
Post-transcriptional cross- and auto-regulation buffer expression of the human RNA helicases DDX3X and DDX3Y
- 1Whitehead Institute, Cambridge, Massachusetts 02142, USA;
- 2Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA;
- 3Departments of Bioengineering, Biology, Chemistry, and Chemical Biology, Single Cell Proteomics Center, and Barnett Institute, Northeastern University, Boston, Massachusetts 02115, USA;
- 4Howard Hughes Medical Institute, Whitehead Institute, Cambridge, Massachusetts 02142, USA
Abstract
The Y-linked gene DDX3Y and its X-linked homolog DDX3X survived the evolution of the human sex chromosomes from ordinary autosomes. DDX3X encodes a multifunctional RNA helicase, with mutations causing developmental disorders and cancers. We find that, among X-linked genes with surviving Y homologs, DDX3X is extraordinarily dosage sensitive. Studying cells of individuals with sex chromosome aneuploidy, we observe that when the number of Y Chromosomes increases, DDX3X transcript levels fall; conversely, when the number of X Chromosomes increases, DDX3Y transcript levels fall. In 46,XY cells, CRISPRi knockdown of either DDX3X or DDX3Y causes transcript levels of the homologous gene to rise. In 46,XX cells, chemical inhibition of DDX3X protein activity elicits an increase in DDX3X transcript levels. Thus, perturbation of either DDX3X or DDX3Y expression is buffered: by negative cross-regulation of DDX3X and DDX3Y in 46,XY cells and by negative auto-regulation of DDX3X in 46,XX cells. DDX3X–DDX3Y cross-regulation is mediated through mRNA destabilization—as shown by metabolic labeling of newly transcribed RNA—and buffers total levels of DDX3X and DDX3Y protein in human cells. We infer that post-transcriptional auto-regulation of the ancestral (autosomal) DDX3X gene transmuted into auto- and cross-regulation of DDX3X and DDX3Y as these sex-linked genes evolved from ordinary alleles of their autosomal precursor.
DDX3X and DDX3Y are homologous but nonidentical genes on the human X and Y Chromosomes (Lahn and Page 1997). They encode pleiotropic RNA helicases implicated in multiple aspects of RNA metabolism, including splicing, export, stability, translation, and stress response (Soto-Rifo and Ohlmann 2013). DDX3X is widely conserved across eukaryotes, with orthologs in mammals, flies, worms, and yeast (Johnstone et al. 2005; Elbaum-Garfinkle et al. 2015; Sharma et al. 2017). Human DDX3X mutations are associated with several neurodevelopmental disorders and cancers (Snijders Blok et al. 2015; Valentin-Vega et al. 2016). DDX3X is expressed throughout the body from the “inactive” Chr X (Xi) in females as well as from the “active” Chr X (Xa) in males and females (Lahn and Page 1997; Tukiainen et al. 2017). Like DDX3X, its Y-linked homolog DDX3Y is expressed in a wide array of human tissues (Godfrey et al. 2020), but studies of its organismal function have focused on roles in spermatogenesis (Ramathal et al. 2015). The X- and Y-encoded helicases are 91% identical at the amino acid level (Lahn and Page 1997). Although they have significantly diverged in their N- and C-terminal regions, the RNA binding and helicase domains are largely conserved (Rosner and Rinkevich 2007). Early experiments showed that the DDX3Y protein was functionally interchangeable with DDX3X in vitro (Sekiguchi et al. 2004). More recent work has shown that the proteins have partially overlapping functions, with similar effects on protein synthesis (Venkataramanan et al. 2021) but differing capacities for stress granule formation and translational repression (Venkataramanan et al. 2021; Shen et al. 2022).
DDX3X and DDX3Y constitute one of only 17 human X–Y gene pairs that survived the sex chromosomes’ evolution from ordinary autosomes (Lahn and Page, 1999; Skaletsky et al. 2003). Although human Chr X retains 98% of the genes that were present on the ancestral autosomes, Chr Y retains only 3% of these genes (Bellott et al. 2014). Most of these surviving Chr Y genes were preserved by natural selection to maintain the ancestral dosage of regulators of key cellular processes. Among this select group of X-linked genes with surviving Y homologs, we recently noticed a distinguishing feature of DDX3X: Although the gene is robustly expressed from both Xa and Xi in human cells (and in this sense resembles other X-linked genes with surviving Y homologs), steady-state levels of DDX3X transcripts were only modestly higher in 46,XX cells than in 46,XY cells (San Roman et al. 2023), suggesting that DDX3X (and possibly DDX3Y) might be subject to dosage constraints and regulatory mechanisms not seen with other X–Y gene pairs. Accordingly, we decided to examine closely the dosage sensitivity and regulatory mechanisms that govern expression of DDX3X and DDX3Y.
Results
DDX3X and DDX3Y are especially dosage sensitive compared with genes with a similar evolutionary trajectory
We first asked if DDX3X and DDX3Y are more dosage sensitive than other human X–Y gene pairs. For each of the 17 gene pairs, we tallied whether dosage sensitivity had necessitated (1) expression from Xi in human females and (2) maintenance of a Y-homolog in males of diverse species—both features of highly dosage-sensitive genes (Bellott et al. 2014). We addressed the first point by reanalyzing Xi expression data recently generated from cultured human cells (San Roman et al. 2023). We addressed the second point by examining the survival of each Y-linked gene across 14 species of therian mammals for which high-quality, contiguous sequence assemblies of the sex chromosomes are available. Specifically, for each Y-homolog, we calculated a phylogenetic branch length, the sum of all branch lengths connecting species in which the gene is present, and thus a measure of the gene's longevity on Chr Y in therian mammals. We also calculated, for each Y-homolog, the survival fraction: the ratio of observed phylogenetic branch length to maximum possible branch length across the set of species examined (Bellott and Page 2021).
Among X–Y gene pairs, those with the highest dosage sensitivity should be expressed from Xi in females and be long-lived and universally retained on the Chr Y across species, that is, have a survival fraction of one. We find that, among the 17 human X–Y gene pairs, only DDX3X(Y), KDM6A(UTY), ZFX(Y), and NLGN4X(Y) are expressed from Xi in human females and survive in all possible lineages (Table 1).
Dosage-sensitivity of human X–Y pair genes across therian mammalian lineages
We further profiled the sensitivity of DDX3X to dosage changes using gene-wise metrics of constraint on overexpression or loss of function: (1) PCT scores, which measure the evolutionary conservation of microRNA targeting sites in a gene's 3′ UTR (Friedman et al. 2009), and (2) LOEUF values, the ratio of observed to expected loss-of-function variants in a gene in human populations (Supplemental Table S1; Karczewski et al. 2020). Consistent with the role of miRNAs tuning gene dosage by lowering target mRNA levels, high conservation of miRNA targeting is a feature of genes sensitive to dosage changes in humans, particularly increases in gene dosage (Naqvi et al. 2018), whereas a low LOEUF value demonstrates sensitivity to diminished function. We rank-ordered all non-PAR genes on the human Chr X by each of these two metrics (San Roman et al. 2023), from least to most constrained. Among X–Y pair genes expressed from Xi, DDX3X has the highest combined sensitivity to overexpression and diminished function, implying that its level of expression is especially constrained (Fig. 1A; Supplemental Table S1).

DDX3X is highly dosage-sensitive and expressed broadly among human tissues. (A) Among human X–Y pair genes, DDX3X ranks highest in combined sensitivity to overexpression (as judged by PCT percentile among all Chr X genes) and diminished function (as judged by LOEUF percentile among all Chr X genes). (B,C) DDX3X (B) and DDX3Y (C) and their chicken ortholog display the highest expression breadth among, respectively, the X and Y members of human X–Y pairs. Note that expression breadth data were not available for the chicken ortholog of KDM5C/D.
We also assessed whether DDX3X and DDX3Y are expressed more broadly across the body than other X–Y gene pairs, another feature of highly dosage-sensitive genes (Bellott et al. 2014), and whether this breadth was present ancestrally. The ancestral state of sex-linked genes can be inferred from analyses of birds such as chickens, in which the orthologs of human sex chromosomal genes are found on autosomes 1 and 4 (Bellott et al. 2010). For each gene pair for which expression data were available in humans (GTEx Consortium, 2017) and chickens (Merkin et al. 2012; Bellott et al. 2014), we measured how broadly the chicken gene and human gene pair were expressed across the body's various tissues. DDX3X, DDX3Y, and their autosomal chicken ortholog display the highest combined expression breadth across the two species, suggesting that their dosage is critical throughout the body (Fig. 1B,C; Supplemental Table S2).
DDX3X and DDX3Y transcript levels fall, respectively, as Chr Y and Chr X copy numbers rise
To identify mechanisms that regulate DDX3X and DDX3Y expression in human cells, we reanalyzed RNA sequencing data from primary skin fibroblasts of human donors with sex chromosome aneuploidies (San Roman et al. 2023). We first assessed DDX3X and DDX3Y transcript levels in cells with a single Chr X and increasing copies of Chr Y (Supplemental Table S3). As expected, DDX3Y transcript levels rise with increasing copy numbers of Chr Y. However, DDX3X expression from the single Chr X falls significantly (Fig. 2A,B). Conversely, in cells with a single Chr Y and increasing numbers of Chr X, DDX3X transcript levels rise, as expected given the gene's expression from both Xa and Xi. However, DDX3Y expression from the single Chr Y falls significantly (Fig. 2C,D).
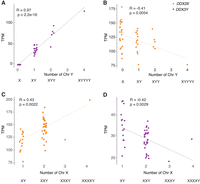
DDX3X and DDX3Y transcript levels are negatively responsive to Chr Y and Chr X copy numbers, respectively. Scatterplots show DDX3X and DDX3Y transcript levels in cultured fibroblasts with the indicated sex chromosome constitutions. Each point represents a primary fibroblast culture from one individual. (A,B) DDX3Y transcript levels are significantly elevated and DDX3X transcript levels significantly reduced in fibroblasts with multiple Chr Y. (C,D) DDX3X transcript levels are significantly elevated and DDX3Y transcript levels significantly reduced in fibroblasts with multiple Chr X. R-values and statistical significance were calculated using Pearson's correlation.
We asked whether this inverse relationship is shared across all X–Y gene pairs or is a unique feature of DDX3X and DDX3Y. For each X–Y pair gene, we obtained values for the change in its transcript levels per added Xi and for the change in its transcript levels per added Chr Y (San Roman et al. 2023). In both fibroblasts and lymphoblastoid cell lines (LCLs), DDX3X transcript levels fall significantly as the Chr Y copy number increases; conversely, DDX3Y transcript levels fall as the Chr X copy number increases (Supplemental Table S3). This response is not observed with other X–Y pair genes; it is unique to DDX3X and DDX3Y (Supplemental Table S3).
We considered the possibility that these decreases in DDX3X and DDX3Y transcript levels in response to changes in sex chromosome copy number might reflect a general cellular response to aneuploidy. To test this, we examined data from individuals with trisomy 21 (San Roman et al. 2023). We observed no change in DDX3X or DDX3Y transcript levels in response to Chromosome 21 copy number (Supplemental Fig. S1). We conclude that DDX3X and DDX3Y transcript levels are inversely related to Chr Y and Chr X copy numbers, respectively.
Perturbing DDX3X elicits an opposing response in DDX3Y and vice versa
We asked whether these effects of altering sex chromosome copy number are owing to DDX3X and DDX3Y expression changes. We profiled cells with naturally occurring mutations that affect DDX3X or DDX3Y expression and performed experimental knockdowns to capture the effects of perturbing DDX3X and DDX3Y transcript levels (Supplemental Tables S4–S9).
First, we quantified DDX3X transcripts in LCLs from azoospermic (infertile) males with AZFa microdeletions. AZFa microdeletions result from homologous recombination between endogenous retroviral elements on the human Chr Y, and they remove the DDX3Y and USP9Y genes without affecting other genes (Fig. 3A; Sun et al. 2000). We found that DDX3X transcript levels were significantly higher in LCLs from AZFa-deleted males compared with males with intact Chr Y (Fig. 3B; Supplemental Table S4). To test whether DDX3X transcript levels are elevated upon deletion of other regions of Chr Y, we analyzed data from XY individuals whose Chr Y retains DDX3Y but is missing several other genes, including the sex-determining gene SRY (Schiebel et al. 1997). DDX3X transcript levels were unaltered in these individuals (Supplemental Fig. S2; Supplemental Table S5; San Roman et al. 2023), demonstrating that DDX3X levels are specifically elevated in response to DDX3Y deletion.
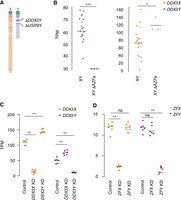
DDX3X and DDX3Y each respond to perturbations in the other's expression. (A) Schematic diagram of naturally occurring Chr Y (AZFa) microdeletion of DDX3Y and USP9Y. (B) DDX3X transcript levels are significantly higher in AZFa-deleted 46,XY LCLs compared with 46,XY LCLs with intact Chr Y. Each point represents a sample from one individual. Statistical significance determined by Mann–Whitney U-test: (***) P < 0.0001, (*) P < 0.05 (C) CRISPRi-mediated knockdown of DDX3Y using two independent gRNAs in three unrelated 46,XY fibroblast cultures results in significantly elevated DDX3X transcript levels. Conversely, DDX3X knockdown results in significantly elevated DDX3Y transcript levels. (D) Reanalysis of CRISPRi knockdown of ZFX or ZFY (San Roman et al. 2024) demonstrates that knockdown of either gene does not result in significant elevation of the homolog's transcripts. Statistical significance determined by ANOVA: (**) P < 0.001.
We then used CRISPRi to target DDX3X or DDX3Y for knockdown in primary 46,XY fibroblasts. DDX3X transcript levels rose significantly upon knockdown of DDX3Y (DDX3Y KD), and DDX3Y transcript levels responded in a reciprocal fashion to DDX3X KD (Fig. 3C; Supplemental Table S6). This negative cross-regulation across X and Y homologs was specific to DDX3X and DDX3Y; data from CRISPRi knockdowns of ZFX and ZFY, another broadly expressed, dosage-sensitive X–Y gene pair, did not show this pattern (Fig. 3D; Supplemental Table S7; San Roman et al. 2024). We validated these findings in an independent data set, the Cancer Cell Line Encyclopedia (CCLE), which catalogs mutational and expression data from hundreds of cancer cell lines (Ghandi et al. 2019). There we identified 491 different XY cell lines that retained the Chr Y and, among these, a set of 11 lines that harbored point mutations in DDX3X predicted to cause loss of function, either by introducing premature stop codons or by ablating helicase function (Supplemental Table S8). DDX3Y transcript levels are significantly higher in these 11 cell lines compared with lines in which DDX3X is intact (Supplemental Fig. S3; Supplemental Table S9). Thus, knockdowns or loss of function in either DDX3X or DDX3Y is consistently buffered by compensatory increases in the homolog's expression, demonstrating that DDX3X and DDX3Y are negatively cross-regulated.
Negative cross-regulation of DDX3X buffers total levels of DDX3X and DDX3Y
We hypothesized that negative cross-regulation of DDX3X and DDX3Y maintains the combined expression of the two genes in a narrow range, buffering total transcript levels against changes in gene dosage. To test this, we summed transcript levels for the two genes in our knockdown models. We observed that, in the setting of DDX3Y knockdown, the increase in DDX3X transcript levels fully compensates and maintains the summed transcript levels of DDX3X and DDX3Y at control levels (Fig. 4A; Supplemental Table S6). However, in the setting of DDX3X knockdown—a larger perturbation—the increase in DDX3Y transcript levels does not fully compensate.
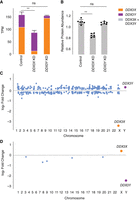
Increased expression of DDX3X fully compensates, at transcript and protein levels, for CRISPRi knockdown of DDX3Y, but the inverse is not true. (A) Stacked bar graph showing summed TPM of DDX3X and DDX3Y transcripts in knockdowns using two independent gRNAs in three independent 46,XY fibroblast cultures. Statistical significance calculated by ANOVA: (**) P < 0.001. (B) Bar graph showing abundance of shared DDX3X and DDX3Y peptides in CRISPRi knockdowns with three technical replicates in two independent 46,XY fibroblast cultures. (C) Differential gene expression analysis of control versus DDX3X knockdown reveals significant expression changes in 397 target genes across the genome, including DDX3X. Genes with P < 0.05 (after multiple hypothesis correction) are indicated in blue, with exception of DDX3X (orange) and DDX3Y (purple). (D) Differential gene expression analysis of control versus DDX3Y knockdown reveals only six genes, including DDX3Y, that change significantly.
We confirmed these results at the protein level using a mass spectrometry framework that enables sensitive protein quantification by multiplexing peptides and samples (Derks et al. 2022). To measure the summed expression of DDX3X and DDX3Y protein, we quantified peptides shared by DDX3X and DDX3Y (Fig. 4B; Supplemental Table S10).
Given these results, we predicted that the incomplete compensation of summed DDX3X + DDX3Y protein levels seen with the DDX3X KD would result in transcriptome-wide changes, whereas such changes would not occur with the DDX3Y KD. Indeed, the DDX3X KD significantly altered the expression of 379 genes (Fig. 4C; Supplemental Table S11). In contrast, the DDX3Y KD significantly altered the expression of only six genes genome-wide, indicating nearly complete compensation through elevated DDX3X expression (Fig. 4D; Supplemental Table S12). We asked if the far-reaching consequences of DDX3X knockdown were because of the limited compensatory upregulation of the lower expressed DDX3Y or because DDX3Y is nonfunctional. The effects of DDX3Y knockdown are positively correlated with those of DDX3X knockdown but do not reach significance, suggesting that DDX3Y and DDX3X share a common function but that DDX3Y’s lower share of combined DDX3X/Y expression can more readily be replaced by upregulation of DDX3X (Supplemental Fig. S4).
We then asked whether negative cross-regulation of DDX3X and DDX3Y dampens differences in genome-wide gene expression that might otherwise be observed in individuals with sex chromosome aneuploidies. We found no significant overlap between (1) the set of genes differentially expressed in our DDX3X KD and (2) the set of genes transcriptionally responsive to increasing numbers of Chr X in the aneuploidy data set (Supplemental Fig. S5A; San Roman et al. 2024). We hypothesize that, unlike ZFX, which drives a large portion of the genome-wide response to Chr X copy number (San Roman et al. 2024), DDX3X expression that is elevated upon addition of Xi does not drive significant gene expression changes in the aneuploid lines. An alternative hypothesis, which is not mutually exclusive with DDX3X auto-regulation, is that DDX3X is negatively modulated by other genes expressed from Xi. Regardless, the increase in summed DDX3X and DDX3Y transcript levels per additional Chr X or Y is more modest than that of similarly constrained X–Y pairs (Supplemental Fig. S5B–D), consistent with the concept that DDX3X and DDX3Y are not prominent drivers of gene expression differences associated with sex chromosome aneuploidy.
DDX3X is negatively auto-regulated in 46,XX cells
We hypothesized that negative cross-regulation of the DDX3X–DDX3Y gene pair evolved from an earlier system of negative auto-regulation in the autosomal ancestor of this X–Y pair. Indeed, DED1, the yeast ortholog of DDX3X, appears to be negatively auto-regulated (Silvia Marina 2015). If negative cross-regulation in human XY cells evolved from negative auto-regulation, we might expect to observe negative auto-regulation of DDX3X in human 46,XX cells. We set out to test for this and, if present, to ask whether it might be unique among the 17 human NPX genes with NPY homologs. For each X–Y pair gene for which informative SNPs could be identified, we obtained its allelic ratio (AR), the ratio of Xi- and Xa-derived transcripts (San Roman et al. 2023). For each gene, we then compared its AR value to its ΔEX value, the increment of change in a gene's expression per additional X, relative to Xa (San Roman et al. 2023). If an X-linked gene's expression from Xi and Xa are independent and additive, then the gene's AR should approximate its ΔEX. We found this to be true for other NPX genes with NPY homologs. In contrast, whereas DDX3X has an AR of 0.55 in LCLs and 0.42 in fibroblasts, it has a significantly lower ΔEX of 0.26 in LCLs and 0.16 in fibroblasts (Fig. 5A; Supplemental Table S13). In other words, although Xi contributes 55% or 42% as many DDX3X transcripts as Xa does, DDX3X transcript levels increase by only 26% or 16% with each additional Xi. In the context of our other findings, this strongly suggests that DDX3X is negatively auto-regulated in the absence of DDX3Y.
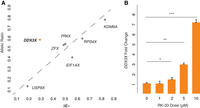
DDX3X is negatively auto-regulated in 46,XX cells. (A) DDX3X’s allelic ratio (AR) is significantly higher than its ΔEX value in LCLs, setting it apart from all other Xi/Xa/Y-expressed X–Y pair genes, whose AR values approximate their ΔEX values. Statistical significance determined via one sample t-test: P = 0.02. (B) DDX3X transcript levels (by qPCR) in 46,XX fibroblasts are significantly elevated in a dose-responsive manner upon treatment with DDX3 helicase inhibitor RK-33. Statistical significance determined by one-sided t-test on delta Ct values. Error bars indicate the standard deviation of three technical replicates. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
We also hypothesized that chemical inhibition of DDX3X protein activity could lead to increased DDX3X transcript levels. To test this, we treated 46,XX fibroblasts with RK-33, an inhibitor designed to occupy the DDX3X ATP-binding cleft and disrupt helicase function (Bol et al. 2015). DDX3X transcript levels were significantly elevated, in a dose-dependent manner, in cells treated with RK-33, consistent with negative auto-regulation of DDX3X in 46,XX cells (Fig. 5B; Supplemental Table S14). Increasing duration of RK-33 treatment also increased DDX3X transcript levels in a time-dependent manner (Supplemental Fig. S6; Supplemental Table S15). Although it is possible that RK-33 may affect the function of other helicases and alter DDX3X levels by a non-auto-regulatory pathway, these results are consistent with the allele-specific analysis of transcription in 46,XX cells and provide additional evidence for negative auto-regulation of DDX3X.
In theory, our observations concerning auto- and cross-regulation could be explained by independent, parallel evolution of negative cross-regulation of DDX3Y by DDX3X and of DDX3X by DDX3Y, but such convergence seems unlikely, especially given the absence of crossing-over as an evolutionary enabler in the case of DDX3Y. A simpler hypothesis is that reciprocal cross-regulation of DDX3X and DDX3Y derives directly from a post-transcriptional mechanism that negatively auto-regulated the ancestral (autosomal) DDX3X gene. We suggest that this regulatory scheme governed the DDX3X ortholog in our amniote ancestors before the autosome carrying it became part of today's (eutherian) mammalian sex chromosomes.
DDX3X response is mediated by mRNA stability
DDX3X encodes an RNA-binding protein known to bind its own transcripts (Van Nostrand et al. 2020). Yeast DED1 auto-regulation is dependent on its 3′ UTR (Silvia Marina 2015), indicating that mRNA stability is being modulated. We reasoned that the negative cross-regulation we observed between human DDX3X and DDX3Y may also involve mRNA stability. If DDX3Y destabilizes DDX3X transcripts, we would expect the half-life of DDX3X transcripts to decrease in response to increasing DDX3Y dosage. We tested this prediction by labeling nascent mRNAs in 46,XY and 49,XYYYY LCLs with 5-EU and sequencing the resultant mRNA populations at discrete intervals to quantify the half-life (Fig. 6A). We calculated the ratio of nascent mRNA/total mRNA normalized to steady-state levels across time points, and we observed a striking difference in DDX3X mRNA half-life between the two conditions. DDX3X mRNAs have a half-life of 0.5h in 49,XYYYY cells compared with 1.3 h in XY cells (Fig. 6B; Supplemental Table S16), implying that high DDX3Y levels lead to a marked destabilization of DDX3X mRNAs, reducing steady-state levels of DDX3X transcripts. We replicated this finding, including the half-life values, in an independent metabolic labeling trial with double the time resolution (Supplemental Fig. S7; Supplemental Table S17).

DDX3X mRNA stability is regulated. (A) Schematic of experiment to determine half-lives of mRNAs. 46,XY and 49,XYYYY LCLs were incubated with 5-ethyl uridine (5-EU) to obtain nascent mRNAs. (B) DDX3X has an mRNA half-life of 0.5 h in 49,XYYYY versus 1.3 h in 46,XY LCLs.
These results support a model where the ancestral (autosomal) DDX3 gene in amniotes destabilized its own transcripts to negatively auto-regulate its expression, foreshadowing the ability of mammalian DDX3X and DDX3Y to destabilize their own and each other's transcripts.
Discussion
As described here, DDX3X and DDX3Y are negatively, post-transcriptionally cross-regulated (Fig. 3), and DDX3X is negatively, post-transcriptionally auto-regulated (Fig. 5), such that perturbations to one allele (of DDX3X or DDX3Y) can be buffered by upregulation of the other allele. This is the first observation of an X–Y gene pair having cross-regulatory capabilities, and it helps explain certain human phenotypes associated with loss-of-function mutations as well as diverse observations in the literature.
In 46,XY males, rare constitutional (germline) mutations in DDX3X cause a neurodevelopmental disorder (“DDX3X syndrome”) (Kellaris et al. 2018; Nicola et al. 2019). In contrast, constitutional mutations of DDX3Y in 46,XY males cause a subtle phenotype. De novo deletions of the entire DDX3Y gene (so-called AZFa deletions) cause spermatogenic failure and, thereby, infertility but otherwise have no reported impact on somatic development, function, or health (Fig. 3; Sun et al. 2000). In vitro, in LCLs, we find that elevated DDX3X transcript levels compensate for the absence of DDX3Y (Fig. 4A,B). If the same holds in the brain (and other somatic tissues) of AZFa-deleted males, it would explain why males with germline DDX3Y deletions display no neurodevelopmental consequences. In testicular germ cells, in which DDX3Y predominates (Ramathal et al. 2015), DDX3X cannot compensate for the loss of DDX3Y, making DDX3Y essential for male fertility and fitness. We propose that the ratio of DDX3X to DDX3Y in various tissues determines the potential for compensatory regulation and the impact of each homolog's loss of function. DDX3Y may be required for the development of tissues in which DDX3Y expression predominates (e.g., pituitary or cerebellum) (Godfrey et al. 2020). In euploid individuals, tissues with skewed ratios of DDX3X:DDX3Y could be a prominent source of sex differences.
Our data indicate that negative cross-regulation of DDX3X and DDX3Y operates broadly and potentially universally across human somatic cell types. We observe this phenomenon in multiple human cell types: in LCLs, in primary fibroblasts, and in cancer cell lines originating from five different tissues (Fig. 3; Supplemental Table S2; Supplemental Fig. S4). The generality of these findings allows us to reinterpret and better comprehend diverse observations regarding DDX3X and DDX3Y reported in the literature. Negative post-transcriptional regulation offers a unifying explanation for the following observations:
-
In DDX3X-mutant lymphomas in human males, Gong et al. (2021) reported that DDX3Y transcript levels were elevated compared to wild-type lymphocytes (B cells). Gong et al. (2021) speculated that DDX3Y upregulation in these DDX3X-mutant lymphomas reflected an aberrant, oncogenically adaptive gene expression program. A simpler explanation is provided by negative cross-regulation that operates universally in human somatic cell types, including cancers of somatic origin.
-
In the brains of male mice bearing various conditional Ddx3x knockouts designed to model either human DDX3X syndrome (Hoye et al. 2022) or medulloblastoma (Patmore et al. 2020), investigators noted that Ddx3y transcript levels were elevated compared with wild-type. Viewed in light of our current findings, these observations suggest that negative cross-regulation of DDX3X and DDX3Y occurs not only in humans but also in mice. DDX3X and DDX3Y loss of function may also present with different phenotypes in mice and other species, depending on the ratios of each homolog in that species.
These post-transcriptional regulatory connections between DDX3X and DDX3Y are unique, to our knowledge, among human X–Y gene pairs. How could such a system evolve? We considered this question in the context of the evolution of Chr X and Chr Y. We reasoned that tight delimiting of DDX3 gene expression likely predated the divergence of the homologous genes DDX3X and DDX3Y on the (eutherian) mammalian sex chromosomes, as this would most economically explain the presence of both auto- and cross-regulation of the human genes. Yeast DED1 auto-regulation (Silvia Marina 2015) suggests that this regulation has been preserved in both the eutherian and yeast lineages during the 1.3 billion years since their divergence (Kumar et al. 2022). The DED1 3′ UTR is necessary and sufficient for auto-regulation, suggesting that a similar mechanism potentially operates on human DDX3X and DDX3Y. We infer that DDX3 was already highly dosage sensitive when, as a single-copy gene, it resided on an amniote autosome that later gave rise to much of the sex chromosomes of eutherian mammals. DDX3X and DDX3Y evidently retained this high dosage sensitivity and the associated negative regulatory scheme that had governed their common autosomal ancestor.
Combined with other recent discoveries, our present findings illuminate the breadth and diversity of gene regulatory mechanisms and networks that were selectively preserved as Chr X and Chr Y evolved from ordinary autosomes during the past 200 million years (Fig. 7). For example, our recent studies of the genome-wide consequences of human sex chromosome aneuploidy showed that the X- and Y-linked transcriptional activators ZFX and ZFY modulate expression of large and similar sets of autosomal genes (San Roman et al. 2024). Given the scale of these gene regulatory networks, their similarity is unlikely to be the result of convergent evolution. A more economical explanation is evolutionary preservation of preexisting gene regulatory networks centered on the single autosomal forebear of the eutherian ZFX and ZFY genes. Another example involves our recent observation that expression of the Y-linked translation initiation factor EIF1AY is enhanced (relative to its X-linked homolog, EIF1AX) in the human heart (Godfrey et al. 2020). This was explained through our recent discovery of (1) a miR-1 (cardiac microRNA) binding site in the 3′ UTR of the ancestral (autosomal) EIF1A gene, (2) preservation of that ancestral binding site in the 3′ UTR of EIF1AX, and (3) loss of the ancestral binding site in the 3′ UTR of EIF1AY, resulting in its enhanced expression in the human heart. In sum, the cases of DDX3X/Y, ZFX/Y, and EIF1AX/Y illustrate the diversity and reach of ancestral (autosomal) gene regulatory mechanisms preserved or, in some cases, lost during the 200-million-year evolution of the eutherian sex chromosomes from ordinary autosomes.
Not only protein-coding sequences but also gene regulatory mechanisms were preserved during the evolution of sex chromosomes from ordinary autosomes. The auto- and cross-regulation of DDX3X and DDX3Y reported here likely originated from the auto-regulation of ancestral (autosomal) DDX3X. Together with published studies of two other X–Y gene pairs—EIF1AX-EIF1AY and ZFX-ZFY (Godfrey et al. 2020; San Roman et al. 2024)—our findings suggest that an array of gene-specific regulatory schemes operative on the ancestral autosomes persist today on human Chr X and Chr Y.
Methods
Analysis of total branch length and survival fraction
For each gene, total branch length and survival fraction values in therian species were obtained from Bellott and Page (2021). To obtain a gene's total branch length, all branch lengths in the most parsimonious tree connecting all species in which the gene is present are summed from the last common ancestor. The survival fraction is the observed total branch length divided by the maximum possible branch length. Survival fractions range from zero (lost in all lineages) to one (retained in every lineage).
Analysis of constraint metrics
We downloaded LOEUF (loss-of-function observed/expected upper fraction) scores from gnomAD (v2.1.1.lof_metris.by_gene.txt; https://gnomad.broadinstitute.org/) and only used scores with a minimum of 10 expected LoF variants. For sensitivity to an increase in gene dosage, we used the per-gene average probability of conserved miRNA targeting scores (PCT) (Friedman et al. 2009). We computed a percentile rank score for each metric, from most constrained to least constrained (San Roman et al. 2023). Pythagorean sum of ranks was used to calculate a combined metric for dosage sensitivity (Supplemental Table S1).
Calculation of expression breadth
Human expression breadth was calculated from GTEx v8 using male samples. For each gene, expression breadth was calculated using TPM values as follows: sum of expression across tissues/(maximum expression in a tissue × number of tissues). For each X–Y gene pair, expression breath values for the X-homolog and Y-homolog were averaged to generate a mean score. Chicken expression breadth values were obtained from Bellott et al. (2010) using data from Merkin et al. (2012). Pythagorean sum of breadths was used to calculate a combined metric for dosage sensitivity (Supplemental Table S2).
Aneuploidy data
RNA sequencing data from cultured cells of individuals with sex chromosome aneuploidy (San Roman et al. 2023) were downloaded from https://doi.org/10.1016/j.xgen.2023.100259.
Cell culture
All LCLs were cultured in complete RPMI at 37°C. Fibroblasts were cultured in high-glucose DMEM (GIBCO), 20% FBS, L-glutamine (MP Biomedicals), MEM nonessential amino acids (GIBCO), and 100 IU/mL penicillin/streptomycin (Lonza).
CRISPRi
Three independent, unrelated 46,XY fibroblast cultures stably expressing a nuclease-dead Cas9 fused with a repressive KRAB domain (dCas9-KRAB) were obtained from Adrianna San Roman. gRNAs for control (intergenic), DDX3X, and DDX3Y were chosen from the human CRISPRi v2 library (Horlbeck et al. 2016) and cloned into the sgOpti lentiviral expression vector. Viral particles were generated and frozen as described in San Roman et al. (2023). Guide sequences were as follows:
-
Control 1: GACATATAAGAGGTTCCCCG
-
Control 2: AACGGCGGATTGACCGTAAT
-
DDX3X 1: GTCCCGTGAGAGGGCCTTCG
-
DDX3X 2: GCCCGGGACGAGCACAATGG
-
DDX3Y 1: GTTCGGTCTCACACCTACAG
-
DDX3Y 2: GAGTACTGGGCCTCACGCAA
Control and DDX3X- or DDX3Y-targeting gRNAs were transduced into the stably-expressing dCas9-KRAB fibroblasts, and cells were selected using 2 μg/mL puromycin (Sigma-Aldrich) beginning 24 h post infection. Cells were washed once with PBS and collected 72 h post infection. RNA was extracted with the RNeasy mini kit (Qiagen). RNA sequencing libraries were prepared using the KAPA mRNA HyperPrep kit V2 (Roche). Paired-end 100 × 100 bp sequencing was performed on a NovaSeq 6000 (Illumina). Reads were pseudoaligned with kallisto and imported into R using tximport. Differential gene expression analysis was performed using DESeq2 (Love et al. 2014). RNA sequencing data from ZFX and ZFY knock-down experiments (San Roman et al. 2024) were downloaded from the NCBI database of Genotypes and Phenotypes (dbGAP; https://www.ncbi.nlm.nih.gov/gap/) under accession number phs002481.v2.p1.
Treatment with RK-33
46,XX fibroblast cultures were treated with 0, 1, 2, 5, or 10 μM RK-33 in DMSO for 24 h. For the time course, they were treated with 2 μM RK-33 for 0, 1, 2, 4, or 24 h.
qPCR
Cells were washed once with PBS and collected 72 h post treatment. RNA was extracted with the RNeasy mini kit (Qiagen) and cDNAs prepared with SuperScript Vilo master mix (Thermo Fisher Scientific). DDX3X levels were quantified by qPCR using fast SYBR Green master mix (Thermo Fisher Scientific). Primers for DDX3X and reference gene ACTB were as follows:
-
DDX3X F: GTGGAAGTGGATCAAGGGGA
-
DDX3X R: TGATTTGTCACACCAGCGAC
-
ACTB F: CACCAACTGGGACGACAT
-
ACTB R: ACAGCCTGGATAGCAACG
Analysis of cancer cell line expression data set
Expression and mutation data for cancer cell lines were downloaded from the DepMap 22Q2 release (https://depmap.org/portal/download/all/). Analysis was restricted to 46,XY cells by applying a log2TPM filter of >0.2 DDX3Y, >0.2 RPS4Y1, <2 XIST.
Sample preparation for mass spectrometry
Samples were prepared for proteomic analysis by minimal proteomic sample preparation (mPOP) as described by Specht et al. (2018). Briefly, cells were resuspended in MS-grade water and frozen. They were then heated for 10 min at 90°C to lyse cells. Proteins were reduced and treated with trypsin gold (Promega). The peptide abundance of each sample was measured, and each sample was labeled with nonisobaric mass tags, mTRAQ Δ0, Δ4, or Δ8 (SciEx: 4440015, 4427698, 4427700) following the manufacturer's instructions. Reactions were quenched and pooled as a three-plex with relative mass offsets of Δ0, Δ4, and Δ8.
Mass spectrometry data acquisition
mTRAQ-labeled peptide sets were separated by reversed-phase UHPLC in 1 µL injections by a Dionex UltiMate 3000 using a 25 cm × 75 µm IonOpticks Aurora series UHPLC column (AUR2-25075C18A). Buffer A was 0.1% formic acid in MS-grade water. Buffer B was 80% acetonitrile (ACN) with 0.1% formic acid, mixed in MS-grade water. The gradient was as follows: 4% buffer B (minutes 0–11.5), 4%–7% buffer B (minutes 11.5–12), 7%–32% buffer B (minutes 12–75), 32%–95% buffer B (minutes 75–77), 95% buffer B (minutes 77–80), 95%–4% buffer B (minutes 80–80.1), and 4% buffer B until minute 95. The flowrate was 200 nL/min throughout.
Mass spectrometry data were acquired using a DIA method which utilizes frequent MS1-scans for quantitation, as previously described (Derks et al. 2022). The duty cycle consisted of five subcycles of (1 MS1 full scan × 5 MS2 windows) for a total of 25 MS2 windows to span the full m/z scan range (380–1370 m/z). MS1 and MS2 scans were performed at 140k and 35k resolving power, respectively.
Mass spectrometry data analysis
Raw plexDIA data were processed with DIA-NN (version 1.8.1 beta 16) (Demichev et al. 2019) using the following settings and additional commands: {‐‐window 1}, {‐‐mass-acc 10.0}, {‐‐mass-acc-ms1 5}, {‐‐reanalyse}, {‐‐rt-profiling}, {‐‐peak-height}, {‐‐fixed-mod mTRAQ, 140.0949630177, nK}, {‐‐channels mTRAQ,0,nK,0:0; mTRAQ,4,nK,4.0070994:4.0070994; mTRAQ,8,nK,8.0141988132:8.0141988132}, {‐‐peak-translation}, {‐‐original-mods}, {‐‐report-lib-info}, {‐‐ms1-isotope-quant}, {‐‐mass-acc-quant 5.0}.
The resulting data were filtered at 1% FDR for precursors and protein-groups (DDX3X;DDX3Y). Precursors were further filtered for Translated.Q.Value < 0.01. MaxLFQ (Cox et al. 2014) was used to perform protein group–level quantification for all samples. Each protein group was normalized to the mean value in each LC-MS run, as each LC-MS run contained three technical replicates from each of the three conditions (control, DDX3X knockdown, and DDX3Y knockdown). Each sample was then normalized to its own respective median protein group value to account for differences in absolute protein abundances between samples. Finally, each protein group was normalized to the mean value of the protein group across all samples, for each cell line. For each cell line, batch correction was performed using Combat (Leek et al. 2012) with missing data imputed with a kNN algorithm (k = 3) to correct biases produced by using different mass-tag offsets (e.g. Δ0, Δ4, and Δ8).
5-EU labeling and cell collection
LCLs were thawed and allowed to grow in T175 flasks. Cells were split with fresh LCL media and 5EU (Jena Bioscience) was added to a final concentration of 400 µM. Cells were collected 0, 0.5, 1, 1.5, 2, 3.5, and 7 h later; washed with PBS; and pelleted prior to addition of TRIzol (Thermo Fisher Scientific) reagent. Cells were snap-frozen at −80°C. RNA was precipitated with isopropanol, and 1 ng of 5-EU EGFP positive control was added.
Biotinylation and pulldown
Biotinylation and pulldown were performed as previously described (Kingston and Bartel 2019). Briefly, biotin was attached to metabolically labeled RNAs in a 10 µL reaction protected from light. The reaction was quenched
and RNA-precipitated. RNA was then incubated with blocked and prewashed streptavidin bead slurry. Beads were washed once more,
and RNA was eluted with tris(2-carboxyethyl) phosphine (TCEP) and water. RNA was precipitated, and libraries were then prepared
using the Smart-seq v4 ultralow input RNA kit and sequenced on a NovaSeq 6000. Input RNAs were also sequenced to measure total
RNA. TPMs were normalized to 5-EU positive EGFP spike-in. Normalized fraction of nascent/total DDX3X mRNA was fit to the equation 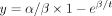 to obtain β (half-life).
to obtain β (half-life).
Statistical methods
Various statistical tests were used to calculate P-values as indicated in the Methods section, figure legends, or text, where appropriate. Results were considered statistically significant when P < 0.05 or FDR < 0.05 when multiple hypothesis correction was applied, unless stated otherwise. All statistics were calculated using R software (R Core Team 2013), version 4.2.1 or Prism version 9.4.1 unless stated otherwise.
Data access
All cell lines used in this study are listed in Supplemental Table S18. Raw reads for sequencing data generated in this study have been submitted to the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) under accession number phs002481.v5.p1. Original code for the analyses in this paper is available as Supplemental Code and at GitHub (https://github.com/shruthi3195/DDX3X_SR_2023).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the Page Laboratory, especially Jennifer Hughes and Adrianna San Roman, for comments on the manuscript and Jorge Adarme and Susan Tocio for laboratory support. We thank the Whitehead Genome Technology Core facility for library preparation and sequencing and Caitlin Rausch for illustration. This work was supported by grants from Simons Foundation Autism Research Initiative Collaboration Award (D.C.P.), Howard Hughes Medical Institute (D.C.P.), and the National Institutes of Health (NIH) (R35GM148218 to N.S.), as well as philanthropic support from The Brit Jepson d'Arbeloff Center on Women's Health, Arthur W. and Carol Tobin Brill, Matthew Brill, Charles Ellis, The Brett Barakett Foundation, Howard P. Colhoun Family Foundation, Seedlings Foundation, and Knobloch Family Foundation. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Author contributions: S.R., N.S., and D.C.P. designed the experiments. S.R. and J.D. performed the experiments. S.R., J.D., and D.W.B. performed computational analyses. S.R. and D.C.P. wrote the manuscript with edits from N.S., D.W.B., and J.D.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279707.124.
-
Freely available online through the Genome Research Open Access option.
- Received July 8, 2024.
- Accepted November 26, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution 4.0 International), as described at http://creativecommons.org/licenses/by/4.0/.








