
Dynamic dysregulation of retrotransposons in neurodegenerative diseases at the single-cell level
- 1Center for Precision Health, McWilliams School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, Texas 77030, USA;
- 2MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, Texas 77030, USA;
- 3Faillace Department of Psychiatry and Behavioral Sciences, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, Texas 77030, USA;
- 4Department of Biomedical Informatics, Vanderbilt University Medical Center, Nashville, Tennessee 37203, USA
-
↵5 These authors contributed equally to this work.
Abstract
Retrotransposable elements (RTEs) are common mobile genetic elements comprising ∼42% of the human genome. RTEs play critical roles in gene regulation and function, but how they are specifically involved in complex diseases is largely unknown. Here, we investigate the cellular heterogeneity of RTEs using 12 single-cell transcriptome profiles covering three neurodegenerative diseases, Alzheimer's disease (AD), Parkinson's disease, and multiple sclerosis. We identify cell type marker RTEs in neurons, astrocytes, oligodendrocytes, and oligodendrocyte precursor cells that are related to these diseases. The differential expression analysis reveals the landscape of dysregulated RTE expression, especially L1s, in excitatory neurons of multiple neurodegenerative diseases. Machine learning algorithms for predicting cell disease stage using a combination of RTE and gene expression features suggests dynamic regulation of RTEs in AD. Furthermore, we construct a single-cell atlas of retrotransposable elements in neurodegenerative disease (scARE) using these data sets and features. scARE has six feature analysis modules to explore RTE dynamics in a user-defined condition. To our knowledge, scARE represents the first systematic investigation of RTE dynamics at the single-cell level within the context of neurodegenerative diseases.
Transposable elements (TEs) are DNA elements that are capable of moving and reinserting themselves into the eukaryotic genomes (Wells and Feschotte 2020). They account for >45% of the human genome (International Human Genome Sequencing Consortium 2001). The vast majority of TEs are retrotransposable elements (RTEs; class I TEs, also known as endogenous retroelements [EREs]), which comprise ∼42% of the human genome. RTEs can transpose via an RNA intermediate and primarily rely on the reverse transcriptase for their integration in the genome (Wells and Feschotte 2020; Copley and Shorter 2023). As their original copy remains intact in the genome, they are also known as “copy-and-paste elements” (Wells and Feschotte 2020). In humans, RTEs are further subdivided into long terminal repeats (LTRs) and non-LTRs, including long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) (Li et al. 2022). However, LTRs and LINEs facilitate transposition by encoding their own reverse transcriptase, whereas SINEs rely on other TEs to retrotranspose their mRNAs (Rodríguez-Quiroz and Valdebenito-Maturana 2022). Notably, although endogenous retroviruses (ERVs) in other species, such as mice, are known to be replication-competent, human endogenous retroviruses (HERVs), a significant subset of LTRs, are not recognized for their ability to replicate (Vargiu et al. 2016; Sankowski et al. 2019). HERVs comprise 8% of the human genome, and these elements are formed when an exogenous retrovirus is integrated into an infected host germline via an endogenization event (Dopkins et al. 2022; Dopkins and Nixon 2024). Throughout evolution, they have acquired multiple mutations or deletions, rendering them noninfectious and incapable of retrotransposition (Dopkins and Nixon 2024). In most HERVs, these mutations also suppress their transcriptional activity under physiological conditions, whereas several HERVs provide retroviral genes and promoters to the genome that can actively transcribe and influence overall transcription (Jern and Coffin 2008; She et al. 2022).
Despite their discovery over 70 years ago (McClintock 1950), the specific functions and mechanisms of TEs in the cellular context remain largely unknown. This lack of knowledge is because of the low complexity of their sequences and abundance in the genome (Copley and Shorter 2023), making their precise mapping of short reads to the genome challenging (Zhao and Han 2009; Wells and Feschotte 2020). TEs have long been categorized as “junk” sequences in the genome (Orgel and Crick 1980). However, advancements in sequencing technologies have revealed that RTE-derived sequences, either with or without transposition activities, are involved in various biological processes and diseases, including development (Peaston et al. 2004; Fadloun et al. 2013; Göke et al. 2015; Robbez-Masson and Rowe 2015; Rodriguez-Terrones and Torres-Padilla 2018), cancer (Hancks and Kazazian 2016; Burns 2017), neurodegenerative diseases (Li et al. 2013; Guo et al. 2018; Sun et al. 2018), as well as other diseases (Brouha et al. 2002; Geister et al. 2015; Hu et al. 2015). More specifically, derepression or inhibition of RTE expression has been observed in various neurodegenerative diseases, indicating their potential regulatory impact on disease progression (Kremer et al. 2013; Guo et al. 2018; Copley and Shorter 2023).
The recent emergence of single-cell sequencing technologies, particularly single-cell RNA-seq (scRNA-seq) and single-nucleus RNA-seq (snRNA-seq), has enabled investigation of cellular heterogeneity in gene expression at the single-cell level (Lake et al. 2016; Habib et al. 2017). Computational pipelines and tools have emerged to quantify RTEs from sc/snRNA-seq data. For instance, Shao and Wang (2021) developed a pipeline to quantify RTEs at the transcript level, whereas scTE (He et al. 2021) and IRescue (Polimeni et al. 2024) provide subfamily level quantification of RTEs. Additionally, several tools, including SoloTE (Rodríguez-Quiroz and Valdebenito-Maturana 2022), MATES (Wang et al. 2024), CELLO-Seq (Berrens et al. 2022), SCIFER (Stow et al. 2022), and Stellarscope (Reyes-Gopar et al. 2023), can quantify RTEs at the locus level. Leveraging these tools, researchers have uncovered numerous aspects of RTE regulation and function across various conditions and diseases (Shao and Wang 2021; McKerrow et al. 2023). For example, studies have reported that LINE-1 is expressed in tumor cells but is absent in immune cells (McKerrow et al. 2023), and ncRNA has substantial tissue specificity during gastrulation and early organogenesis (Shao and Wang 2021).
With the growth of lifespan and expansion of the aged population, neurodegenerative diseases, particularly Alzheimer's disease (AD), have become a significant public health challenge (Mayeux and Stern 2012). Extensive research has been conducted to unravel cellular heterogeneity in neurodegenerative diseases including AD, Parkinson's disease (PD), and multiple sclerosis (MS) (Grubman et al. 2019; Mathys et al. 2019; Schirmer et al. 2019; Lau et al. 2020; Zhou et al. 2020; Absinta et al. 2021; Leng et al. 2021; Morabito et al. 2021; Otero-Garcia et al. 2022; Sadick et al. 2022; Smajić et al. 2022; Xu et al. 2023). These studies have significantly advanced our knowledge of the molecular mechanisms underlying neurodegenerative diseases at the cellular level. It has been suggested that RTE alterations are associated with genomic instability in tau-mediated AD (Guo et al. 2018). Additionally, various studies have reflected the potential role of RTEs in neurodegenerative diseases at the bulk level (Krug et al. 2017; Guo et al. 2018; Tam et al. 2019; Chang and Dubnau 2023). However, no systematic investigation of RTE dynamics in multiple neurodegenerative diseases at the single-cell level has been conducted so far.
To bridge this gap, our study aimed to examine the cell type–specific dynamics of RTEs in different neurodegenerative diseases using a systematic approach. Specifically, we analyzed the publicly available single-cell/nucleus transcriptome profiles in AD, PD, and MS to unveil RTE heterogeneity across different cell types in the human brain. Furthermore, we applied machine-learning approaches to detect AD stages using RTE features. Finally, we built a comprehensive web portal, namely, single-cell atlas of retrotransposable elements in neurodegenerative disease (scARE; https://bioinfo.uth.edu/scARE), for searching, browsing, and analysis of RTEs and their dynamics in neurodegenerative diseases. This study provides new insights into the role of RTEs in the cell type– and disease-specific dynamics of neurodegenerative diseases and offers a valuable resource for further research in this area.
Results
scARE workflow and web portal
To investigate RTE dynamics at the single-cell level in neurodegenerative diseases, we collected and curated publicly available single-cell/nucleus transcriptome profiles from recent studies (Supplemental Table S1) encompassing studies on AD, PD, and MS (Grubman et al. 2019; Mathys et al. 2019; Schirmer et al. 2019; Lau et al. 2020; Zhou et al. 2020; Leng et al. 2021; Morabito et al. 2021; Otero-Garcia et al. 2022; Smajić et al. 2022). In total, we retrieved 12 data sets from 10 single-cell studies, including eight AD, two PD, and two MS data sets. By including multiple neurodegenerative diseases, we aimed to explore the common and disease-specific RTE dynamics in these diseases. A unified pipeline was developed to perform alignment, quantification, normalization, quality control, clustering, cell type annotation, and downstream analysis of these data sets (Fig. 1). Cell types were reannotated by mapping to the reference data set from the Allen Institute for Brain Science; only those cells with a cell identity score above 0.95 were retained (for details, see Methods). After gene and RTE quantification, we examined the RTE dynamics in various dimensions, including their landscape in neurodegenerative diseases, cell type specificity, and disease-stage specificity. Furthermore, we built a comprehensive web portal, scARE, for searching, browsing, and analyzing RTE dynamics and regulation in neurodegenerative diseases at the single-cell level (Fig. 1).
An overview of the single-cell atlas of retrotransposable elements in neurodegenerative disease (scARE) framework: pipeline, analysis, and web portal. The retrieved single-cell transcriptome profiles underwent systematic processing via a customized pipeline, followed by analysis focusing on three aspects: the landscape of retrotransposable elements (RTEs) in neurodegenerative diseases, their cell type specificity, and disease specificity (discovery component). The scARE database provides search and browse options, along with six analytical modules, including highly expressed RTEs, differential expression between disease conditions, differential expression between cell types, correlated expression between RTE and gene, correlated expression between RTE and gene list, and coexpression networks.
Landscape of RTE expression in the human brain
In this study, we downloaded TE annotations from the RepeatMasker track of the UCSC Table Browser (Karolchik et al. 2011), focusing on repeat elements in the classes of LINEs, SINEs, and LTRs for the following analysis. Approximately 42% of the human genome comprises RTEs. Among the RTE types, SINEs were the most frequently observed, as measured by copy number (Fig. 2A), whereas LINEs had the greatest total genomic length (Fig. 2B; Supplemental Table S2).
Landscape of RTE expression. (A) Proportion of copy number of each RTE class (inner circle), family (middle circle), and subfamily (outer circle). (B) Proportion of the sequences of each RTE class (inner circle), family (middle circle), and subfamily (outer circle) in the human genome. (C) Correlation between mean expression of RTE subfamilies across all data sets and their copy number, colored by the RTE class. (D) Correlation between the mean expression of RTE subfamilies across all data sets and their total length, colored by the RTE class. (E) Proportion of RTE reads mapped to short interspersed nuclear element (SINE), long interspersed nuclear element (LINE), and long terminal repeat (LTR) families in each data set. (F) RTE subfamilies with the highest expression levels in the LINE class. The size of the circle indicates the percentage of cells expressing RTE. The extent of color reflects the mean expression across all cells in each data set.
We then performed a correlation analysis of the mean expression (averaged from all brain cells in this study), including normal and diseased cells of RTEs in the brain, with the number of copies or total length of RTE subfamilies (Fig. 2C,D). As expected, RTEs with a greater number of copies or longer total length exhibited higher mean expression (Fig. 2C,D). Furthermore, we observed a stronger correlation between RTE expression and copy number (R2 = 0.76, P < 2.2 × 10−16) than between RTE expression and RTE sequence length (R2 = 0.73, P < 2.2 × 10−16) (Fig. 2C,D). Notably, certain RTE subfamilies, such as L1M4c, exhibited expression levels (4.88 in data set AD_HS_00001) that significantly deviated from the predicted values based on linear regression (1.65 and 1.87 for copy number and total length, respectively). The proportion of reads mapped to SINEs was disproportionately high in the human brain, as evidenced by our findings (Fig. 2E; Supplemental Table S3). In 10 out of the 12 data sets (AD_HS_00003.1 and AD_HS_00003.2 originating from the same study), >50% of the reads were aligned to SINEs. SINE elements represented 44.48% of the total number of RTE copies and 30.43% of the total length of RTEs in the human genome.
To further explore the characteristics of RTEs in the human brain, we extracted LINEs, SINEs, and LTRs with the highest average expression in each data set and selected the top 10 from each of these classes for further analysis (Fig. 2F; Supplemental Fig. S1). As shown in Figure 2F and Supplemental Figure S1, our analysis revealed that the expression patterns of the most highly expressed RTEs exhibited considerable consistency across diverse samples. Although the majority of these highly expressed RTEs were characterized by high copy numbers and longer lengths, distinctively characteristic RTEs were identified within each class. For example, L1M4c, despite being ranked the 77th in total length among 185 LINEs and the 80th in copy number, was among the top 10 most expressed RTEs in nine data sets (Fig. 2F). These results indicate that although copy number and total length had a significant influence on RTEs expression, such factors may not universally govern this regulatory process.
In this study, we used default parameters of scTE to detect RTE expression. scTE also provides a no-intron mode to exclude all TEs inside the intronic regions of genes (He et al. 2021). We explored this feature in our analysis using two data sets, AD_HS_00001 and AD_HS_00002 (Supplemental Fig. S1D–F). We first compared the cell numbers identified in our pipeline with or without no-intron mode (Supplemental Table S4). In AD_HS_00001, there was a substantial decrease of 31.7% in detected cell numbers, whereas in AD_HS_00002, the decrease was 10.9%. We further inspected the proportion of reads mapped to SINEs, LINEs, and LTRs. A slight difference was observed between the two modes (Supplemental Fig. S1C). Regarding the most expressed RTEs, SINEs showed the highest consistency between the two modes, with eight commonly expressed SINEs in the data set AD_HS_00001 and nine in the data set AD_HS_00002. In contrast, LINEs exhibited a least consistency, with six commonly expressed LINEs in both data sets (Supplemental Fig. S1D–F). This preliminary analysis indicated the potential variability in analyzing RTE in different categories. Therefore, we only reported the results using the default parameters (without no-intron mode) for RTE detection below.
Cell type specificity of RTE expression in normal brain samples
Various studies have reported the site-specificity of RTEs, including ERVs and L1, in the normal brain, suggesting their potential roles in brain development (Suarez et al. 2018; She et al. 2022). To determine RTE dynamics in various cell types, different gene markers for brain-specific cells were retrieved from previous studies to identify distinct expression profiles. For example, SYT1 has been used as a marker for neurons (Geppert et al. 1994), MBP for oligodendrocytes (Mathys et al. 2019), PDGFRA expression for oligodendrocyte precursor cells (OPCs) (Traiffort et al. 2016), and AQP4 for astrocytes (Fig. 3A,B; Mathys et al. 2019). To explore the cell type specificity of RTE expression across diverse cell types in the human brain, we first examined data set 1 (AD_HS_00001, source data from ROSMAP) as an example. We chose four cell types—neurons (including excitatory and inhibitory neurons), astrocytes, oligodendrocytes, and OPCs—because they had good number of cells for the analysis. Furthermore, we exclusively incorporated control samples to avoid the potential confounding effects of disease status. As shown in Figure 3C, RTEs were dynamically expressed across these cell types. L1M3d was found to have higher expression in neurons than in other cell types (Fig. 3C; Supplemental Fig. S2A), whereas MER31B had higher expression in oligodendrocytes (Fig. 3C; Supplemental Fig. S2B), MER101B showed higher expression in the OPCs (Fig. 3C; Supplemental Fig. S2C), and HUERS-P2 was significantly expressed in astrocytes (Fig. 3C; Supplemental Fig. S2D). The above results revealed that RTEs are dynamically expressed in different brain cells, indicating their potential roles in shaping cell function and identity.
Cellular heterogeneity of RTEs. (A) Uniform manifold approximation and projection (UMAP) plot of cells in normal patients in data set AD_HS_00001 colored by cell type. Excitatory and inhibitory neurons were combined as neurons. (Ast) Astrocytes, (Oli) oligodendrocytes, (OPC) oligodendrocyte precursor cells. (B) Examples of well-known gene markers of neurons, Oli, OPCs, and Ast. Colors represent the expression of genes in the corresponding cell types. (C) RTE markers identified in this study for neurons, Oli, OPCs, and Ast. (D) Expression of L1MD2 in AD_HS_00001. (E) UMAP of cells in normal individuals in data set AD_HS_00003.2, colored by cell type. (F) Expression of L1MD2 in data set AD_HS_00003.2. (G) Boxplot of expression of L1MD2 in data sets AD_HS_00001 and AD_HS_00003.2. Color represents cell type (neurons vs. nonneurons). (H) Dot plot showing signature RTE expression and percentage of cells for neurons, OPCs, Oli, and Ast in the data set AD_HS_00001.
Because of the strong dynamic nature of RTE expression and the drop-out issue in the data sets generated by 10× Genomics platforms, RTEs identified as highly expressed in one data set did not always demonstrate consistently significant expression levels in other data sets (Supplemental Fig. S3A–H). For example, L1MD2 was identified to be upregulated in neurons in data set AD_HS_00003.2 (entorhinal cortex) (Fig. 3E,F), but it had no significant difference in data set AD_HS_00001 (dorsolateral prefrontal cortex) (Fig. 3D,G).
To further explore RTE expression patterns in different brain cells, we performed differential expression analysis of RTEs across cell types in most of the data sets included in this study, but excluding the samples from the whole brain and midbrain (MS_HS_00001, MS_HS_00002, and PD_HS_00001) (Supplemental Table S1). We ranked upregulated RTEs in astrocytes, neurons, OPCs, and oligodendrocytes for each data set and then calculated the average ranking of RTEs across different data sets. The top 20 differentially expressed RTEs in each of the four cell types were considered cell type signature RTEs (Fig. 3H; Supplemental Fig. S3). Most of the signature RTEs were from L1, ERV1, ERVL, and Alu subfamilies (Fig. 3H), which is largely consistent with the number of copies of these subfamilies (Fig. 1A,B).
Previous studies have highlighted the important role of L1s in diseased and healthy human brains. TDP-43 regulates genomic L1 accessibility and expression in neurons (Liu et al. 2019; Copley and Shorter 2023) and is widely observed to accumulate in neurodegenerative diseases (Jo et al. 2020). Genome-wide L1 insertion studies of neurons in the healthy human brain suggested that neuronal L1s might contribute to phytopathology and susceptibility to different neurological disorders and also contribute to somatic mosaicism (Coufal et al. 2009; Evrony et al. 2012; Zhao et al. 2019). Our results supported the significance of L1s in neurons, as all the 20 signature RTEs of neurons are L1s of the LINE class (Fig. 3H). Furthermore, 18 ERV1s were found in the signature list, which is the second most abundant RTE family among the identified cell type–specific RTEs (Fig. 3H). A previous systematic analysis of physiologically expressed HERVs using bulk RNA sequencing data revealed site-specific expression in human brain subregions, especially in the cerebellum (She et al. 2022). Additionally, bulk RNA sequencing studies have reported the substantial expression of HERVK (HML-2) in different regions, including the frontal cortex, in the normal brain. This finding implies immunomodulatory effects that contribute to inflammation in neurodegenerative diseases (Burn et al. 2022; Nevalainen et al. 2023). This list also encompasses other RTE families such as HERVK (Sun et al. 2018; Copley and Shorter 2023) and Gypsy (Guo et al. 2018), which may be linked to neurodegenerative diseases. Despite the predominantly high expression of most signature RTEs across the corresponding cell types in majority of the data sets, discrepancies in the significance of RTE expression across different data sets were also noted (Supplemental Fig. S3). This variability underscores the intricate regulation and expression of RTEs, indicating the need for additional research to elucidate their underlying mechanisms. Notably, the expression patterns of HERVs vary across different human body sites based on age, sex, and ethnicity (She et al. 2022), which underscores the need to consider various covariates, including brain region, age, history of drug treatment, ethnic background, and genetic factors, to offer a more comprehensive understanding of RTE dynamics within the framework of neurodegenerative diseases.
RTEs are dynamically expressed in neurodegenerative diseases
We further examined the dynamics of RTEs across three neurodegenerative diseases. We focused on excitatory neurons because of their relatively high abundance (e.g., number of cells with measurable RTE level) and their frequent role as primary targets in the pathogenesis of neurodegenerative diseases. To mitigate the potential confounding effects of various brain regions and cell types, our analysis focused on excitatory neurons in three data sets to explore RTE dynamics, which included AD_HS_00003.1, PD_HS_00001, and MS_HS_00002 in AD, PD, and MS, respectively.
We identified 23 upregulated RTEs in the excitatory neurons of AD, including AluYa5, AluYb8, L1HS, and LTR29 (Fig. 4A; Supplemental Table S5). Notably, three of the top five most upregulated RTEs were primate specific (AluYa5, AluYb8, and L1HS) (Storer et al. 2021), indicating human-specific dysregulation of RTEs in AD. The Alu family, including AluYa5 and AluYb8, is reported to have the most inserted RTEs in AD (Ramirez et al. 2024), which are among the most active members. In addition, 311 RTEs were downregulated in the neurons of AD brain (Fig. 4A; Supplemental Table S5). Among the 20 neuron-signature RTEs identified previously, 12 were notably downregulated in AD, whereas the remaining eight showed no significant differential expression. (Fig. 4A; Supplemental Table S5). Further analysis suggested a potential association between the expression of certain RTEs and AD stage; for instance, L1PBa was less highly expressed in severe patients (Fig. 4D; Supplemental Fig. S4A), whereas AluYb8 expression level increased by symptom severity (Fig. 4A,D; Supplemental Fig. S4B), suggesting the potential regulatory role of RTEs in AD progression.
RTEs are dynamically regulated in neurodegenerative diseases. (A–C) Volcano plots showing differentially expressed RTEs in excitatory neurons of three diseases—(A) AD, (B) PD, and (C) MS—versus their matched normal condition. (D) L1PBa expression decreases as AD progressed, whereas AluYb8 expression shows the opposite trend. (E) MSTA-int shows higher expression in the neurons of the PD brain. (F) L3 has lower expression in neurons from the MS brain, and AluYa5 displays the opposite trend.
Furthermore, in the excitatory neurons of MS, the RTE expression showed different dynamics (Fig. 4C,E; Supplemental Fig. S4D). A total of 24 RTEs were upregulated in MS (Fig. 4C). Among them, the top five most significantly upregulated RTEs belonged to the Alu family, including AluYa5 and AluYb8, which were also upregulated in AD (Fig. 4A,C). Similar to the RTE dynamics in AD, 370 RTEs were found to be downregulated in MS (Fig. 4C; Supplemental Table S6). For example, L3 was significantly downregulated, whereas AluYa5 was upregulated in cells from the MS patients (Fig. 4F; Supplemental Fig. S4A). In contrast to the extensive changes observed in AD and MS, only three RTEs were found to be significantly altered in PD. Among these three RTEs, MSTA-int, a member of the LTR family, was the most upregulated (Fig. 4B,E).
Overall, our analysis revealed substantial RTE alterations within the context of AD and MS brain samples but not much in PD. Notably, the majority of the differentially altered RTEs were downregulated in the neurons of AD and MS patients. Furthermore, our findings highlighted the differential RTE expression landscapes not only between AD patients and healthy controls but also among AD patients at different stages of the disease (Fig. 4A,D).
The significant alteration of gene expression in AD and the identification of gene markers of neurons with AD suggested that the AD stage could be inferred from gene expression patterns. Similarly, we hypothesized that the disease stage could be deduced from RTE dynamics if RTE expression is correlated with the AD stage. To test this hypothesis, we initially employed the most differentially expressed genes as features to predict the AD stage using machine learning approaches. We employed four machine learning algorithms without the assumption of Gaussian distribution, including radial basis function (RBF) kernel support vector machine (SVM), random forest (RF), adaptive boosting (AdaBoost), and multiple layer perceptron (MLP). Considering that 730 RTEs were detected in excitatory neurons of data set AD_HS_00003.1, we extracted the top 730 most differentially expressed genes as the features in this step to maintain a consistent feature dimension. To calculate the confusion matrix, we randomly selected 80% (20,821) cells as the training data set and 20% (5206) cells as the testing data set. Meanwhile, all cells were used for 10-fold cross-validation. As a result, RBF SVM, RF, AdaBoost, and MLP demonstrated notable prediction performance with area under the ROC curve (AUC) values of 0.96, 0.95, 0.90, and 0.96, respectively, based on 10-fold cross-validation (Supplemental Fig. S5A,C).
Next, we extended our analysis by using the RTE expression as a predictive feature in the RBF SVM, RF, AdaBoost, and MLP. Our results demonstrated the potential of RTEs in distinguishing AD stages. When we used RTE expression as a feature, the models correctly predicted sample labels (stage 0, stage 2, and stage 6) for most cells, with minimal mislabeling observed between stages 0 and 6 (Fig. 5A). Notably, among the 1861 stage-0 cells and 1425 stage-6 cells, only 100 cells were mislabeled between stage 0 and stage 6 by the RBF SVM prediction (Fig. 5A). The prediction performance measured by AUC for identifying AD stage-based RTE expression reached 0.93, 0.90, 0.86, and 0.92 by RBF SVM, RF, AdaBoost, and MLP, respectively (Fig. 5B). Moreover, we extracted the mean decrease in impurity from the trained RF model as the feature importance (Fig. 5C). As expected, most of the top-ranked RTEs were significantly differentially expressed between the AD and control groups (Fig. 4A,B).
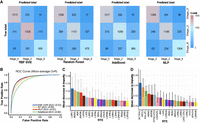
RTE expression profile predicts the disease stage using machine learning algorithms. (A) Confusion matrix for predicting the disease-stage label of excitatory neurons using RTE expression profile by radial basis function kernel support vector machine (RBF SVM), random forest (RF), adaptive boosting (AdaBoost), and multiple layer perceptron (MLP). (B) Tenfold cross-validation receiver operating characteristic (ROC) curve for predicting the disease-stage label of excitatory neurons using RTE expression profile by RBF SVM, RF, MLP, and AdaBoost. (C) Feature importance extracted from the RF model in A. (D) Feature importance extracted from the RF model built with a combination of RTE and gene expression selected by recursive feature elimination (RFE). Only excitatory neurons were considered in this figure.
We further investigated whether comparison of RTE expression with gene expression could improve the inference of the disease stage of AD. We applied recursive feature elimination (RFE) with an RF estimator tailed with RF predictor to select the most informative genes/RTEs from the most variable genes/RTEs. We investigated the optimal number of features to select (n) and the selection pool, which includes N genes and N RTEs (Supplemental Fig. S5D). We observed that the prediction accuracy evaluated by 10-fold cross-validation started to converge when n ≥ 200 and N ≥ 200, and it reached a median accuracy of >0.82 (Supplemental Fig. S5D). With this parameter setting, we extracted feature importance from the RF model and had an intriguing finding: Among the top 20 most significant features extracted from the RF model, 12 were RTEs. This emphasizes the notable contribution and relevance of RTEs in the prediction process (Fig. 5D). Our findings also highlighted the important role of noncoding RNAs, including BCYRN1, MALAT1, and MEG3, in AD, which has been reported in previous studies (Yao et al. 2016; Zhang et al. 2021; Balusu et al. 2023). MEG3 was recently reported to activate necroptosis in AD neurons (Balusu et al. 2023). These studies support the important role of RTEs in AD, especially RTEs that are ranked top in the importance list (Fig. 5D).
scARE: a web portal for exploring and discovering RTE expression patterns
In this study, we quantified genes and RTEs expression using single-cell transcriptome profiles from 12 studies related to neurodegenerative diseases, including AD, PD, and MS. Our analysis revealed systematic dysregulation of RTEs in neurodegenerative diseases, especially in AD and MS. To facilitate the exploration of RTE dynamics in neurodegenerative diseases, we implemented scARE, a single-cell atlas of RTEs in neurodegenerative diseases. scARE offers extensive functionalities for users to search, browse, and analyze RTE expression patterns (Fig. 6A).
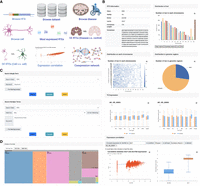
scARE for exploration of RTE information and heterogeneity in three neurodegenerative diseases. (A) The home page of scARE displays prompt access to the major functions. (B) RTE view of the scARE. It contains seven major components: basic information, distribution of loci across chromosomes, distribution of loci within each chromosome, distribution of loci in different genomic regions, mean expression across data sets, and analytical module of expression correlation. (C) The search page of the scARE. Users can search for RTEs by using single or multiple terms in the fields of RTE, gene, data set, and disease. (D) Browse by RTE options. Four different options to browse RTEs by “RTE,” “cell type,” “disease,” and “data set.”
The scARE web portal has three main functions for exploring RTEs: RTE view (Fig. 6B), data set view (Supplemental Fig. S5A), and cell type view (Supplemental Fig. S5B). In the RTE view, we provide a summary of RTEs, including their classification, number of loci, consensus sequence (if available), and distribution in the genome and genomic regions (Fig. 6B). Beyond the basic information, we compiled an overview of the expression patterns of RTEs across eight primary cell types within all data sets and a module for conducting expression correlation analysis between user-selected RTEs/genes (Fig. 6B). Additionally, the data set view summarizes the sample information from the studied data sets, total number of cells in these samples, cell clustering, RTE, and gene expression measured by the uniform manifold approximation and projections (UMAPs) in various cell types (Supplemental Fig. S5A). Moreover, the cell type view consolidates information on cell types, providing links to data sets containing specific cell types, along with the total number of cells categorized by disease, data set, and RTEs (Supplemental Fig. S5B). To access these data views, we implemented various search and browse options (Fig. 6C,D). As depicted in Figure 6C, users can search for different data views by choosing search fields including “RTEs,” “genes,” “diseases,” and “data sets.” Furthermore, we implemented four browse functions: “RTE,” “cell type,” “disease,” and the “data set” (Fig. 6D).
We implemented six analysis modules to explore highly expressed RTEs, differentially expressed RTEs, and expression correlation and coexpression networks (Supplemental Figs. S6, S7). The highly expressed RTE module was designed to explore the RTEs with the highest expression in user-defined cell types and disease conditions (Supplemental Fig. S7A). The results can be easily exported to tables for further analysis. We implemented two differential expression analysis modules to perform differential expression analysis of RTEs between disease conditions or cell types (Supplemental Fig. S7B,C). In addition, two modules were implemented to explore the expression correlation of a RTE with a gene or a list of genes (Supplemental Fig. S7D,E). In these two modules, users can define RTE and gene(s) in comparison, disease condition, cell type, and specific data set to calculate the correlation coefficient. Consequently, scatter plots and boxplots for the defined parameters are generated and available for downloading. Finally, we built two coexpression networks based on differentially expressed genes in AD versus the control, in one cell type versus other cell types in AD. More details are provided in the Methods section (Supplemental Fig. S7F).
Discussion
Neurodegenerative diseases pose a significant health challenge. Understanding the molecular mechanisms underlying these conditions is crucial for the development of therapeutic strategies (Gammon 2014; Heemels 2016; Gitler et al. 2017). Several studies have illuminated the potential significance of RTEs in the context of neurodegenerative diseases, although such evidence requires extensive validation (Krug et al. 2017; Guo et al. 2018; Tam et al. 2019; Chang and Dubnau 2023). Recently, nanopore sequencing technology has highlighted the involvement of various RTE loci in modulating structural variants and methylation patterns within repetitive sequences in AD that can contribute to its pathology (Ramirez et al. 2024). However, single-cell dynamics of RTEs in neurodegenerative diseases remain largely unexplored. In this study, we analyzed single-cell transcriptome profiles from 12 data sets covering AD, PD, and MS, aiming to elucidate the dynamics of RTEs across different cell types and disease states. Our results highlight cell type–specific and disease-specific regulation of RTEs, offering a detailed single-cell-level landscape of RTE expression in normal and diseased brain cells. Our analysis highlighted the cell type–specific expression patterns of three ERV families of RTEs, MER31B in oligodendrocytes, MER101B in OPCs, and HUERS-P2 in astrocytes, underscoring their potential relevance to cellular functions. Previous studies have shown an association between the ERV and the pathology of amyotrophic lateral sclerosis (ALS) (Douville et al. 2011; Li et al. 2015; Chang and Dubnau 2023), MS (Bhetariya et al. 2017; Morandi et al. 2017), and AD (Guo et al. 2018; Sun et al. 2018).
We further identified and ranked differentially expressed RTEs across different data sets of AD and highlighted the delineation of cell type specificity of RTE subfamilies. Our analysis, which considered all control samples from the cortex regions, prioritized the marker RTEs uniquely associated with neurons, oligodendrocytes, OPCs, and astrocytes. Notably, L1s were prominently featured in neurons, as all signature RTEs identified in neurons belong to the L1 family. The direct targeting of L1 promoters revealed that elevated expression of L1s in ataxia-telangiectasia patients caused severe neurodegeneration and was found to be sufficient to cause the disease (Takahashi et al. 2022b). This observation might strengthen the regulatory role of L1 subfamilies through tau pathology in neurons, considering the role of L1 elements in clinical–pathological conversion was recently reported in AD by Macciardi et al. (2022) and Takahashi et al. (2022a).
Next, we investigated RTE expression dynamics in three neurodegenerative diseases, with a focus on excitatory neurons. Our initial analysis revealed a substantial decrease in RTE expression in excitatory neurons in AD. This finding aligns with the previous research and suggests a potential association between epigenetic regulation and cellular responses specific to RTEs in this particular cell type (Dembny et al. 2020; Jönsson et al. 2021; Pascarella et al. 2022; Copley and Shorter 2023). Furthermore, our examination revealed varying expression patterns of RTEs within excitatory neurons in both PD and MS. The decreased expression of L1 subfamilies and increased expression of Alu within neurons of MS postmortem brains align with observations in AD. This consistency may suggest the similar regulation of some RTE expression in neurodegenerative diseases. Our comparative analysis of the disease-specific expression of RTEs in AD, PD, and MS offers insights into the intricate dysregulation of RTEs under these conditions. Additionally, our machine learning prediction of AD stages using features derived from RTE and gene expression provided further evidence supporting a correlation between RTE expression and AD progression.
In summary, our study proposes a comprehensive single-cell-level landscape of RTE expression in neurodegenerative diseases, especially AD. Our findings revealed the cell type specificity of RTE expression and underscored the importance of investigating RTEs in neurodegenerative diseases. The scARE web portal offers a valuable resource for the community to further investigate RTE expression patterns across multiple neurodegenerative diseases, potentially guiding future research and therapeutic development. We examined RTE expression at the subfamily level. It is worth noting that recent tools, such as MATES (Wang et al. 2024) and SoloTE (Rodríguez-Quiroz and Valdebenito-Maturana 2022), have unique features for identifying dynamically expressed RTEs at the locus level. Additionally, because of the limited availability of snRNA-seq data sets generated from postmortem brains of MS and PD individuals, we were not able to examine and compare the dynamic of RTEs in their multiple brain regions. With rapid advancements in single-cell sequencing technology and analytical tools, investigating loci-specific RTE expression dynamics holds great promise for unveiling the hidden roles of RTEs in neurodegenerative diseases. Although our study provides insights into cell type–specific RTE dysregulation in neurodegenerative diseases, we envision further directions and enhancements in our work. First, because the current brain sc/snRNA-seq data were primarily generated from a European-American population, our results should be interpreted with caution. Systematic analysis of sc/snRNA-seq data from other ethnic populations is required to assess population-specific features. Second, we plan to incorporate other modalities at the single-cell level, such as single-cell ATAC sequencing (scATAC-seq), to investigate the potential regulatory roles of RTEs. Finally, our future endeavors will include leveraging a novel algorithm to delve deeper into the locus-specific expression dynamics of RTEs in neurodegenerative diseases.
Methods
Data collection and processing
We collected sc/snRNA-seq data sets from human postmortem brains of three major neurodegenerative diseases. Specifically, we obtained eight data sets for AD (GEO/synapse accession: syn18485175, GSE138852, GSE147528, syn21125841, GSE157827, GSE129308, GSE174367, GSE167494), two data sets for PD (GEO accession: GSE157783, GSE161045), and two data sets for MS (GEO/NCBI BioProject accession: GSE180759, PRJNA544731) from the synapse (https://adknowledgeportal.synapse.org) and NCBI (Supplemental Table S1; https://www.ncbi.nlm.nih.gov/). 10× Genomics Cell Ranger 7.0.0 (Zheng et al. 2017) was utilized to align the sc/snRNA-seq reads to the human genome (hg38) obtained from GENCODE (Frankish et al. 2021).
Genes and RTE quantification
The annotations for human genes and TEs were obtained from GENCODE (Frankish et al. 2021) and UCSC Genome Browser RepeatMasker track (Karolchik et al. 2011), respectively, to build the genome index for Cell Ranger and scTE. For each scRNA-seq data set, the binary alignment map (BAM) file obtained from Cell Ranger was used to quantify human genes and RTEs at the subfamily level (interchangeably used as RTEs in this paper) using the scTE pipeline with default parameters (He et al. 2021).
Quality filtering and cell annotations
To ensure data quality and reliability, we performed quality filtering of the expression matrix obtained from the quantification step using Seurat (v.4.3.0) (Hao et al. 2021). Specifically, we retained cells with 200 to 2500 features and with <5% mitochondrial reads. To ensure comparability across data sets, we applied standardized cell annotation procedures to all collected sn/scRNA-seq data sets with RTE quantification. The predicted cell types were determined for each cell using the Seurat reference-mapping pipeline (Hao et al. 2021). The SCT-normalized gene expression of each query data set was mapped to a reference snRNA-seq data set of five healthy control samples of the middle temporal gyrus (MTG) from SEA-AD (Gabitto et al. 2023). The reference data set contained eight major cell types: astrocytes, endothelial, excitatory neurons, inhibitory neurons, microglia, oligodendrocytes, OPCs, and vascular and leptomeningeal cells (VLMCs). We excluded cells with a predicted cell type score of less than 0.95 for the downstream analysis.
Normalization and differential analysis
The quality-filtered object was log-normalized and scaled using the NormalizeData and ScaleData functions of Seurat. UMAP dimension reduction was performed using the RunUMAP function of Seurat. Because the backend of our website was built with Python CGI, we extracted normalized and raw read counts, along with UMAP dimension reduction plots, to CSV tables and transferred them to SCANPY 1.9.2 for the following process (Wolf et al. 2018). Differential expression analysis between cell types and diagnoses was performed using the rank_genes_groups function of SCANPY.
Identification of cell type–specific RTEs
To obtain cell type–specific RTEs, we merged two neuron cell types, excitatory and inhibitory neurons, as neurons. Moreover, a previous study indicated that snRNA-seq data are not suitable for the detection of microglial activation genes in humans (Thrupp et al. 2020). To avoid potential artifacts, microglia were excluded from this analysis. Moreover, endothelial cells and VLMCs were excluded because of insufficient cell numbers. This procedure led to follow-up analysis of four cell types: neurons, astrocytes, OPCs, and oligodendrocytes. To minimize the effect caused by different disease conditions and brain regions, we only considered control samples while excluding samples from the whole brain and midbrain. To identify cell type–specific RTEs, we extracted a score table generated by the rank_genes_groups function of SCANPY, grouped them by cell type, and ranked the RTEs for each cell type in each data set. Finally, we summarized the top-ranked RTEs by averaging the ranks across different data sets, and generated a signature RTE list for each cell type.
Identification of differentially expressed RTEs in disease versus control
For each of the three diseases—AD (AD_HS_00003.1), PD (PD_HS_00001), and MS (MS_HS_00002)—differential expression was determined by running rank_genes_groups function of SCANPY grouped by diagnosis. In particular, Braak stage-2 and stage-6 samples in data set AD_HS_00003.1 were marked as AD when calculating differentially expressed RTE between AD and control. The adjusted P-value and log2 fold change were extracted from the output. RTEs with an adjusted P-value < 0.01 and |log2 fold change| > 0.5 were regarded as statistically significant.
Prediction of disease stage from RTE and gene expression
Because AD_HS_00003.1 contained disease-stage information, we extracted the gene and RTE expression in excitatory neurons from this data set. To calculate the confusion matrix, we randomly selected 80% of the cells for the training data set and the remaining 20% for the evaluation data set, whereas all cells were used for 10-fold cross-validation. We utilized four machine learning algorithms without Gaussian distribution assumption, including SVM, RF, AdaBoost, and MLP. Python package scikit-learn 1.2.1 was used to construct the machine learning models. We initially explored the predictive power of RTE expression and gene expression by building a model with two types of features: (1) the expression profile of 730 detected RTEs and (2) the expression profile of the top 730 most differentially expressed genes. We set robust parameters for each algorithm to ensure optimal performance and accuracy: SVM—kernel set to RBF; decision function shape set to “ovr”; AdaBoost—the estimator number set at 100; RF—estimator number set at 100; and MLP—alpha = 1, max_iter = 10000, hidden_layer_sizes = (1000,1000). The confusion matrix is then calculated using the test data set. The micro-average one versus rest (OvR) ROC curve of 10-fold cross-validation was calculated as a performance evaluation. RFE was performed using the RFE function of the sklearn.feature_selection package. The feature importance (mean decrease in impurity) was extracted from the RF models using the RandomForestClassifier.feature_importances_ function. The standard deviation of feature importance was calculated from the feature importance in each tree of the RF model.
Construction of web portal of scARE
The scARE web portal was built using JavaScript, Python, and MySQL. Bootstrap V4.0 was used to organize the webpage components. jQuery 3.7 was utilized for query backend, event handling, and DOM manipulation. Highcharts JS v11.0.1 and plotly.js v2.20.0 were employed to generate and display charts; D3 v7.8.4 was used to generate RTE family tree. Python 3.6 CGI was used to build the backend of the scARE, and all data sets were stored in MySQL 5.5.56.
Software availability
All codes for data processing and analysis can be found in the Supplemental Code and at GitHub (https://github.com/bsml320/scARE).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank those investigators who generated and shared the scRNA-seq data sets used in this work. We thank all the members of the Bioinformatics and Systems Medicine Laboratory, especially Dr. Hyun-Hwan Jeong, Dr. Yulin Dai, and Mr. Wendao Liu, for the initiation of the project, valuable discussion, and insightful suggestions in this work. This research was supported by National Institutes of Health (NIH) grant R03AG077191. Z.Z. was partially supported by NIH grants (U01AG079847, R01LM012806, and R01LM012806-07S1). We thank the technical support from the Cancer Prevention and Research Institute of Texas (CPRIT RP180734 and RP240610). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: Conceptualization was by Z.Z. Methodology was by W.D., C.C., and Z.Z. Formal analysis was by W.D., C.C., and A.L. Investigation was by W.D. and C.C. Resources were by W.D. and C.C. Data curation was by C.C. Writing of the original draft was by W.D. and C.C. Reviewing and editing were by W.D., C.C., and Z.Z. Visualization was by W.D. Supervision was by Z.Z. Project administration was by Z.Z. Funding acquisition was by Z.Z.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279363.124.
- Received March 17, 2024.
- Accepted September 18, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








