland Clinic, Cleveland, Ohio 44195, USA;
- 2Department of Genomic Sciences and Systems Biology, Cleveland Clinic Research, Cleveland Clinic, Cleveland, Ohio 44195, USA;
- 3Department of Quantitative Health Sciences, Cleveland Clinic Research, Cleveland Clinic, Cleveland, Ohio 44195, USA;
- 4Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, Ohio 44195, USA;
- 5Department of Neurosurgery and Genetics, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 6Department of Psychiatry, Case Western Reserve University, Cleveland, Ohio 44106, USA;
- 7Brain Health Medicines Center, Harrington Discovery Institute, University Hospitals Cleveland Medical Center, Cleveland, Ohio 44106, USA;
- 8Geriatric Psychiatry, GRECC, Louis Stokes Cleveland VA Medical Center, Cleveland, Ohio 44106, USA;
- 9Institute for Transformative Molecular Medicine, School of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, USA;
- 10Department of Pathology, Case Western Reserve University, School of Medicine, Cleveland, Ohio 44106, USA;
- 11Department of Neurosciences, Case Western Reserve University, School of Medicine, Cleveland, Ohio 44106, USA;
- 12Chambers-Grundy Center for Transformative Neuroscience, Department of Brain Health, Kirk Kerkorian School of Medicine, University of Nevada–Las Vegas, Las Vegas, Nevada 89154, USA;
- 13Case Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, Ohio 44106, USA
Abstract
Alzheimer's disease (AD) is a complex and poorly understood neurodegenerative disorder that lacks sufficiently effective treatments. Computational and integrative analyses that leverage multiomic data provide a promising strategy to uncover disease mechanisms and identify therapeutic opportunities. Here, we develop a cell type–specific regulatory atlas of the human middle temporal gyrus via leveraging single-nucleus RNA-seq (1,197,032 nuclei) and ATAC-seq (740,875 nuclei) data sets from 84 donors across four stages of AD neuropathological change (ADNC). We observe differential gene expression for six major cell types intensified at severe ADNC. Integrating peak-to-gene linkages and motif enrichment analyses, we reconstruct transcription factor (TF)–target gene networks across six major brain cell types. By integrating genome-wide association study (GWAS) loci with cell type–specific cis-regulatory DNA elements (CREs), we pinpoint 141 ADNC-associated genes. Using gene set enrichment analysis (GSEA) and network proximity analysis, we further identify nine candidate repurposable drugs that were associated with these ADNC-related genes. In summary, this cell type–specific multiomic atlas provides a comprehensive resource for mechanistic understanding, target prioritization, and therapeutic hypothesis generation in AD and AD-related dementia if broadly applied.
Alzheimer's disease (AD) is a complex, age-related neurodegenerative disorder and the leading cause of dementia worldwide (Long and Holtzman 2019). The accumulation of amyloid-β (Aβ) plaques and tau protein tangles are a hallmark of AD pathology. However, it is increasingly recognized that additional neuropathological processes, such as neuroinflammation, lipid metabolism dysfunction, mitochondrial impairment, axonal degeneration, impaired postnatal neurogenesis, and synaptic dysfunction, also play crucial roles in disease progression (Ballard et al. 2011; Long and Holtzman 2019; Murdock and Tsai 2023). This intricate pathology poses significant challenges for developing effective therapies. Recent advances in single-cell multiomics have illuminated cell type–specific dynamic changes of transcriptome and epigenome in AD heterogeneities and disease progression (Mathys et al. 2019, 2023, 2024; Corces et al. 2020; Murdock and Tsai 2023; Sziraki et al. 2023; Xiong et al. 2023; Gabitto et al. 2024), revealing new complexities in disease mechanisms. Furthermore, intercellular heterogeneity complicates therapeutic efforts, and substantial differences in the molecular alterations between laboratory animal models and human patients exist, highlighting the limitations of traditional laboratory-based approaches to therapeutic discovery (Frozza et al. 2018; Tatulian 2022; Li et al. 2023). Given the magnitude of the problem, innovative strategies for discovering treatments for AD are essential.
In this study, we develop a cell type–specific multiomic regulatory atlas to model AD progression and explore therapeutic opportunities. Using single-nucleus RNA-seq (snRNA-seq) and ATAC-seq (snATAC-seq) data from the middle temporal gyrus of 84 donors across four stages of AD neuropathological changes (ADNCs; non-AD, low, intermediate [inter], and high), we construct stage-resolved transcriptomic and epigenomic profiles for six major brain cell types. By integrating transcription factor (TF)–target gene relationships, chromatin accessibility, and genetic variation, this framework aims to elucidate the cell type–specific regulatory mechanisms underlying AD progression, link genetic risk variants to cis-regulatory elements and target genes, and prioritize potential therapeutic targets and drug candidates for AD.
Results
Study design
We obtained snRNA-seq and snATAC-seq data from 84 human brain middle temporal gyrus samples provided by the Seattle Alzheimer's Disease Brain Cell Atlas (SEA-AD) study (Gabitto et al. 2024). Understanding and defining AD progression remain significant challenges. Currently, AD progression is commonly assessed using neuropathologic criteria such as Braak stage, Thal phase, Consortium to Establish a Registry for Alzheimer's Disease (CERAD) score, and the more comprehensive Alzheimer's disease neuropathologic change (ADNC) metric (DeTure and Dickson 2019). ADNC is an integrated measure that combines multiple neuropathologic indicators, including neurofibrillary tangles (Braak), neurotic plaque scores (CERAD), and Aβ plaques (Thal) (Trejo-Lopez et al. 2022). Thus, to optimally capture the distinct stages of AD progression, we categorized the 84 samples into four groups based on ADNC information from the metadata (Gabitto et al. 2024): non-AD (n = 9), low ADNC (n = 12), inter ADNC (n = 21), and high ADNC (n = 42) (Fig. 1A; Supplemental Table S1).
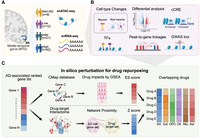
Overview of study design. (A) Collection and preprocessing of snRNA-seq and snATAC-seq data sets in physical space. (B) Multiple analysis to identify an AD-associated gene list including differential gene expression analysis; identification of cCRE; cell type–specific, AD-associated risk variants; and peak-to-gene linkages. (C) Two in silico perturbation methods for drug repurposing. Network proximity analysis identifies potential drugs by assessing the proximity between the drug target set and AD-related gene set. Drug efficacy is evaluated based on the proximity distance and Z-score. GSEA leverages drug–gene signatures from the CMap database and differentially expressed genes to calculate the enrichment score (ES). The ES reflects the drug's potential to reverse the observed gene expression patterns within the given network.
To construct the cell type–specific regulatory atlas, we performed comprehensive bioinformatics analyses encompassing (1) differential gene expression analysis; (2) characterization of candidate cCREs; and (3) inference of cell type–specific, TF–target gene networks; and (4) integrative analyses linking peaks to genes and associating AD genome-wide association study (GWAS) loci with cCREs. This enabled prioritization of ADNC-associated variants and genes within a unified multiomic framework (Fig. 1B). Based on the resulting set of AD-associated genes, we next performed in silico perturbation and drug repurposing analyses, combining gene set enrichment analysis (GSEA) and network proximity methods integrated with the Connectivity Map (CMap) database and the drug–target interactome (Fig. 1C). This approach facilitated the identification of candidate repurposable drugs for each major brain cell type across AD progression.
Overview of snRNA-seq and snATAC-seq data across ADNC progression
AD progression staging based on ADNC does not fully align with individual neuropathologic staging metrics such as Thal phase, Braak stage, or CERAD score (Fig. 2A) or with clinical cognitive status. These findings underscore the importance of defining AD progression through neuropathology to better understand the molecular changes occurring in the early stages of the disease and to inform the development of therapeutics aimed at halting AD progression. Additionally, we presented the distribution of limbic-predominant age-related TDP-43 encephalopathy (LATE) stage, apolipoprotein E (APOE) gene carrier status, sex, and age at death across ADNC stages in this cohort (Fig. 2A; Supplemental Table S1).
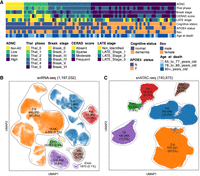
Overview of snRNA-seq and snATAC-seq data sets. (A) Heatmap displaying metadata for 84 individual samples. The rows of the heatmap, ordered from top to bottom, represent ADNC, Thal phase, Braak stage, CERAD score, LATE stage, cognitive status, APOE4 status, sex, and age at death. (B,C) UMAP representation of snRNA-seq (B) and snATAC-seq (C) is shown. The figure also shows the cell count number and proportion of each cell type.
We annotated 1,197,032 snRNA-seq cells from 84 matched individuals into eight major cell types based on known marker genes (Fig. 2B; Supplemental Fig. S1A). Inhibitory neurons (312,903 cells, 26.1%) and excitatory neurons (640,285 cells, 53.5%) together constitute the majority, accounting for more than one-half of the total cells. We further annotated 18 neuronal cell subtypes based on the classifications described in the original paper (Supplemental Fig. S2; Gabitto et al. 2024). Among these, we observed that the L2/3 IT subtype of excitatory neurons showed a significant decrease in cell counts at the high ADNC stage compared with other stages, whereas the L4 IT subtype of excitatory neurons exhibited a significant increase at the high ADNC stage (Supplemental Fig. S3). Additionally, the major glial populations included oligodendrocytes (105,058 cells, 8.8%), oligodendrocyte progenitor cells (OPCs; 29,602 cells, 2.5%), astrocytes (65,462 cells, 5.5%), and microglia (37,843 cells, 3.2%). Notably, microglia cell counts showed a significant increase at the high ADNC stage compared with the non-AD and intermediate ADNC stages (Supplemental Fig. S4). Two additional cell types, endothelial cells and vascular leptomeningeal cells (VLMCs), make up a smaller fraction, with 5879 cells (0.4%).
For snATAC-seq, we identified 740,875 cells distributed across six major cell types using the same marker genes as for snRNA-seq annotation (Fig. 2C; Supplemental Fig. S1B). Inhibitory neurons (184,686, 24.9%) and excitatory neurons (370,341, 50.0%) showed a similar trend, comprising the largest proportions of the total cells. The remaining glial cell groups included oligodendrocytes (100,965, 13.6%), OPCs (13,935, 1.9%), astrocytes (46,816, 6.3%), and microglia (24,132, 3.3%); the cell-type proportion also showed similar trends with snRNA-seq.
Cell type–specific gene expression changes across AD progression
To identify differentially expressed genes (DEGs) across AD progression for each cell type, we conducted six pairwise stage comparisons (low vs. non-AD, inter vs. non-AD, inter vs. low, high vs. non-AD, high vs. low, high vs. inter). A greater number of significant DEGs were observed in the high stage compared with non-AD (4942 DEGs), low (4470 DEGs), and intermediate stages (3393 DEGs) (Fig. 3A; Supplemental Table S2). The transcriptional patterns we observed across ADNC stages are consistent with those reported by Gabitto et al. (2024) using pseudoprogression scores, both showing marked pathological escalation in late stages (Gabitto et al. 2024). Notably, ADNC strongly correlated with the continuous pseudoprogression score in their study (R = 0.75, P = 2.08 × 10−16) (Supplemental Fig. S5; Gabitto et al. 2024). We further examined gene expression changes between adjacent stages from non-AD to high ADNC for each cell type. In most cell types, including astrocytes, excitatory neurons, inhibitory neurons, microglia, and oligodendrocytes, the largest proportion of upregulated genes occurs in the transition from non-AD to low ADNC (Fig. 3B). Oligodendrocytes, OPCs, astrocytes, and microglia retain relatively high proportions of upregulated genes even from inter to high ADNC stages, whereas excitatory neurons and inhibitory neurons show a decline (Fig. 3B). This observation indicates that there are distinct gene expression dynamics among cell types in the different stages of AD.
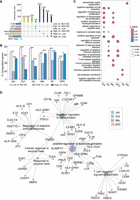
Gene expression changes across AD progression. (A) UpSet plot illustrating the number of DEGs across AD progression. The left bar plots display the total number of DEGs for each stage comparison group, and the top bar plots represent the number of DEGs that are either overlapping or unique to different groups. Colored dots connected by lines indicate the stage groups that have overlapping DEGs, and a single-colored dot represents the stage group without overlapping DEGs with other groups. (B) Percentage of upregulated DEGs number for each cell type across adjacent stages of AD progression. Statistical significance was assessed using the Chi-squared test, with P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). (C) GO functional enrichment analysis of each cell for upregulated DEGs during high-versus-inter stage. (D) Selected GO terms from C with corresponding genes. Different colors indicate distinct cell types, and the pie charts represent GO terms and associated genes shared across various cell types.
We performed pathway enrichment analysis of upregulated genes from the non-AD–to–low ADNC transition. In microglia, significantly enriched pathways included hypoxia response, chemotaxis, antigen presentation via MHC-II, and lipid/lipoprotein metabolism (Supplemental Fig. S6), overall consistent with a previous study (Gabitto et al. 2024). In astrocytes, enriched GO terms encompassed hypoxia response, angiogenesis (e.g., ITGA5, ADTRP), axonogenesis (e.g., BSG, ITGA4), glycolysis, and retinoic acid signaling, reflecting similar biological processes to those described previously (Gabitto et al. 2024), including early astrogliosis, enhanced adhesion and axon guidance signaling, and metabolic reprogramming.
We further investigated the functions of upregulated genes from the inter to high stage using Gene Ontology (GO) analysis (Fig. 3C). For each cell type, we identified the DEGs contributing to the enriched GO terms (Fig. 3D). Our results revealed that upregulated genes in excitatory and inhibitory neurons were significantly enriched in pathways related to the response Aβ, including key genes such as triggering receptor expressed on myeloid cells 2 (TREM2), ADRB2, and GJA1 (Fig. 3D). TREM2 is involved in Aβ clearance by microglia, playing a pivotal role in mitigating Aβ plaque accumulation in AD (Gratuze et al. 2018; Zhao et al. 2018). Similarly, ADRB2 is associated with signaling pathways that regulate Aβ metabolism and confer neuroprotective effects (Evans et al. 2020), and GJA1 contributes to mechanisms that modulate Aβ toxicity (Kajiwara et al. 2018). Furthermore, DEGs expressed in excitatory neurons, microglia, OPCs, and inhibitory neurons were also significantly enriched in pathways related to immune-related pathways, such as FYN, HLA-E, LCK, and SYK (Fig. 3D). These findings suggest that upregulated genes from the inter to high stage are potentially associated with Aβ-related processes, alongside the inflammatory and immune mechanisms underlying AD progression.
Identifying TF–target gene networks involved in AD progression
The accessibility of chromatin regions to TFs is a major determinant of cellular transcriptional profile (Zhu et al. 2019; Coux et al. 2020; Ren et al. 2021). To identify lineage-defining TFs enriched in cell type–specific accessible regions, we first defined cell type–specific peaks using the stringent criteria of Wilcoxon false-discovery rate (FDR) ≤ 0.05 and log2(fold change [FC]) ≥ 0.5, followed by TF motif enrichment analysis. For example, well-characterized TFs such as SPI1 were enriched in microglial cells as supported by foot-printing evidence, whereas SOX4 and SOX9 were enriched in oligodendrocytes, and JUND and FOSB were enriched in excitatory neurons (Fig. 4A,B; Supplemental Table S3).
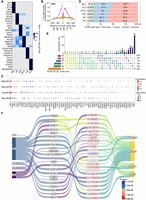
Positive TF-regulators and cell type–specific regulatory landscapes of GWAS loci across AD progression by integrative analysis. (A) Top enriched TF motifs for each cell type. The color scale represents −log10 (adjusted P-value) of the normalized enrichment score. (B) Tn5 bias-corrected TF footprinting for SPI1. (C) Positive TF-regulators for all cell type across AD progression. (D) The percentage of genomic region annotation for cell type–specific peaks. (E) Numbers of candidate genes associated with significant AD-related SNPs for each cell type. The left bar plot shows the total number of genes per cell type, and the top bar plot highlights genes that overlap between cell types or are unique to specific cell types. (F) Sankey plot illustrating the cell type–specific AD-associated lead SNPs and their potential target genes. The first column represents cell types; the second column indicates potential target genes linked to AD-associated lead SNPs; the third column lists AD-associated lead SNPs with P-values represented by scale bar color and indicated by different text colors; and the fourth column displays genomic region annotations for peaks associated with these lead SNPs.
Some TF families share highly similar binding motifs, which can complicate identification of specific TFs associated with chromatin accessibility changes (Granja et al. 2021). To address this challenge, we identified TFs with gene expression that correlated positively with the accessibility of their corresponding motifs by integrational analysis with snRNA-seq and snATAC-seq, designating them as “positive TF-regulators.” Across AD progression, we identified 64 positive TF-regulators based on stringent cut-off values (correlation > 0.5, adjusted P-value < 0.01, Z-score > 0.75) (Fig. 4C; Supplemental Table S4). Specifically, the microglial-specific TF known as SPI1 was present across all stages of AD progression. In contrast, the excitatory neuron-specific TF known as JUND was predominantly enriched at the high AD stage, whereas STAT2 was primarily enriched at the inter AD stage. These findings highlight the dynamic and cell type–specific regulatory roles of positive TF-regulators in modulating chromatin accessibility during AD progression.
Identifying AD progression–associated targets enriched by cCREs with AD GWAS
We further identified cell type–specific cCREs. In total, 52,565 cCREs were identified in astrocytes, 67,253 in excitatory neurons, 22,198 in inhibitory neurons, 42,241 in microglia, 25,943 in OPCs, and 24,186 in oligodendrocytes. Among these, the proportions of cCREs annotated as promoters (defined as regions 2000 bp upstream of and 100 bp downstream from a transcription start site [TSS]) and distal regions (located > ± 2 kb from a TSS) were much greater in microglia compared with other cell types, accounting for 6% and 38.2%, respectively (Fig. 4D). We also observed that excitatory neurons exhibited a comparable proportion of distal CREs, accounting for 37.2%, relative to those observed in microglia (Fig. 4D).
We additionally identified significant GWAS loci (P-value < 5 × 10−8) from the GWAS summary statistics reported by Bellenguez et al. (2022). To prioritize AD-associated genes, we linked cell type–specific cCREs with AD-associated SNPs by assessing overlaps between GWAS loci and peak regions. This enabled identification 141 AD-associated genes (Supplemental Table S5). Of these, 78 genes were associated with microglia, including 43 genes that were specific to microglia, including such as FCER1G, APOC4-APOC2, and INPP5D (Fig. 4E). Additionally, 44 AD-associated genes were linked to astrocytes, with 13 genes being astrocyte specific, such as SEC61G and CLU, and 41 genes were associated with excitatory neurons, including 15 genes that were excitatory neuron specific, such as DGKQ and GRN (Fig. 4E).
Next, we retrieved 88 lead SNPs directly from Bellenguez et al. (2022) and linked these lead SNPs to cCREs (see Methods) (Supplemental Table S6). This analysis further highlighted 23 AD-associated genes enriched in a cell type–specific manner (Fig. 4F). Specifically, we identified 13 AD-associated genes in microglia, including well-known AD genes, such as TREM2 and BIN1. TREM2 was associated with the rs143332484 locus (AD GWAS P-value 6.035 × 10−19), with its corresponding CRE mapped to an exonic region, whereas BIN1 was linked to the rs6733839 locus (AD GWAS P-value 6.48 × 10−90), with its CRE located in a distal regulatory region (Fig. 4F). Additionally, AD-associated genes such as ACE, DGKQ, GRN, and MIR8085 were enriched in excitatory neurons, and genes such as SNX31, SEC61G, and FOSB were observed in astrocytes. Furthermore, ABCA1 and NKPD1 were associated with inhibitory neurons, and DTWD1 was linked to oligodendrocytes. Collectively, we identified and prioritized cell type–specific AD-associated genes through integrating regulatory landscapes associated with cCREs and AD GWAS data across diverse cellular populations.
Cell type–specific TF regulatory network in AD progression
To gain further insight into TF-mediated gene regulation across AD progression, we constructed cell type–specific positive TF regulatory networks. We identified candidate target genes for positive TF-regulators by assessing the correlation between motif activity and gene expression, as well as the linkage score for each gene–TF pair (Supplemental Table S7). We selected several TFs specific to microglia (SPI1, IRF1, and CEBPB) (Fig. 5A), excitatory neurons (JUN, JUND, FOSB, NEUROD2, and NEUROD6) (Fig. 5B), and oligodendrocytes (SOX4, SOX9, SOX12, and SOX30) (Fig. 5C) to construct the TF–target networks. Within these networks, we highlighted TF targets that are annotated as DEGs across AD progression and genes located at AD GWAS loci. For instance, APOE as the candidate target of CEBPB is a well-established AD-associated gene that was also found to have significantly upregulated DEGs during the transition from the inter to high stage of AD. Additionally, it is located within an AD-associated GWAS locus (Fig. 5A). These findings underscore the pivotal role of cell type–specific TFs in regulating key genes associated with AD progression, linking transcriptional changes to genetic risk loci and highlighting potential mechanisms underlying disease pathology.
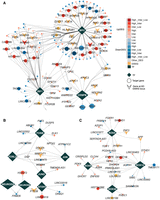
Cell type–specific TF regulatory network. (A) TF regulatory networks in microglia for SPI1, IRF1, and CEBPB, highlighting predicted candidate target genes that exhibit significant expression change during AD progression or are annotated in AD-associated GWAS locus. (B) TF regulatory networks in excitatory neurons for the TFs JUN, JUND, FOSB, NEUROD2, and NEUROD6, showing the predicted candidate target genes that were significantly changed during AD progression or annotated in AD GWAS locus. (C) TF regulatory networks in oligodendrocytes for the TFs SOX4, SOX9, SOX12, and SOX30, showing the predicted candidate target genes that were significantly changed during AD progression or annotated in AD GWAS locus. In all panels, the shape of diamonds represents TFs; circles represent target genes of TFs; and arrowheads indicate the target genes of TFs located within AD-associated GWAS loci. The red gradient denotes target genes annotated with upregulated DEGs at different stages of AD progression, and the blue gradient represents target genes annotated with downregulated DEGs. Genes with inconsistent DEG change across stages are labeled in gray.
Discovery of cell type–specific repurposable drugs for AD progression
In total, we identified 141 potential AD-associated genes based on prior analyses (Fig. 6A). Among these, 23 genes were associated with AD lead SNPs (e.g., ACE, TREM2, B1N1); 54 genes were associated with significant DEGs across AD progression (e.g., ABCA1, APOE, APOC1, APOC2); 52 genes were target genes of positive TF-regulators (TG; e.g., DGKQ, APOE, SNX31); and 36 genes (e.g., ACE, APP, APOE, ABCA1, CRHR1) were known drug targets with FDA-approved or clinically investigational therapies (Fig. 6A).
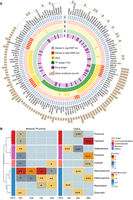
Highlighted repurposable drug candidates in a cell type–specific manner across AD progression. (A) Prioritized AD-associated genes by combining cCREs and significant AD-associated variants. The first circle with blue rectangles highlights genes identified by overlapping AD-associated significant SNPs loci with cell type–specific cCREs. The second circle with dark red rectangles represents genes associated with AD lead SNPs loci and cCREs. The third circle with dark yellow rectangles is DEGs. The fourth circle with dark green rectangles represents the target genes regulated by TFs. The fifth circle with dark purple rectangles comprises target genes annotated as known drug targets. The outer bar plot provides the evidence counts for each gene. (B) Predicted repurposable drug candidates by network proximity and GSEA clustered by ATC first-level class. Left panel shows enriched drug candidates identified by network proximity method, in which gradient colors reflect absolute Z-scores. Right panel shows enriched drug candidates identified by GSEA method, in which gradient colors represent enrichment scores. Statistical significance is denoted by (*) FDR < 0.05, (**) FDR < 0.01, (***) FDR < 0.001.
To predict cell type–specific drug candidates across AD progression, we utilized 141 potential AD-associated genes as input for a network proximity analysis. This approach evaluates the network relationship between AD-associated genes and drug targets within the human PPI network. We subsequently identified enriched drug candidates by performing GSEA integrating drug–gene signatures from the CMap database (Lamb et al. 2006) and information from the DEGs (see Methods). The enrichment score (ES) indicates each drug's potential to reverse the observed gene expression patterns. By integrating these methods, we identified nine overlapping candidate drugs that met the criteria for both network proximity (Z-scores < −2, FDR < 0.05) and GSEA (ES > 0, FDR < 0.05) (Fig. 6B; Supplemental Table S8), including galantamine, resveratrol, imatinib, deferoxamine, captopril, staurosporine, quercetin, procaine, and probucol. These nine drugs were categorized into four pharmacological classes based on the first level of the Anatomical Therapeutic Chemical (ATC) classification system, with the most candidate drugs targeting the nervous and cardiovascular systems.
Among these, galantamine, which emerged as a potential drug candidate in inhibitory neurons, is an acetylcholinesterase inhibitor approved for the treating mild to moderate AD (Wilcock et al. 2000). By inhibiting acetylcholine breakdown and modulating nicotinic receptors, it enhances cholinergic transmission, thereby improving cognitive function. However, galantamine does not halt AD progression (Raskind et al. 2004). Beyond galantamine, other identified drugs have also shown promise in preclinical studies. For instance, resveratrol, highlighted as a potential drug candidate in astrocytes, oligodendrocytes, and inhibitory neurons, is a polyphenolic compound found in grapes and red wine with antioxidant properties (Bastianetto et al. 2015; Jia et al. 2017; Gomes et al. 2018). Notably, it has been extensively studied for its ability to modulate Aβ aggregation and reduce neuroinflammation in AD (Bastianetto et al. 2015; Jia et al. 2017; Gomes et al. 2018; Zhang et al. 2023), both critical in AD pathology.
Captopril is another promising candidate that targets ACE to modulate the renin–angiotensin system. This is consistent with experimental evidence that ACE inhibitors can attenuate amyloid pathology, oxidative stress, and neurodegeneration in AD models (AbdAlla et al. 2013). In addition to ACE, our analysis identified REN, BDKRB1, and MMP2 as potential targets of captopril, suggesting that its effects may include regulation of neurovascular integrity and inflammatory signaling.
Another promising candidate, imatinib, was identified in inhibitory neurons, astrocytes, and microgliosis. Known for its ability to modulate gamma-secretase activity (Weintraub et al. 2013; Olsson et al. 2014), imatinib has been investigated for its potential to reduce Aβ production. Additionally, gabapentin, identified in excitatory neurons, is an anticonvulsant that modulates voltage-gated calcium channels that has been studied for off-label use in managing behavioral symptoms associated with dementia, such as agitation (Buskova et al. 2011).
To provide deeper insights into the mechanism-of-action of the drug candidates, we constructed drug–target networks for nine prioritized drugs (Fig. 7). For example, galantamine was found to target multiple subunits of the nicotinic cholinergic receptor—CHRNA2, CHRNA5, CHRNA10, and CHRND, thereby potentially enhancing cholinergic signaling, a critically impaired pathway in AD (Buckingham et al. 2009), further emphasizing the role of these receptors in AD pathophysiology. Furthermore, resveratrol and deferoxamine target APP, a key gene involved in the pathophysiology of AD (O'Brien and Wong 2011). These findings highlight the potential of drug candidates to modulate critical pathways in AD progression in a cell type–specific manner. However, more functional and clinical observations are highly warranted for those top predicted candidate drugs in the future.
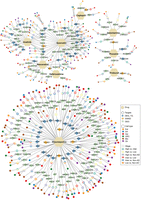
Drug–target network illustrating mechanisms of action for nine candidate drugs. Hexagon denotes drugs, and diamonds shape denote target genes. Green diamonds indicate targets that are differentially expressed genes (DEGs); blue represents targets are both DEGs and TFs’ targets; and yellow denotes genes located within AD-associated GWAS loci. When multiple criteria apply, the most specific class is displayed (blue → yellow → green). An annular ring around each target indicates the six comparisons of ADNC stage(s) at which the gene is a DEG; ring sectors correspond to high versus inter, high versus low, high versus non-AD, inter versus low, inter versus non-AD, and low versus non-AD. Small colored circles attached to stage(s) indicate the related six cell type(s) of DEGs. Edges represent curated drug–target interactions.
To further investigate the temporal specificity of drug effects, we first categorized genes from the input set into three temporal expression stages based on differential expression profiles: low (low vs. non-AD), intermediate (inter vs. non-AD and inter vs. low AD), and high (high vs. non-AD, high vs. low AD, and high vs. inter AD). Using GSEA, we then assessed the enrichment of drug-associated gene signatures across these temporal categories. The analysis revealed that the nine candidate drugs—galantamine, resveratrol, imatinib, deferoxamine, captopril, staurosporine, quercetin, procaine, and probucol—were predominantly enriched in the high stage (Supplemental Table S9; Supplemental Fig. S7), indicating that these compounds primarily target gene programs associated with later stages of AD progression.
In addition, we visualized the top enriched drugs across the low, intermediate, and high stages to highlight their stage-specific activity patterns (Supplemental Fig. S8). The results reveal distinct temporal preferences of drug effects across cell types. For example, chlorprothixene and mephentermine show enrichment at the intermediate stage in microglia, whereas captopril and cyproheptadine are predominantly enriched at the high stage, suggesting stronger effects in later disease progression. Similarly, in excitatory neurons, drugs such as nitrofuraltone and flucloxacillin are enriched at the intermediate stage, whereas nifedipine and mefenamic acid appear at more significant enrichment during the low stage. Together, these findings illustrate that different drugs may exert different responses across distinct temporal phases of AD progression, providing insights into potential personalized treatment in the future.
Discussion
Despite significant efforts to discover effective drugs for AD, the complex neuropathological mechanisms, cellular heterogeneity, and challenges associated with early diagnosis continue to hinder drug development (Long and Holtzman 2019; Murdock and Tsai 2023; Zhang et al. 2023; Cummings et al. 2024). To address these challenges, we developed a single-cell multiomic analytic framework that integrates a network-based approach with snRNA-seq and snATAC-seq data for therapeutic target identification and drug repurposing in AD progression. Our framework categorizes AD progression into four distinct stages based on ADNC that is a robust classification of neuropathology. This dynamic staging allows for detailed observation of cellular and molecular changes during disease progression, enabling identification of precise therapeutic targets to inform drug development.
Although ADNC stages do not capture longitudinal progression within individuals, we still observed clear cell type–specific transcriptional changes across stages. The transition from intermediate to high ADNC showed the strongest gene expression shifts, with a greater proportion of downregulated genes in both excitatory and inhibitory neurons. These patterns align with previous reports (Mathys et al. 2023; Gabitto et al. 2024). In contrast, earlier stage transitions showed far fewer DEGs. Together, these results indicate that gene expression changes vary by cell type and become most pronounced in late-stage AD, underscoring the heterogeneous nature of AD progression.
By integrating cell type–specific CREs, TF regulatory networks, and AD GWAS data, we identified 141 AD-associated genes, including 36 linked to approved or investigational therapies. These genes span key AD pathways—amyloid/tau biology (Hampel et al. 2021), lipid metabolism (Wahrle et al. 2008; Wildsmith et al. 2013; Chen et al. 2020), and neuroinflammation (de la Tremblaye et al. 2017; Liu et al. 2021; Wu et al. 2021b; Chou et al. 2023)—and include both well-established genes (e.g., APP, MAPT, APOE) and emerging candidates. Overall, these findings highlight diverse molecular mechanisms underlying AD and point to multiple potential therapeutic targets.
Using the 141 AD-associated genes, we identified nine candidate drugs supported by both network proximity and GSEA analyses. These drugs act through diverse mechanisms. Imatinib interacts with multiple targets, including ABCB1, ABCG2, SYK, LCK, FYN, KIT, CSF1R, CA7, and CA12, which target transporters and signaling molecules involved in amyloid clearance, microglial activation, and synaptic function (Phamluong et al. 2017; Wanek et al. 2020; Baltira et al. 2024; Zaman et al. 2024). Deferoxamine, an iron chelator, has documented neuroprotective effects in AD models and may act through APP and G-protein–related pathways (Guo et al. 2013, 2015). Probucol targets genes involved in lipid metabolism and amyloid clearance (Lam et al. 2022; Sharif et al. 2024), whereas quercetin's multitarget profile suggests roles in reducing amyloid burden, oxidative stress, and inflammation (Sabogal-Guáqueta et al. 2015). Staurosporine, a broad kinase inhibitor, has shown cognitive benefits in earlier animal studies (Nabeshima et al. 1991). Together, these candidates highlight multiple therapeutic avenues aligned with AD molecular mechanisms.
Despite the promising findings, we acknowledge several limitations in this study. First, the data are derived from a cross-sectional sampling of brains at different ADNC stages, rather than longitudinal tracking of disease progression within the same individuals. As such, the observed molecular changes across stages may be influenced, at least in part, by intersubject variability such as differences in genetic background. Although we applied data normalization and batch correction and included covariates to mitigate these effects, we recognize that these efforts may not be fully eliminate confounding. Our interpretation of “progression” is based on neuropathological severity rather than true temporal dynamics. Further integration of longitudinal data sets, when available, will be essential for confirming disease trajectories and refining our models. Second, although CMap provides a valuable resource for transcriptomic-based drug repurposing, it is based primarily on cancer cell lines (Lamb et al. 2006), which do not fully recapitulate the cellular context of the human brain. Thus, predicted drug effects need be further validation experimentally. Moreover, some predicted candidates, such as resveratrol, have previously failed in clinical trials for AD, highlighting the need for functional and translational validation of prioritized drugs further.
To address these limitations, future studies will expand our training corpus with single-cell multiomics from the Religious Orders Study and Memory and Aging Project (ROSMAP) and explicitly model disease severity using a continuous pseudoprogression score. Leveraging generative AI and large-scale foundation models, we could develop a gene regulatory network–based framework that dynamically aligns molecular states with disease progression in a continuous manner. Such a stage-aware and data-driven gene regulatory network-based framework could ultimately enable real-time modeling of disease trajectories, enhancing precision in therapeutic target discovery and drug development.
Methods
Resources of snRNA-seq and snATAC-seq data
The snRNA-seq and snATAC-seq data used in this study are available from the SEA-AD consortium (syn26223298) (Gabitto et al. 2024). We obtained the gene expression count matrices in the h5ad format and the snATAC-seq raw fragments files from 84 human brain middle temporal gyrus samples. We also acquired the corresponding metadata manifest (Supplemental Table S1), which includes comprehensive neuropathological staging metrics such as Thal phase, Braak stage, CERAD score, and overall ADNC. The metadata also provide demographic and clinical information, including sex and the presence of copathologies such as Lewy body disease and LATE.
snRNA-seq data quality control, dimensionality reduction, and clustering
The snRNA-seq data were processed using SCANPY (v1.9.5) in Python 3.10 (Wolf et al. 2018). Quality-control metrics were computed with calculate_qc_metrics, and low-quality cells (fewer than 200 detected genes) and genes (expressed in fewer than 10 cells) were removed. Outliers for total counts, detected genes, and mitochondrial or ribosomal content were excluded based on interquartile range (IQR) thresholds combined with fixed cutoffs (10% mitochondrial and 20% ribosomal) (for details, see Supplemental Methods). Potential doublets were identified and removed using Scrublet (Wolock et al. 2019).
Filtered count matrices were normalized using a shifted logarithmic transformation, followed by principal component analysis (PCA) for dimensionality reduction and Leiden clustering for community detection. Low-dimensional visualization was performed with UMAP. Major cell types were annotated according to established marker genes (Morabito et al. 2021; Mathys et al. 2023; Xiong et al. 2023; Emani et al. 2024; Gabitto et al. 2024) and verified for consistency with prior studies (Supplemental Fig. S1; Gabitto et al. 2024). Neuronal subtypes were assigned following the classifications reported in the original reference data set (Gabitto et al. 2024).
DEG and gene functional enrichment analysis
Differential expression analysis was performed using the muscat R package by aggregating single-cell data into pseudobulk profiles while adjusting for sex, APOE4 status, and age as covariates (Crowell et al. 2020). Expression changes were assessed within each cluster across six pairwise comparisons representing AD progression stages (low vs. non-AD, intermediate vs. non-AD, high vs. non-AD, intermediate vs. low, high vs. low, and high vs. intermediate). Genes with an adjusted P < 0.05 and log2FC| > 0.5 were considered differentially expressed. Functional enrichment analysis of DEGs was conducted using clusterProfiler (v4.10.1) (Yu et al. 2012; Wu et al. 2021a), focusing on GO biological process and molecular function categories, with results corrected by the Benjamini–Hochberg method (adjusted P < 0.05).
snATAC-seq data quality control, dimensionality reduction, and clustering
The snATAC-seq data were processed using the ArchR package (v1.0.3) (Granja et al. 2021) in an R 4.4.2 environment (R Core Team 2024) with the hg38 reference genome. Quality-control filtering was applied based on fragment count, TSS enrichment, and fragment size distribution, and potential doublets were removed using built-in ArchR functions. Dimensionality reduction and clustering were performed using the iterative latent semantic indexing (LSI) and Leiden algorithms implemented in ArchR (cf. Supplemental Methods). Low-quality clusters were excluded, and cell types were annotated based on canonical marker genes (Supplemental Fig. S1). Gene activity scores were visualized using UMAP (Ober-Reynolds et al. 2023), with additional smoothing applied via the MAGIC algorithm (van Dijk et al. 2018).
Peak calling and annotation
Chromatin accessibility peaks were identified using MACS2 (Zhang et al. 2008) with default parameters, and cell type–specific marker peaks were determined using the getMarkerFeatures function in ArchR. Candidate cis-regulatory elements (cCREs) were annotated based on hg38 genome features, and TF motif enrichment was assessed with cisBP database (Weirauch et al. 2014) using the addMotifAnnotations and peakAnnoEnrichment functions. Motif footprinting was further performed with getFootprints to evaluate motif accessibility and regulatory activity (Granja et al. 2021).
Integration of snRNA-seq and snATAC-seq data sets
Integrated analysis of snATAC-seq and snRNA-seq data across AD progression stages was carried out using the addGeneIntegrationMatrix function in ArchR, which employs Seurat's canonical correlation analysis framework (Satija et al. 2015; Butler et al. 2018; Stuart et al. 2019; Hao et al. 2021, 2024). This integration enabled joint mapping of chromatin accessibility and gene expression profiles across major brain cell types (for details, see Supplemental Methods).
Linked AD-associated GWAS loci with cCRE
GWAS summary statistics from Bellenguez et al. (2022) were used to identify AD-associated loci (P < 5 × 10−8). Lead SNPs were obtained from the EBI GWAS catalog, excluding 23andMe samples. These loci were intersected with cell type–specific cCREs (FDR ≤ 0.05, |log2FC| > 0.5) to link genetic variants with putative regulatory elements.
Identification of positive TF-regulators and putative targets
To infer TF-regulators, we correlated motif accessibility (chromVAR deviation Z-scores) with TF gene expression across aggregated cells using ArchR (Granja et al. 2021). TFs showing strong positive correlations (cor > 0.5, adjusted P < 0.01) and high intercluster variability were defined as positive regulators. Regulatory targets were inferred by integrating motif activity with gene expression correlations, summarized into linkage scores, and filtered using stringent correlation and FDR thresholds (for details, see Supplemental Methods). Functional enrichment of the target genes was performed using topGO (https://bioconductor.org/packages/topGO).
Building human protein–protein interactome and drug–target network
The human protein–protein interactome (PPI) and drug–target network were constructed as previously described (Cowley et al. 2012; Goel et al. 2012; Licata et al. 2012; Breuer et al. 2013; Orchard et al. 2014). Briefly, the PPI network was compiled by integrating experimentally validated interactions from multiple public databases, including yeast two-hybrid (Luck et al. 2020), kinase–substrate, signaling, and protein complex data sets, resulting in a comprehensive network of 351,444 interactions among 17,706 proteins. The drug–target network was built by aggregating data from DrugBank (version 4.3) (Law et al. 2014), BindingDB (Liu et al. 2007), ChEMBL (version 20) (Gaulton et al. 2012), the Therapeutic Target Database (Yang et al. 2016), PharmGKB (Whirl-Carrillo et al. 2012), and the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al. 2014), retaining high-confidence interactions with binding affinities ≤10 µM. Both networks are available through the AlzGPS platform (https://alzgps.lerner.ccf.org).
Network proximity-based drug repurposing
We applied a network-based proximity approach to identify potential AD drug candidates by quantifying the shortest distances between drug targets and AD-associated genes within the human interactome (Xu et al. 2021; Zhou et al. 2023). Statistical significance was assessed using permutation testing to calculate Z-scores, with significant candidates defined by Z < –2 and FDR < 0.05 (for details, see Supplemental Methods).
Gene set enrichment analysis
Complementary to network proximity, we performed GSEA-based drug enrichment analysis using transcriptional signatures from CMap (Lamb et al. 2006; Zhou et al. 2020). AD-associated DEGs were used as input to identify compounds capable of reversing disease-related expression patterns. ESs and FDRs were computed using established pipelines, and positively enriched compounds (ES > 0, FDR < 0.05) were prioritized as repurposing candidates (for details, see Supplemental Methods).
Code availability
All source code and custom scripts used in this study are available at GitHub (https://github.com/ChengF-Lab/AD-digitaltwins) and as Supplemental Code.
Data access
Raw snRNA-seq and snATAC-seq data used in this study and processed data have been submitted to the AD Knowledge Portal (https://adknowledgeportal.synapse.org/) under study number syn26223298 and can be downloaded from SEA-AD website (https://brain-map.org/consortia/sea-ad/our-data).
Competing interest statement
J.C. has provided consultation to Acadia Pharmaceuticals, Acumen Pharmaceuticals, ALZpath, Annovis Bio, APRINOIA Therapeutics, Artery Therapeutics, Biogen, Biohaven, BioXcel Therapeutics, Bristol Myers Squibb, Eisai, Fosun Pharma, Global Alzheimer's Platform (GAP) Foundation, Green Valley Pharmaceuticals, Janssen Janssen Pharmaceuticals, Karuna Therapeutics, Kinoxis Therapeutics, Lighthouse Pharma, Lilly, Lundbeck, EQT Life Sciences (formerly LSP), Mangrove Therapeutics, Merck, MoCA Cognition, NewAmsterdam Pharma, Novo Nordisk, OncoC4, OptoCeutics, Otsuka, Oxford Brain Diagnostics, Praxis Bioresearch, Prothena Biosciences, remynd, Roche, Scottish Brain Sciences, Signant Health, Simcere Pharmaceutical Group, Sinaptica Therapeutics, T-Neuro Pharma, TrueBinding, and Vaxxinity pharmaceutical, assessment, and investment companies. The other authors declare no competing interests.
Acknowledgments
This work was primarily supported by the National Institute on Aging (NIA) under award numbers U01AG073323, R01AG066707, R01AG084250, R01AG076448, R01AG082118, R01AG092462, R01AG092591, RF1AG082211, R33AG083003, R21AG083003, and P30AG072959; by the National Institute of Neurological Disorders and Stroke (NINDS) under award number RF1NS133812; by the Alzheimer's Association award (ALZDISCOVERY-1051936); the Alzheimer's Disease Drug Discovery Foundation (ADDF); and the Dr. Keyhan and Dr. Jafar Mobasseri Endowment to F.C. This work was supported in part by the Brockman Foundation, project 19PABH134580006-AHA/Allen Initiative in Brain Health and Cognitive Impairment, the Elizabeth Ring Mather & William Gwinn Mather Fund, the S. Livingston Samuel Mather Trust, and the Louis Stokes VA Medical Center resources and facilities to A.A.P. This work was supported in part by National Institute of General Medical Sciences (NIGMS) grant P20GM109025, NIA R35AG71476, NIA R25AG083721-01, NINDS R01NS139383, the Alzheimer's Disease Drug Discovery Foundation (ADDF), the Ted and Maria Quirk Endowment, and the Joy Chambers-Grundy Endowment to J.C.
Author contributions: F.C. conceived the study. Y.R. designed and performed all data analyses and experiments. M.H., Y.E.L., A.A.P., and J.C. interpreted the data analysis. J.C. provided clinical context of the outcomes. Y.R. drafted the manuscript. Y.R., F.C., A.A.P., and J.C. critically revised the manuscript. All authors gave final approval of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280436.125.
-
Freely available online through the Genome Research Open Access option.
- Received January 14, 2025.
- Accepted January 15, 2026.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








