
Binding profiles for 961 Drosophila and C. elegans transcription factors reveal tissue-specific regulatory relationships
- Michelle Kudron1,
- Louis Gevirtzman2,
- Alec Victorsen3,
- Bridget C. Lear4,
- Jiahao Gao5,6,
- Jinrui Xu7,8,
- Swapna Samanta1,
- Emily Frink2,
- Adri Tran-Pearson2,
- Chau Huynh2,
- Dionne Vafeados2,
- Ann Hammonds9,
- William Fisher9,
- Martha Wall10,11,
- Greg Wesseling4,
- Vanessa Hernandez4,
- Zhichun Lin4,
- Mary Kasparian4,
- Kevin White12,
- Ravi Allada4,
- Mark Gerstein5,6,13,
- LaDeana Hillier2,
- Susan E. Celniker9,
- Valerie Reinke1 and
- Robert H. Waterston2
- 1Department of Genetics, Yale University, New Haven, Connecticut 06520, USA;
- 2Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA;
- 3Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota 55455, USA;
- 4Department of Neurobiology, Northwestern University, Evanston, Illinois 60208, USA;
- 5Program in Computational Biology and Bioinformatics, Yale University, New Haven, Connecticut 06520, USA;
- 6Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06520, USA;
- 7Department of Biology, Howard University, Washington, District of Columbia 20059, USA;
- 8Center for Applied Data Science and Analytics, Howard University, Washington, District of Columbia 20059, USA;
- 9Division of Biological Systems and Engineering, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA;
- 10Institute for Genomics and Systems Biology, University of Chicago, Chicago, Illinois 60637, USA;
- 11Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA;
- 12Department of Biochemistry and Precision Medicine Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, 117597 Singapore;
- 13Department of Statistics and Data Science, Yale University, New Haven, Connecticut 06520, USA
Abstract
A catalog of transcription factor (TF) binding sites in the genome is critical for deciphering regulatory relationships. Here, we present the culmination of the efforts of the modENCODE (model organism Encyclopedia of DNA Elements) and modERN (model organism Encyclopedia of Regulatory Networks) consortia to systematically assay TF binding events in vivo in two major model organisms, Drosophila melanogaster (fly) and Caenorhabditis elegans (worm). These data sets comprise 605 TFs identifying 3.6 M sites in the fly and 356 TFs identifying 0.9 M sites in the worm, and represent the majority of the regulatory space in each genome. We demonstrate that TFs associate with chromatin in clusters termed “metapeaks,” that larger metapeaks have characteristics of high-occupancy target (HOT) regions, and that the importance of consensus sequence motifs bound by TFs depends on metapeak size and complexity. Combining ChIP-seq data with single-cell RNA-seq data in a machine-learning model identifies TFs with a prominent role in promoting target gene expression in specific cell types, even differentiating between parent–daughter cells during embryogenesis. These data are a rich resource for the community that should fuel and guide future investigations into TF function. To facilitate data accessibility and utility, all strains expressing green fluorescent protein (GFP)-tagged TFs are available at the stock centers for each organism. The chromatin immunoprecipitation sequencing data are available through the ENCODE Data Coordinating Center, GEO, and through a direct interface that provides rapid access to processed data sets and summary analyses, as well as widgets to probe the cell-type-specific TF–target relationships.
Precise deployment of gene expression across space and time drives the successful development and sustained health of all organisms. To understand the underlying causes of developmental disorders, polygenic diseases, and cancer, we must decipher the code that directs gene expression in diverse cell types and environments. Transcription factors (TFs) bind specific genomic sequences while interacting with regulatory proteins, RNA polymerase II, and the chromatin environment to direct gene expression programs. The set of expressed TFs and their activity in a cell determines which genes are expressed and to what level, thereby dictating cellular fate and function. Despite the diverse body plans, lifespans, and habitats of different species, the fundamental mechanisms by which TFs control gene regulation are highly conserved. Moreover, mutations in TFs are estimated to contribute to almost one-third of human developmental disorders (Vaquerizas et al. 2009), highlighting their central role in fundamental cellular processes.
Building a catalog of regulatory sequences—genomic sites bound by each TF—is, therefore, a critical step toward understanding gene regulation. Much of the work defining TF–chromatin interactions has been led by multilaboratory consortia including ENCODE (Encyclopedia of DNA Elements) for humans and mice (The ENCODE Project Consortium et al. 2020) and modENCODE (model organism Encyclopedia of DNA Elements) and modERN (model organism Encyclopedia of Regulatory Networks) for Caenorhabditis elegans and Drosophila (Celniker et al. 2009; Gerstein et al. 2010; The modENCODE Consortium et al. 2010; Boyle et al. 2014; Kudron et al. 2018). These efforts have resulted in the systematic collection of millions of genome-scale measurements of TF binding, RNA transcripts, and chromatin accessibility and organization in a variety of organisms.
The nematode C. elegans and the fruit fly Drosophila melanogaster were selected for the modENCODE Project in 2007 because of their experimental strengths and prominence as model organisms as well as for their compact, sequenced genomes, which simplifies the identification of TF binding sites and candidate target genes. Critically, they also permit the mapping of TF binding in the living organism, which is not straightforward for human TFs. Many TFs in these two species are orthologous to human TFs. To a significant extent, they encompass the same broad classes of DNA-binding domains, exhibit similar functions, respond to the same signaling inputs, and bind to similar consensus sequence motifs as human TFs. Over several decades, investigations of worm and fly TFs have illuminated their function during development and provided important insights into the function of human disease genes and human biology.
Working first as members of the worm and fly modENCODE consortia (Gerstein et al. 2010; The modENCODE Consortium et al. 2010), and subsequently together as the modERN consortium (Kudron et al. 2018), we have generated hundreds of strains with tagged TFs, representing most of the TFs in each species. With these strains, we have characterized TF expression patterns and performed ChIP-seq analysis. These data sets define hundreds of thousands of binding sites throughout development for each species. Now, at the conclusion of this comprehensive effort, we report on the cumulative resources available to the community. We also analyze the global data to examine important TF features and relationships, including the correlation of cobinding by TFs at target genes, the frequency of high TF occupancy sites in the genome, and the impact of underlying sequence motifs and chromatin accessibility on TF binding. Finally, we begin to decipher tissue-specific codes of gene regulation for individual and groups of TFs using available single-cell gene expression data in both species. Studying key conserved factors in worm and fly greatly will enhance analysis, interpretation, and the broader relevance of data gathered in ENCODE and other consortia-scale projects. Together the results demonstrate the value and richness of these available data, which will be crucial to further developing detailed network maps in model organisms. In turn, these maps will accelerate the understanding of how the cognate genes function in analogous networks in humans.
Results
Strain generation and resource
The initial step in obtaining ChIP-seq data in our pipeline was to generate strains with green fluorescent protein (GFP)-tagged TFs. For this large-scale project, it was not feasible to generate TF-specific antibodies, and early experiments showed that representative GFP-tagged TFs rescued null mutants (Venken et al. 2009; Gerstein et al. 2010).
To generate strains in the two species, we exploited the specific features and resources of each organism. For flies, available bacterial artificial chromosome (BAC) clones that contained the TF and sufficient flanking sequence from two different libraries (Venken et al. 2009) were tagged using recombineering. Clones were introduced into flies using the ϕC31 integrase system and attP docking sites on either the second or third chromosome. PCR was used to confirm the location and presence of the targeted TF. As reported previously, these constructs almost always can rescue their corresponding mutant phenotypes demonstrating proper expression of the TF (Venken et al. 2009). In the latter stages of the project, 151 lines were obtained commercially from Genetivision (https://www.genetivision.com/). Altogether strains were obtained for 616 TFs and 56 chromatin or nucleic acid-binding proteins (Table 1; Supplemental File S1). (Of these TF strains, 189 were generated after Kudron et al. [2018]). The tagged TF strains capture all but 43 of the estimated 659 TFs (93.5%) in our curated list. The number of estimated TFs in the fly genome has fallen from 708 in 2013 (Hammonds et al. 2013), for a variety of reasons, including revised gene models and reassignment to other nucleic acid-associated proteins. Strains were not generated for TFs with complex genomic organization where there was no BAC containing the predicted TF transcriptional unit or when no MIMIC line (Venken et al. 2011) was available to produce a tagged TF.
Summary of strains, ChIP-seq data sets, and stages for worm and fly TFs
For worms, we primarily utilized the recombineered fosmid resource TransgeneOme to generate carboxy-terminal GFP-tagged TFs (Sarov et al. 2012). These tagged fosmids, generally selected so that the gene of interest was in the middle of the fosmid and flanked by other genes on both sides, were introduced into the worm by microparticle bombardment, which results in low copy (∼1–10) integration events at random sites in the genome. With only rare exceptions, the tagged strains have been viable and without readily detectable phenotypes. More recently, we switched to introducing GFP directly into the endogenous gene via CRISPR for TFs that did not yield strains by bombardment, that lacked a fosmid construct, or did not pass ChIP-seq metrics after recombineering (Paix et al. 2017; Dokshin et al. 2018; Ghanta and Mello 2020). The CRISPR approach has the advantage of being a single copy and retaining the native regulatory context of the TF gene. To test whether this approach impacted TF function, we compared expression pattern and ChIP-seq results for fosmid-based and CRISPR-generated strains directly for five different TF genes and found no consistent differences (Supplemental Table S1). In total, strains with GFP tags were generated for 514 TFs and 45 nucleic acid-associated proteins. (Of these TF strains, 111 were generated after Kudron et al. [2018]). TFs exhibited a wide range of expression patterns from ubiquitously expressed to highly tissue-specific in both soma and germline (Supplemental Fig. S1). The total number of predicted worm TFs has fallen from an estimated 934 genes early in the project (Reece-Hoyes et al. 2005) to 833 total TFs in our curated list, due to factors such as revised gene models and reassignment to other nucleic acid-associated proteins (Supplemental File S1). Of the 833 total TFs, we targeted 640, and of these, 80.3% were successful, including 81 by CRISPR. Tagging was not successful, using either fosmids or CRISPR, for the remaining 126 targeted TFs lacking strains.
These fly and worm strains are available through the Bloomington Drosophila Stock Center (BDSC) and the Caenorhabditis Genetics Center (CGC), respectively, and have been an extremely popular resource with the community. By 2023, the respective stock centers reported filling requests for 17,293 and 4334 of these strains, respectively.
ChIP-seq resource
The GFP-tagged strains of both species were used to perform whole animal ChIP-seq using a highly specific, validated anti-GFP antibody (Kudron et al. 2018). In each case, synchronized populations of either flies or worms were grown to the desired stage, harvested, and subjected to ChIP, using previously described methods (Gerstein et al. 2010; The modENCODE Consortium et al. 2010; see Methods). The bulk of fly experiments were performed on embryos because ChIP-seq data were more readily obtained at this stage and most TFs were well expressed at this stage, while for the worm, the experiments were distributed more broadly across the life cycle and based on maximal expression predicted by RNA expression profiles and observed peak GFP fluorescence (Table 1; Supplemental Fig. S1). We elected not to perform ChIP-seq on some tagged worm strains because, based on prior experience, the TF was expressed in so few cells and/or at such low levels that a successful experiment was unlikely, or the expression was predominantly cytoplasmic. For a few TFs in both species, ChIP-seq failed to meet quality criteria despite repeated attempts (see Supplemental File S1 for TFs that could not be assessed with ChIP-seq). In total, we successfully performed ChIP on 605 fly TFs (92% of total TFs) in 678 different experiments. Of these TF data sets, 326 (379 total data sets) were generated after Kudron et al. (2018). A total of 356 worm TFs (56% of targeted TFs) in 525 different experiments have ChIP-seq data (Table 1), of which 350 were included in later analyses. Of these TF data sets, 166 (174 total data sets) were generated after Kudron et al. (2018). In addition to the ChIP-seq data for TFs, we have also successfully generated ChIP-seq data for 45 additional DNA-associated proteins in the worm in a total of 65 experiments of which 15 (16 total data sets) were done or reannotated as non-TFs since Kudron et al. (2018), and 55 other DNA-associated proteins in 62 experiments in the fly of which five were generated since Kudron et al. (2018). These results are all available at the ENCODE DCC and at https://epic.gs.washington.edu/modERNresource/. The non-TF, DNA-associated proteins were excluded from the further analysis presented here.
ChIP-seq data were processed through a peak-calling pipeline using SPP (Methods and Supplemental Methods in Supplemental File S4; Kharchenko et al. 2008). Data sets were immediately evaluated using the self-consistency ratio and rescue ratio parameters as defined by the ENCODE Consortium (Landt et al. 2012). Passing data sets (see Supplemental File S4 for details) were submitted to the ENCODE Data Coordinating Center, until it stopped accepting submissions in October 2022. Later data sets are available at https://epic.gs.washington.edu/modERNresource/ and through the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/).
Because the SPP peak-calling pipeline evolved over the project lifetime, we reanalyzed all the data using a recent version of SPP and the DCC pipeline at the end of the ENCODE Project to obtain more directly comparable data sets (see Supplemental File S4). In these uniformly processed data sets, in the nuclear genome, we detected 3,628,005 peaks in the fly and 900,852 in the worm, with a median of 4416 peaks and 1130 peaks per TF experiment, respectively (Fig. 1A,B). The basis for this considerable difference in peaks per experiment in the two species is unclear. The rightward distributions of peaks per experiment are broadly similar. Perhaps the fly, which has no more TFs than the worm, may achieve a much more complicated development, cellular composition, and anatomy by more extensive use of each TF. However, technical considerations might play a role. For example, for fly we routinely collected three technical replicates, whereas for worm we collected only two. Validation experiments suggest the use of the extra replicate can increase the number of peaks by ∼20%. However, this amount only accounts for a small fraction of the observed difference.
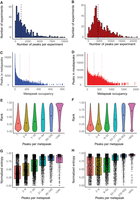
Peaks and metapeaks. (A,B) Number of peaks per experiment (A) for the worm and (B) for the fly. Vertical lines denote the median number of peaks in worm (1130) and fly (4416). (C,D) The total number of peaks in metapeaks declines rapidly with increasing metapeak occupancy (the number of peaks in an individual metapeak) in both worm (C) and fly (D). The vertical bars indicate the thresholds used to define HOT (left) and ultra-HOT (right) sites in each species (84 and 240 peaks, respectively, in the worm and 277 and 602 peaks in the fly). (E,F) In worm (E) and fly (F), the relative rank of the signal strength of peaks within metapeaks increases with metapeak occupancy. (G,H) Both worm (G) and fly (H) targets of high-occupancy metapeaks show a predominance of high entropy genes (more uniform expression), while targets with lower occupancy metapeaks show lower entropy, indicating more cell-type-specific expression. The entropy of fly genes is shifted higher in the fly than in the worm.
Most peaks in both worm and fly individually span fewer than 1000 bases, with a median of 436 bases in the fly and 376 bases in the worm (Supplemental Fig. S2A,B). Inspection of the few peaks more than 1000 bases (4518 in fly [0.12%] and 250 in worm [0.03%]), suggests that these often represent multiple overlapping peaks that SPP failed to separate.
Peaks and metapeaks
As noted previously (Gerstein et al. 2010; Boyle et al. 2014), inspection of the mapped binding sites for each species revealed substantial clustering of peaks from multiple TFs in distinct regions around genes, with vastly differing numbers of peaks in each apparent cluster. To measure peak clustering in a principled manner, we treated each peak call as a sequence read and used MACS2 to call clusters of peaks, which for purposes here we have termed “metapeaks” (see Supplemental File S4; Zhang et al. 2008). This process produced 77,969 metapeaks in the fly, of which 19,136 were stand-alone peaks, or “singletons” (for simplicity, we included singletons as metapeaks in subsequent analyses). For the worm, there were 52,874 metapeaks, including 16,232 singletons. The nucleotide span of these metapeaks increased only modestly with increasing numbers of individual peaks per metapeak (metapeak occupancy) with only 50 and 46 metapeaks spanning more than 3 kb in worm and 4 kb in fly, respectively (Supplemental Fig. S2C,D). The DNA spanned by these sites amounted to 57,515,618 total bases in the fly (40% of the genome) and 27,564,528 bases in the worm (27% of the genome), even though the fly and worm have a very similar number of bases in coding exons (33,650,573 for fly and 29,511,901 for worm). The greater coverage of the genome by the fly metapeaks could reflect the greater fraction of TFs assayed, the greater number of peaks per experiment, and/or greater complexity in the regulatory program. A single metapeak generally spans <3000 bases in the fly and 2000 bases in the worm, even for metapeaks with high TF occupancy (Supplemental Fig. S2C,D), so that despite covering a large fraction of the genome in total, they represent quite discrete regions.
We investigated the overlap of the sites detected by ChIP-seq with recently obtained ATAC-seq data from both worm (Jänes et al. 2018) and fly (Cusanovich et al. 2018; Calderon et al. 2022) and found good agreement, in that the majority of identified ATAC-seq peaks overlap with metapeaks in each species (see Supplemental File S4 for exact numbers). We also compared the metapeaks with worm yeast-one-hybrid data (Fuxman Bass et al. 2016), but found low agreement between the different data sets as previously reported, suggesting the methods may well be assaying different aspects of gene regulation (see Supplemental File S4).
The number of individual peaks (occupancy) within a metapeak declines rapidly, especially for the worm (Fig. 1C,D). Nonetheless, 1408 metapeaks in fly contain more than 500 individual peaks and 491 metapeaks contain more than 250 peaks in the worm, suggesting regions of extremely dense TF binding. High occupied target (HOT) sites were recognized in early modENCODE papers (Moorman et al. 2006; Gerstein et al. 2010) and have been defined subsequently in various ways (Chen et al. 2014; Wreczycka et al. 2019). After exploring several approaches and comparing the HOT sites across the different stages (see Supplemental File S4; Supplemental Fig. S3), we settled on the straightforward criterion of defining the 5% most highly occupied metapeaks as HOT sites and the top 1% as ultra-HOT sites. For the worm, these thresholds are 84 and 240 peaks for HOT and ultra-HOT sites, respectively, and 277 and 602 peaks for the fly (Supplemental Fig. S3B,C). We found that most HOT/ultra-HOT sites were present across developmental stages in the worm, and even those that were not shared were usually only a few percent below the HOT/ultra-HOT threshold, indicating that most sites are quite consistent across development. Because the fly data are primarily from embryos, we did not examine HOT sites across development. For these reasons, we combined the data from all stages to define HOT sites in each of the species for further analysis in this paper. Because of their high occupancy, these HOT/ultra-HOT metapeaks contain a disproportionate number of individual peaks (Table 2).
Number of peaks and metapeaks in worm and fly
Because HOT sites may represent open chromatin in many cell types that lead to nonspecific TF binding, we sought to determine the relationship between metapeak occupancy and peak signal strength as reported by SPP. To normalize signal strength between experiments, we ranked the peaks within each experiment from 0 to 1, with 1 representing the strongest signal in any experiment. The distribution of peak ranks among metapeaks of differing TF occupancy shows that the relative peak signal strength steadily increases with increasing cluster occupancy (Fig. 1E,F). These results argue against HOT sites being locations of low-frequency, spurious binding and are consistent with the notion that they represent highly accessible, open chromatin that facilitates strong TF binding (Partridge et al. 2020). Singletons are enriched in the weakest binding sites, suggesting that they are less likely to be functional.
We postulate that together the regions spanned by metapeaks sample much of the regulatory DNA in these two genomes. As such we would expect these regions on average to have conservation rates intermediate between coding exons and random unannotated regions of the genome. We initially used available multiple alignment groups for the two species. However, the differences in evolutionary distance among fly species relative to worm species (C. elegans has only quite distant relatives) made direct comparison of the results difficult. Instead, we opted to compare pairwise alignments between species selected to be of comparable evolutionary distance—C. elegans versus C. remanei (64% similarity) and D. melanogaster versus D. virilis (62% similarity). In both species, genomic regions covered by metapeaks show levels of conservation intermediate between exons and random segments (Fig. 2A,B).
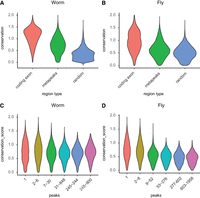
Conservation of metapeak regions. Regions of the genomes spanned by metapeaks show increased conservation compared to random regions but less conservation than coding exons. The fly exons (A) are slightly less conserved than exon sequences in the worm (B). (C,D) Metapeaks with increasing numbers of peaks show decreasing conservation, particularly in the fly (D).
We also evaluated DNA conservation covered by the metapeaks based on the number of TFs per metapeak. In pairwise alignments, both species showed a decrease in conservation with increasing metapeak occupancy, with the trend more pronounced in flies (Fig. 2C,D). The larger span of the HOT and ultra-HOT metapeaks may include more unconstrained bases, with sequence-specific binding of only a subset of TFs critical to the formation of the site, and the presence of many other less consequential TFs associated with open chromatin.
Coassociation of TF metapeaks
Manual inspection of the metapeaks suggested that certain TFs frequently occur together in the same metapeak. We, therefore, derived the Pearson's correlation of each TF pair across the full set of metapeaks with TF membership greater than two and clustered the results. Together, data from worm experiments showed that TFs from the same stage often clustered together, as well as experiments with the same TF across different stages. To avoid these stage-specific or TF-dependent effects, we grouped the experiments into three different sets for the worm (all embryo stages, grouped larval stages L1–L3, and L4/young adult [YA]) (Fig. 3A; Supplemental Fig. S4D,E; Supplemental File S2). Each set contains well-correlated TFs, including some known to be involved in muscle development (hlh-1, rnt-1, and unc-120), hypodermal development (elt-1, elt-3, and blmp-1) in the embryo (Supplemental Fig. S4D,E), as well as other less familiar associations, for example in L4 and YA TFs likely involved in the gonad (D2030.7, F33H1.4, ceh-91, madf-8, efl-1, and madf-6) and some likely involved in the intestine (pqm-1, dve-1, and others) (Fig. 3A).
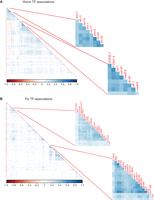
Correlation of TF–TF pairs. Peaks of some TF pairs occur in the same metapeaks more frequently than others as measured by Pearson's correlation for both worm L4/YA experiments (A) and fly embryo experiments (B). Two clusters of TFs with correlated peaks are highlighted for each species but others are also evident. Negative associations are evident on the left side of the worm plot.
For the fly, we examined Pearson's correlations of the TFs in each of the stages assayed (Fig. 3B; Supplemental Fig. S4A–C; Supplemental File S2), but the relatively few experiments in the larva, pupa, and adult stages limit their utility. However, the fly embryo has several high-density clusters, two of which are highlighted (Fig. 3B). The first cluster is composed of 21 genes, of which there is molecular evidence of expression for 18 of the 21 (85%) in the nervous system, four only becoming active in larval and adult stages (Hammonds et al. 2013; Brown et al. 2014; Calderon et al. 2022; Li et al. 2022). The second cluster is composed of 18 TFs of which all are maternally deposited and 10 are maximally expressed in ovaries. These genes are members of chromatin insulator complexes (six TFs; Dref, E(bx), Sry-delta, M1BP, gfzf, and pzg), embryonic development (five TFs; Dfd, ems, Ets97D, MESR4, and crp), the dREAM complexes (four TFs; Dp, E2f2, Myb, and CG17186), chromatin-associated proteins (two TFs; stwl and trem), and one member of the NSL complex (MBD-R2). The dREAM and CP190 chromatin insulator complexes have been proposed to be required for transcriptional integrity, and it has been suggested that the dREAM complex functions in concert with CP190 to establish boundaries between repressed/activated genes (Korenjak et al. 2014). The NSL complex regulates housekeeping genes (Lam et al. 2012). Together the genes of the second cluster appear to be required maternally to set up general zygotic transcription. The correlation values for each TF–TF pair for both species are available in Supplemental File S2.
Motifs
To investigate the relationship between TF binding motifs, individual peaks, and/or metapeaks, we collected the motifs determined by in vitro experiments from the CIS-BP database (Weirauch et al. 2014). In both fly and worm, the number of in vitro motifs in a metapeak region is highly correlated with the number of peaks in the region (Fig. 4A,B), supporting the idea that HOT/ultra-HOT sites do not correspond to spurious binding. On average, each in vitro motif appeared in 21.5% (P < 1 × 10−16, two-sided paired t-test comparing to random sequence) and 11.9% (P < 5 × 10−8) of the peaks in fly and worm, respectively. We then inferred TF binding motifs for each TF from its ChIP-seq peaks and considered it as a successful inference if we observed any significant similarity between the best three inferred motifs and the corresponding in vitro motifs. Compared to using all the peaks, we observed an increase in the number of successes when we only used a subset of peaks after removing HOT and ultra-HOT sites and singletons (Fig. 4C,D). With a more stringent HOT site threshold, further improvement was achieved. Nevertheless, both subsets resulted in a higher success rate than using all or a fixed proportion of peaks, consistent across worm and fly.
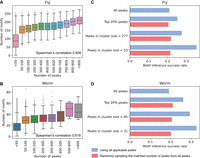
Motif analysis in the metapeaks. (A,B) The number of peaks in a metapeak region was highly correlated with the motifs in the region for both fly (A) and worm (B). The Spearman's correlations for each are shown. Utilizing the ChIP-seq data to obtain motifs, we found using a more stringent peak cluster threshold of 53 for fly (C) and 31 for worm (D) resulted in a better motif inference success rate compared to using all peaks, the top 20% of peaks, or a larger cluster size. For each subset, we randomly sampled the same number of peaks from all peaks for better comparison (shown in red). This sampling process was repeated three times and the average value was shown.
The result above suggested that excluding HOT/ultra-HOT sites may aid downstream motif inference. To test this idea, we inferred motifs for TFs that did not have in vitro motifs, either using all peaks or using only peaks left after stringent filtering of HOT sites. When comparing motifs found using all peaks compared to only non-HOT peaks, inferred motifs were shared in 69% and 60% of experiments for fly and worm, respectively, suggesting very similar capacity for motif inference regardless of the inclusion of HOT sites. These inferred motifs could serve as important resources for further regulatory analysis (Supplemental File S9; modERNresource GitHub, https://github.com/modERNresource).
Distribution of peaks and metapeaks relative to protein-coding genes
To evaluate the correlation between TF binding sites and mRNA abundance as measured in other experiments, we assigned the binding sites to candidate target genes. Binding sites can be at a considerable distance from the genes they regulate (Levo et al. 2022 and references cited therein), but most genes do have promoter-proximal binding sites, and in these compact fly and worm genomes, transgenic reporter experiments indicate endogenous expression patterns can usually be produced from promoter-proximal DNA (≤1–2 kb). Exploiting this simplification, we assigned the apices of peaks and metapeaks to their nearest transcription start site (TSS) as the primary target gene. In most cases, we also assigned the next closest gene as an alternate target (see Supplemental File S4).
A total of 13,109 genes (93.7% of 13,986 total) in the fly and 16,822 genes (84.2% of 19,981 total) in the worm had at least one associated peak, with the number of associated peaks varying widely (Supplemental Fig. S2E,F). This variability is also reflected in the occupancy of the metapeaks associated with target genes. For worms, 2268 genes had one or more associated HOT or ultra-HOT sites, and 2422 genes had only singletons, with 1715 having only one associated singleton (Table 2), while fly had 2848 genes with associated HOT/ultra-HOT sites and only 779 with only singletons, with 494 having only one singleton.
The position of the peaks relative to their targets also varied. Both worm and fly exhibited a strong bias for peaks within 500 bases of the nearest TSS, with substantially more sites upstream than downstream from the TSS (Supplemental Fig. S5), although fly has more peaks in introns than worms. The distribution of peaks in C. elegans is slightly farther upstream of the TSS than in Drosophila, perhaps reflecting the fact that for many C. elegans gene models the annotated TSS is the site of trans-splicing, with the start of transcription occurring further upstream (Fig. 5A,B; Allen et al. 2011; Saito et al. 2013). However, the peak distribution relative to the TSS varies for individual TFs, with some having a median position as much as 1 kb upstream, and others several hundred bases downstream from the TSS. Both species had TFs with biased distributions (Supplemental Fig. S5; Supplemental File S3).
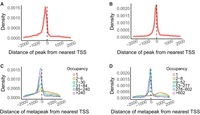
Peak position relative to TSS. (A,B) Peaks in worms (A) and flies (B) predominantly lie close to the TSS of the nearest gene. Worm peaks are slightly farther from the TSS, possibly reflecting the use of splice leaders in worm transcripts, so that the actual start of transcription is further upstream. The hint of two different distributions may reflect those genes with a splice leader and those without. (C,D) Metapeaks with few peaks—less than or equal to six in the worm (C) and less than or equal to eight in the fly (D)—are more broadly distributed.
We next examined the distance relationship from metapeaks to the TSS with respect to the peak number within the metapeak. High-occupancy metapeaks were more tightly distributed near the TSS, with singletons and low-occupancy metapeaks much more broadly distributed (Fig. 5C,D). In fact, in flies, singletons and the lowest occupancy metapeaks were more prevalent downstream from the TSS. If position relative to the TSS is an important indicator of the possibility of transcriptional regulation by the metapeak, this distribution suggests that singletons and very low occupancy metapeaks are less likely to be functional. However, it could also reflect that active promoters tend to be so highly occupied with metapeaks that there is little real estate available for singletons.
Complexity of regulation
Many genes in both worms and flies have complex expression patterns, with expression in multiple cell types at distinct times in the life cycle. This complexity is achieved by particular combinations of TFs in the different cell types at distinct times. To examine the relationship between TF binding and target gene expression in the worm, we exploited available single-cell expression profiles (Cao et al. 2017; Packer et al. 2019; Calderon et al. 2022). Because the resolution of cell types in the published fly data set was limited, we set aside the younger, more transient cells and remapped the older cells, assigning additional and more specific cell types, detecting 83 cell types in all (see Supplemental File S4 for details). To estimate expression complexity, we calculated the entropy of each target gene's expression across cell types and time bins, where entropy here is defined as the sum of the negative fractional expression of a gene multiplied by the log2 of the fractional expression across all cell types (see Supplemental File S4 for Methods). Thus, a gene with uniform expression has a maximum entropy measurement that is the log2 of the total number of cell types, with that value decreasing with more limited expression of the gene, such that the entropy of a gene with expression in a single cell type will be zero. We then subdivided genes into those with ultra-HOT sites, those with HOT sites, down to those with singletons, and assessed the entropy of target genes. Genes with ultra-HOT or HOT sites had the highest entropy, reflecting broad expression, while genes with low occupancy sites had low entropy, with expression in only a limited number of cell types or time bins (Fig. 1G,H). Broad expression may then be the result of the sum of many TFs acting on a target gene. An alternative explanation for such enrichment of high entropy genes with high-occupancy sites may be that broadly expressed genes have a more open chromatin configuration, allowing the binding of many factors, even if they do not play a role in the regulation of that target. Likely, both possibilities occur. In contrast, target genes with more cell-type-specific expression are associated with smaller metapeaks.
We also examined the relationship between the number of peaks associated with a gene and the number of metapeaks. Some target genes have hundreds of associated peaks distributed in only one or two metapeaks, while others have many distinct metapeaks (Supplemental Fig. S6A,B). The median numbers of distinct metapeaks are 15 and 99 for worm and fly, respectively. As might be expected, housekeeping genes (Ghaddar et al. 2023) have predominantly large numbers of peaks in relatively few metapeaks (Supplemental Fig. S6C), as recently demonstrated in yeast (Rossi et al. 2021). In contrast, genes with multiple TSSs tend to have a large number of associated metapeaks of variable occupancy (Supplemental Fig. S6D,E). The TF genes themselves are widely scattered in these plots, with some having only a few associated peaks in one or two metapeaks, while others have multiple TSSs and multiple associated metapeaks, e.g., ceh-38, daf-12, daf-16, egl-44, syd-9, and unc-62 in worms. The regulation of genes with multiple highly occupied metapeaks is likely to be very complex, with different regulatory sites driving expression in different cell types and times.
Target expression patterns can reflect TF expression patterns
A central question is to what extent, if any, the detected peaks or binding events drive gene expression of the target genes of the bound TFs. Addressing this question is complicated by the observation that TF ChIP-seq data often detect binding at many more sites than expected based on known or experimentally validated functional sites (Hu et al. 2007; Cusanovich et al. 2014). Nonetheless, for strong, positive regulators the expectation would be that the expression patterns of TFs would correlate with the expression of targets, whereas negative regulators would be negatively correlated. The emergence of single-cell RNA-seq (scRNA-seq) data for both worm and fly offers an opportunity to investigate these relationships directly. For the worm, we used scRNA-seq data from the embryo, both terminal cells and their progenitors (Packer et al. 2019), L2 (Cao et al. 2017; Packer et al. 2019), and YA data (Ghaddar et al. 2023). For the fly, since most of the ChIP data were collected in embryos, we focused on the recent embryo scRNA-seq data set (Calderon et al. 2022), reanalyzed as described above (see Methods in Supplemental File S4).
As a direct test of the relationship between the TF and its target expression pattern, we aggregated the expression values for each target, summing the values across all cell types. For TF-target pairs in the worm, we matched ChIP-seq data from the embryo with the embryo expression data, ChIP-seq data from L1, L2, and L3 with the L2 expression data, and ChIP-seq data L4 and YA with the YA expression data. For the fly, we only matched the embryo ChIP data with the embryo expression data, since most ChIP-seq experiments were done in embryos.
We used a mean-centered expression matrix to evaluate TF-target expression. As examples, for blmp-1 in worms (Fig. 6A,B) or GATAe in flies (Fig. 6C,D), this matrix results in negative values for cell types with no or very low expression, but still high positive values for other cell types. For genes with broad, uniform expression, most values will be around zero, thus minimizing their impact on the aggregate pattern. We only considered targets that were not associated with HOT sites, recognizing that genes with HOT sites were generally broadly expressed.

TF expression correlates with target gene expression. (A–D) Aggregate target expression reflects the TF expression for the worm blmp-1 (A) TF and (B) targets, and the fly GATAe (C) TF and (D) targets. Cell types are arranged along the x-axis by broad cell class and sorted alphabetically within each class. The worm cell types are further divided into time bins (from Supplemental Files S6, S7; Packer et al. 2019).
The aggregate expression patterns of these non-HOT targets of blmp-1 and GATAe are like the patterns of the respective TFs. blmp-1 in worms is expressed in a variety of cell classes, including hypodermis, seam cells, glia, excretory cells, and rectal cells with the aggregate expression of the target genes closely resembling the TF pattern. GATAe in flies is expressed in several different cells of the intestinal tract, again with its target gene pattern closely reflecting that of the TF.
To evaluate the correlation of TFs more broadly with their cell types, we calculated the cosine of the angle between the expression vector of the TF and the average expression vector of that TF's targets across the different cell states: the embryo TFs against the expression in both progenitor and terminal cell states, the L1, L2, and L3 larval stage TFs against expression in the L2 stage, and the L4 and YA TFs against expression in the YA stage (see Methods in Supplemental File S4). For flies, we examined the embryos TFs against the embryo expression data. The TFs with the most highly correlated targets by this metric include many well-known TFs in both organisms as well as additional less-studied TFs (Supplemental File S5). Manual inspection of many TF–target comparisons showed that, at a cosine of 0.2 or greater, the TF/target patterns still had readily recognizable similarities. In total, 142 worm TFs had an angle ≥0.2 in one or more of the four grouped developmental stages, and 89 fly TFs had an angle ≥0.2 in embryos. Some of the cosine angles are negative, possibly signaling a repressive effect of the TF, with more TFs having more negative values in the fly than the worm. In flies, su(Hw), known to act as a repressor in many circumstances (Geyer and Corces 1992), shows negative values for the aggregate target expression in cell types in which su(Hw) itself is expressed. In worms, zip-5 is highly expressed in the intestine, yet its targets show negative values in the intestine as well as in most of the other cells with high zip-5 expression, suggesting that zip-5 might be a repressor of expression.
To learn which of the targets was contributing to the aggregate signal, we compared each of the targets individually with its TF, with the idea that targets resembling the TF pattern might be the result of functional TF binding. We also compared an equal number of random targets to each TF pattern and compared the two distributions to estimate the likelihood of obtaining such a TF-target distribution by chance, using a Wilcoxon rank-sum test. The results (Supplemental File S5) indicate that most of the cosine angles greater than 0.2 for worms are significant. Those target sets with high cosine angles could be considered the genes regulated by the TF, representing regulons. For flies, the correspondence between the cosine value and the Wilcoxon rank-sum significance is less well correlated, but cosine values above 0.4 are almost all significant.
A limitation of this approach is that for targets expressed in multiple cell types under the control of different regulatory elements (and TFs), the expression pattern of the targets may be quite different from that of the TF. A simple illustration is provided by hlh-1, the worm myoD homolog, with a cosine angle of 0.42. Its targets are highly enriched in body wall muscle expression and the GLR mesodermal cell, where hlh-1 is expressed. But the aggregate targets also show expression in other mesodermal cells, where hlh-1 is not expressed (Supplemental Fig. S7A). Moreover, hlh-8, a homolog of hlh-1, with a cosine angle of 0.39, is expressed specifically in these mesodermal cells and not in body wall muscle, and the aggregate targets of hlh-8 show enrichment in both the mesodermal and body wall muscle (Supplemental Fig. S7B). In this case, the relationship between TF expression and target gene expression is simple enough that it does not depress the cosine angle, but clearly, for complicated situations, very low values could result. Other issues might include TFs that are both positive and negative regulators, effectively canceling any signal. In both analyses, many TFs did not show a correlation between TF expression patterns and target patterns. Despite passing quality metrics, some experiments may not capture these correlations for reasons presently unknown. Other TFs may have only a fine-tuning role, with effects too subtle to measure with these analyses.
Modeling gene expression patterns
We next investigated if the signal from the ChIP sites and their targets could be used to model gene expression. We chose to use a random forest, machine-learning algorithm because it accommodates nonlinear relationships and provides a relative measure of the contribution of the individual TFs in predicting expression. As input, we used the peaks and their targets, with a row for each metapeak–target relationship (targets with multiple metapeaks had multiple rows) and a column for each TF, with values of the matrix being the relative peak strength of the TF binding event (the predictor matrix). The response variable was the cell type gene expression as measured in the scRNA-seq data, which does not distinguish between isoforms. The algorithm then produces an output matrix, giving the relative importance of each TF for determining the target values for each cell type independently. Running the model on multiple cell types produces an output matrix that gives the relative importance of each TF for determining the relative target values for each cell type and an estimate of the goodness of fit.
We modified the input matrix in several ways, testing more than 40 different models in the embryo terminal cell data. We used different filters for the metapeaks to be considered, modified the peak rank by distance from the TSS, considered only peaks from TFs expressed in the cell type above a threshold, and looked at alternate targets as well as primary targets (see Supplemental File S4). Using the estimate of the goodness of fit produced by the algorithm, we compared the performance of the different models across all the cell types and in turn across the different life stages, selecting models that performed well across many cell types. The top model for worms considered only genes that were not targets of HOT sites, required the TF to be expressed in the cell type at least 5% of the maximum across all the cell types, used the peak rank modified by distance to weigh proximal peaks more strongly, removed singleton peaks and used only primary or close alternate targets (see Supplemental File S4). The relative importance of the TFs in each of the cell types defined in the single-cell data in the worm embryo is illustrated in Figure 7A for the model with the best fit across the samples. The model reproduced many known relationships, e.g., in the worm elt-1 and elt-3 were of high importance in the hypodermal cell types, hlh-1 and unc-120 in the body wall muscles, elt-2 in the intestine, and even che-1 in the ASE neurons. But it also suggested the importance of other less well-characterized TFs in these and other cell types (Supplemental Fig. S8).
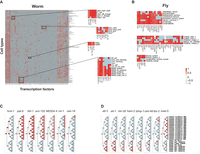
The relative importance of TFs in cell type gene expression. (A) A heatmap of the relative importance of worm TFs in predicting gene expression in the embryo terminal cell types. The importance is indicated by the intensity of the red color from yellow (no importance) to dark red (most important). Light blue indicates that the factor was not expressed above the threshold in that cell type. Clusters of TFs and cell types (black boxes) are blown up on the right, showing the detected relationships of well-studied and novel TFs and the cell types in which they are important. (B) Clusters of fly TFs and cell types in which they are important, selected from the full heatmap in Supplemental Figure S7. Color scale as in A. (C,D) TFs are important in the Ca lineage. (C) TFs are important in the Cap lineage, which produces exclusively body wall muscle cells and are arranged by the onset of their importance. (D) TFs are important in the Caa lineage or in patterned expression in both lineages. The Caa lineage produces primarily hypodermal cells but also some neurons and cell deaths. The lineage names are given on the right, along with the specific cell type in the embryo. Body wall muscle cells are labeled mu_bod followed by letters to indicate Dorsal/Ventral, L/R, and a number indicating the row of the cell, with 24 most posterior. Color scale as in A. Cell types not detected in the single-cell data are indicated by an X.
We also ran the best model on the combined fly ChIP-seq and scRNA-seq data. Because of the low, broad expression of many fly TFs (Supplemental Fig. S8A), we increased the threshold for minimal TF expression to be greater than the mean expression across the cell types plus one standard deviation. Again, the model captured the importance of TFs known to function in certain cell types, such as GATAe in the gut and the TFs involved in muscle (Fig. 7C). However, overall, the fly model appears less discriminating than the worm, either reflecting the different biology of the two species or as a consequence of limitations of the scRNA-seq or ChIP-seq data (see Supplemental Methods in Supplemental File S4; Supplemental File S8; Supplemental Fig. S9 for details on reannotation of embryonic fly cell types in the scRNA-seq data).
Examination of the worm lineage heatmap of the TF importance suggested that the model was capturing some of the dynamics of regulation during development. To facilitate the visualization of such relationships we developed a program that allows the user to view the importance of the various TFs in arbitrary subsets of the lineage and TFs. For example, in the Ca lineage (Fig. 7C), the TF hnd-1 is important in the Ca cell itself, but only in the progeny that gives rise to body wall muscle. As hnd-1 importance wanes, hlh-1 and pat-9 become increasingly important, followed by unc-120 and M03D4.4, each with lesser importance. In the Caa branch, which gives rise predominantly to hypodermis, elt-1 and then elt-3 become important, with nhr-28 later and of lesser importance (Fig. 7D). The ceh-48 TF gene, a known regulator of neural development, only becomes important once the lineage becomes restricted to producing neurons. The restriction of TF expression clearly limits where TFs can be considered important, but the ChIP-seq binding data are central to determining the importance of each factor during development. The program is available as a Java JAR file for users to install on their own computers to explore TF importance in any of the lineages. In addition, we provide the relative importance values for each TF in Supplemental File S6.
Discussion
We describe here the results of a long-term project to discover the TF binding sites in two key model organisms, C. elegans and D. melanogaster. The effort has produced ChIP-seq binding profiles for more than 90% of the fly TFs and more than half of the targeted TFs in the worm. Most of these TFs have been assayed at the stage where the TF is maximally expressed but some have been assayed at multiple stages, with a total of 678 experiments in the fly and 525 in the worm.
Combined, these experiments define 3.6 M peaks in the fly and 0.9 M peaks in the worm. The peaks often overlap in genomic location, creating what we have termed metapeaks, with each metapeak containing anywhere from a single peak to peaks from hundreds of TFs. Together these metapeaks sample much of the regulatory DNA of each genome, based on the overlap with ATAC-seq data and inspection of defined regulatory regions in each species.
The fly and worm data sets have remarkable similarities, but one clear difference is the number of peaks found in each experiment and the number of peaks associated with each gene. Combined with the larger number of TFs assayed in the fly, the fly has about four times as many peaks detected as the worm. This increase is not reflected in a similarly increased number of metapeaks (only a ∼50% increase). While the amount of the genome covered by peak signals is about twofold greater in the fly (57 Mb vs. 27.5 Mb), the fraction covered rises only about by half, from 27% to ∼40%. The result is a considerable increase in the number of peaks per metapeak. Greater cross-reactivity of the GFP antibody with fly DNA-binding proteins might be one explanation, but the pattern of peak signal strength versus metapeak occupancy is almost identical between the fly and worm, using similar fractional cutoffs in grouping. Whatever the reason, a practical consequence has been that more stringent relative thresholds in fly were useful in detecting TF-target site similarities and random forest modeling.
Using a straightforward distance metric, we have assigned each peak and metapeak to a target and a possible alternate target gene. Genes have widely varying numbers of associated peaks and metapeaks, with some genes having tens of regulatory regions as defined by the metapeaks. While the high numbers of metapeaks might result from fortuitous binding at open chromatin, the results also suggest that most genes are regulated by multiple TFs, with the pattern and magnitude of expression reflecting the complex interaction of these multiple factors. Those metapeaks with the largest number of peaks, or HOT sites, contain some of the strongest relative peak signals, are most closely located near TSSs, and have target genes that are broadly expressed. Together these results imply that HOT sites are likely important functional regions at stably open chromatin regions where TFs in almost any cell type are able to bind. In contrast, those metapeaks with fewer peaks (lower occupancy) appear to drive more tissue-specific expression patterns; however, even these sites can have large numbers of TF binding sites. Whether cell-type-specific sites of open chromatin act as similar sinks for TF binding is unclear from these whole animal data sets. Functional redundancy and TF cooperativity could also contribute to this high density of sites (Wu and Lai 2015).
Many of the peaks in worm and fly did not contain motifs. Work from our group and others have studied ways to improve motif inference by filtering the data (Xu et al. 2021, 2024). Nonspecific binding of antibodies and spurious binding to DNA in worm and fly necessitated the development of a method to remove these artifacts (Xu et al. 2021). More recently, improvements have been made to motif inference using signal strength, metapeak occupancy, and genomic length (our work; Xu et al. 2024). These studies highlight the importance of refining the data to uncover modes of gene regulation by sequence-specific TFs. Future improvements to ChIP-seq protocols, antibody specificity, and downstream processing will aid in this process.
Using scRNA-seq data, we find a clear relationship between the expression pattern of a TF with its targets, presumably reflecting functional binding events and perhaps representing regulons. Further, we have used the ChIP-binding sites to drive a random forest model of cell-type-specific regulatory relationships. This analysis identified a subset of TFs that contribute broadly across many cell types to regulate target gene expression. Most importantly, this analysis also identified many TFs with significant contributions to gene expression that is restricted both temporally and spatially. To highlight this specificity, the impact of distinct TFs on gene expression can be distinguished between parent and daughter cells in the embryonic lineage of the worm, as well as for a single neuron pair. In many cases, TFs known to have a role in a specific cell type show similar patterns and scores with TFs that have not yet been linked functionally to that cell type, suggesting worthwhile avenues of investigation into the relationship of these TFs in the development or activity of that cell type. The RNA-seq data were key to determine the cell types in which the TFs might be active, whereas the ChIP-seq data itself specified the importance of each TF in generating the pattern of expression. Although this analysis does not identify individual target genes that depend on a given TF, it increases the overall resolution of the whole animal ChIP-seq data, as well as its relevance for further investigation into TF function.
Beyond the ChIP-seq data, the GFP-tagged strains present a valuable resource to the community. We have produced tagged strains for 514 TFs in the worm and 616 tagged strains in the fly along with additional strains for other genes. These strains have been highly useful to the community, as indicated by their popularity with the stock centers. However, we note that the resource has its limitations. The data come from whole animals, mostly from a single stage. TFs expressed in only a single cell type where that cell type is represented by only a few cells in the animal might not produce robust ChIP signals. Indeed, in the worm, we stopped performing ChIP for strains with very limited expression, which posed challenges in obtaining samples with meaningful peaks. Nonetheless, it is worth noting that the TF che-1, only expressed in the ASE neurons (two cells of that type in the 959 somatic cells of the worm) and known to be a driver of its differentiation (Patel and Hobert 2017), was detected as one of the most meaningful TFs in that cell type in the random forest modeling. In contrast, in the experiments with mec-3, expressed predominantly in four touch cells in the embryo, the targets do not include known genes that are not expressed in mec-3 mutants (Etchberger et al. 2009). Furthermore, TFs may be redeployed driving different genes even in different cell types at different stages in development, and by only assaying one stage, we may have missed these events. Future efforts on an expanded array of developmental stages will reveal the extent of TF redeployment. In aggregate, this resource provides hundreds of worm and fly strains, and millions of in vivo binding events that are already heavily utilized by the model organism community. With increasingly sophisticated computational approaches, we expect that these data will be critical to investigating and understanding complex gene regulatory programs during development.
Methods
Strain generation and resource
For both fly and worm, we revised our lists of TFs, removing those that WormBase or FlyBase had designated as pseudogenes, and reclassifying others after consulting recent publications (Narasimhan et al. 2015; Ma et al. 2021) and considering our own results with factor subcellular localization in transgenic lines. Our revised list contains 658 fly TFs and 833 worm TFs (Supplemental File S1).
For both species, we generated strains with individual TFs tagged with GFP, but the details differed, taking advantage of the different resources available. For the fly, C-terminally GFP-tagged BACs were used to create transgenic lines as described previously (Venken et al. 2009; Kudron et al. 2018). In brief, GFP was recombineered into the smallest P[acman] BAC that also contained either a predicted insulator or a portion of the next nearest gene upstream of the TF. GFP-tagged BACs were integrated into specific attP sites through injection using the phiC31 integration system. Some additional GFP BACs were sent for injection and balancing at GenetiVision (https://www.genetivision.com/). In general, these BACs were either injected into parent strains VK31 or VK37 such that the TF-GFP would integrate into a different chromosome than the endogenous TF. Homozygous lines were able to be generated for ∼90% of the TF-GFP lines. Altogether, tagged strains are available for 616 TFs. Lines were also constructed for an additional 56 non-TF genes (Supplemental File S1). All lines are available from the BDSC (Indiana University Bloomington).
In the worm, for most of the project, GFP-tagged fosmids from the modENCODE TransgeneOme resource (Sarov et al. 2012) were introduced by bombardment (Kudron et al. 2018). Bombardment resulted in a low number of copies of the fosmid (from 1 to 10) at undetermined sites in the genome. However, not all TFs were represented in the library, and by 2017 the remaining available fosmids had repeatedly failed to yield GFP-tagged transformants. Thus, we turned to using CRISPR, and over the course of the project, we used three different methods. In early experiments, we used the SapTrap method (Schwartz and Jorgensen 2016) (strains RW12167–RW12175) but then switched to using Roller plasmid as a coinjection marker (Dokshin et al. 2018; Ghanta et al. 2021). We targeted C-terminal fusions but in the rare cases where these failed, we tagged the N-terminal end. Roller progeny were selected and their progeny were evaluated for GFP expression with fluorescence microscopy. We used this method for the bulk of CRISPR strain collection (strains RW12178–RW12346). Toward the end of the project, we switched to using a dpy-10 repair template as a coinjection marker (Paix et al. 2015, 2017). In our hands, this method seemed to provide the greatest success (strains RW12347 and after).
Because of the potential differences between the strains generated by fosmid bombardment and CRISPR editing, we generated CRISPR lines for TFs that had also been tagged using fosmid bombardment and compared the GFP expression and ChIP-seq results for both strains in detail (Supplemental Table S1). We tested factors at different stages and tissues of expression, looking at the success as measured by IDR, the number of binding sites detected, and the intensity of the GFP signal (expression) in the paired strains. We saw no consistent differences.
Altogether, tagged lines are available for 514 TFs, and another 45 lines were constructed for non-TF genes. Some of these were contributed by the wider community (see Supplemental File S1 for details.) Almost all strains are available from the CGC.
Fly growth and ChIP
As described previously, transgenic flies were expanded in vials and bottles containing standard Drosophila media at ∼22°C. Larvae, white prepupae (WPP), and adults were collected from these expansion bottles. Pupae were obtained by collecting WPP and incubating for 1–2 days at ∼22°C. For embryonic collections, 0- to 8-day-old adult flies were transferred to embryo cages capped with apple juice plates. After a 24 h preincubation, apple juice plates were replaced at appropriate intervals, restricting the collected embryos to their desired developmental stages. For late embryonic stages, collection plates were removed and maintained at room temperature before chromatin collection.
Chromatin was collected and immunoprecipitated (IP) using the goat anti-GFP described previously (Zhong et al. 2010; Niu et al. 2011; Kasper et al. 2014; Kudron et al. 2018). However, due to its limited supply, later experiments used anti-GFP Ab290 (Abcam) (Supplemental File S1). In total, ChIP was successfully performed for 678 (605 different TFs) experiments with TFs and another 62 experiments (55 different genes) for non-TFs. Of the 12 tagged TF strains without ChIP, six failed to yield data and for another six, ChIP was not attempted.
Worm growth and ChIP
Worm growth and/or ChIP were conducted as previously described (Zhong et al. 2010; Niu et al. 2011; Kasper et al. 2014; Kudron et al. 2018) with the following modification. All chromatin samples were purified using either a minElute PCR Purification Kit from Qiagen (Catalog number 28006) or a ChIP DNA Clean and concentrator kit from Zymo (ZD5205).
In total, ChIP was successfully performed for 525 (356 different TFs) experiments with TFs and another 65 experiments (45 different genes) for non-TFs. Of the 158 tagged TF strains without ChIP, 27 failed to yield data despite repeated attempts, 43 were deemed to be poorly expressing or exhibited an aberrant expression pattern, 53 were cytoplasmic, 11 were deemed orphan nuclear hormone receptors and set aside, four had a phenotype, and 17 ChIP-seq experiments were completed since the data freeze in October 2022, and five are pending or were not attempted.
Worm and fly ChIP-seq library prep and sequencing
ChIP and input control samples (genomic DNA from the same sample) for two technical replicates for worm or three for fly were used for library preparation and sequencing as previously described for the modENCODE and modERN projects (Zhong et al. 2010; Nègre et al. 2011; Kasper et al. 2014; Kudron et al. 2018). More recently, samples were processed at the Yale Center for Genomic Analysis, using the following methods (Supplemental File S1): Libraries were generated using ∼5–10 ng of DNA that were end-repaired, A-tailed, adapter ligated and PCR enriched (8–10 cycles) using the KAPA Hyper Library Preparation kit (KAPA Biosystems, Part#KK8504). Indexed libraries were quantified by qPCR using the Library Quantification Kit (KAPA Biosystems, Part#KK4854) and inserts were size distributed using the Caliper LabChip GX system. Samples with a yield of ≥0.5 ng/μL were used for sequencing.
Sample concentrations were normalized to 10 nM and loaded onto an Illumina NovaSeq flow cell at a concentration that yields >5 million reads passing filter clusters per sample. Samples are sequenced using 100 bp paired-end sequencing on an Illumina HiSeq NovaSeq Sequencer according to Illumina protocols. A positive control (prepared bacteriophage Phi X library) provided by Illumina is spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time. Demultiplexed sequencing data passing internal quality controls at YCGA were transferred to the Waterston Lab for downstream processing.
Data analysis
To analyze ChIP-seq data, we employed the standard ENCODE pipeline, which includes the SPP peak caller (Kharchenko et al. 2008). Detailed and extensive description and discussion of this pipeline, and all analytical methods used throughout the paper, including metapeak analysis, HOT site identification, motif inference, cosine calculation, and random forest machine learning, are provided in Supplemental File S4.
Data access
All of the newly acquired data since our prior publications have been submitted to the ENCODE Data Coordinating Center, with the exception of experiments concluded after October 2022, when the Center stopped accepting submissions. All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under the superseries accession number GSE271850. This includes the 17 sets of worm data and 4 sets of fly data collected since October 2022, as well as all the earlier data sets, which were reevaluated using a recent version of SPP and the DCC pipeline at the end of the ENCODE Project. In addition, raw and processed sequencing data for a few additional TFs completed later as well as data for non-TFs have been submitted to GEO under the superseries accession number GSE280245. Additionally, to host these data and the new peak calls, as well as to provide a user-friendly interface, we have developed a website that provides ready access to all the data sets associated with each experiment, including the non-TFs that were not analyzed further in this report (https://epic.gs.washington.edu/modERNresource/). This website also serves links to UCSC Genome Browser displays and has other features. It provides a file of all the peaks with associated metadata for all the TF experiments, such as the target and its distance from the peak, the position of the peak relative to the gene, etc. Interactive heat maps of the importance of each TF in predicting the gene expression pattern of each cell are available as HTML files. Users can also access the Java program that displays the temporal importance of factors in the embryonic lineage of the worm. The custom scripts and unpublished code required to reproduce the work have been deposited at GitHub (https://github.com/modERNresource) as well as in Supplemental File S9.
Competing interest statement
K.P.W. is associated with, and a shareholder in, Tempus Labs and Provaxus, Inc. All other authors do not have external interests.
Acknowledgments
The authors thank: the ENCODE Data Coordinating Center for accepting and providing data access; the Yale Center for Genome Analysis (YCGA), especially Bryan Szewczyk and Irina Tikhonova for library preparation and Christopher Castaldi for sequencing (the YCGA is funded by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number 1S10OD030363-01A1 and the NIH HPC grant (1S10OD030363-01A1 for data storage); the Bloomington Drosophila Stock Center (BDSC) for distributing the fly GFP-tagged TF strains; and the Caenorhabditis Genetics Center (CGC) for distributing the worm GFP-tagged TF strains (the BDSC and CGC are funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs P40 OD-018537 and P40 OD-010440, respectively). We would also like to thank WormBase (https://wormbase.org/) and the UCSC Genome Browser (http://genome.ucsc.edu) for displaying our data; the University of Chicago DNA Sequencing Facility (DNA-seq) for sequencing recombineered BACs (the DNA-seq is supported by the National Cancer Institute, Comprehensive Cancer Center grant P30CA014599); Donald Court for sharing his recombineering plasmids; Jennifer Moran for technical assistance; Krishna S. Ghanta and Craig Mello for providing CRISPR technical advice. This work was supported by NIH grants U41HG007355 (R.H.W.) and NIH grant R01GM76655 (S.E.C.).
Author contributions: M.K. performed worm ChIP-seq, oversaw worm sample processing and sequencing of both fly and worm, and wrote the paper. L.G. analyzed the data, created the website and associated tools, and wrote the paper. A.V. performed worm ChIP-seq and managed fly sample processing. B.C.L. performed fly ChIP and managed fly sample processing. J.G. performed data analysis. J.X. performed data analysis. S.S. performed worm culture. E.F. generated GFP-tagged TF worm strains. A.T.P. generated GFP-tagged TF worm strains. C.H. oversaw worm strain production and edited the paper. D.V. generated GFP-tagged worm strains. A.H. generated GFP-tagged TF fly strains. W.F. generated GFP-tagged TF fly strains. M.W. performed fly ChIP. G.W. performed fly ChIP. V.H. performed fly ChIP. Z.L. performed fly ChIP. M.K. performed fly ChIP. K.P.W. oversaw the fly ChIP. R.A. oversaw the fly ChIP. M.G. performed data analysis. L.D.H. performed data analysis. S.E.C. coordinated fly strain generation and wrote the paper. V.R. coordinated the worm ChIP-seq project and wrote the paper. R.H.W. was the lead coordinator of the entire project, analyzed the data, and wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279037.124.
- Received January 25, 2024.
- Accepted October 17, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








