
Systematic identification of interchromosomal interaction networks supports the existence of specialized RNA factories
- 1Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA;
- 2Paul G. Allen School of Computer Science and Engineering, University of Washington, Seattle, Washington 98195, USA;
- 3Molecular Biotechnology Center “Guido Tarone,” Department of Molecular Biotechnology and Health Sciences, University of Turin, 10126 Torino, Italy
Abstract
Most studies of genome organization have focused on intrachromosomal (cis) contacts because they harbor key features such as DNA loops and topologically associating domains. Interchromosomal (trans) contacts have received much less attention, and tools for interrogating potential biologically relevant trans structures are lacking. Here, we develop a computational framework that uses Hi-C data to identify sets of loci that jointly interact in trans. This method, trans-C, initiates probabilistic random walks with restarts from a set of seed loci to traverse an input Hi-C contact network, thereby identifying sets of trans-contacting loci. We validate trans-C in three increasingly complex models of established trans contacts: the Plasmodium falciparum var genes, the mouse olfactory receptor “Greek islands,” and the human RBM20 cardiac splicing factory. We then apply trans-C to systematically test the hypothesis that genes coregulated by the same trans-acting element (i.e., a transcription or splicing factor) colocalize in three dimensions to form “RNA factories” that maximize the efficiency and accuracy of RNA biogenesis. We find that many loci with multiple binding sites of the same DNA-binding proteins interact with one another in trans, especially those bound by factors with intrinsically disordered domains. Similarly, clustered binding of a subset of RNA-binding proteins correlates with trans interaction of the encoding loci. We observe that these trans-interacting loci are close to nuclear speckles. These findings support the existence of trans-interacting chromatin domains (TIDs) driven by RNA biogenesis. Trans-C provides an efficient computational framework for studying these and other types of trans interactions, empowering studies of a poorly understood aspect of genome architecture.
Mammalian interphase chromosomes are exquisitely folded in three dimensions to enable precise regulation of gene expression (for review, see Hafner and Boettiger 2023). The study of such organization has been greatly advanced by sequencing-based chromosome conformation capture (3C) technologies, chiefly Hi-C (Lieberman-Aiden et al. 2009), and by orthogonal imaging approaches (Jerkovic and Cavalli 2021). A rapidly growing body of evidence indicates that although a sizeable portion of 3D genome architecture is relatively invariant across cell types, specific dynamic changes play a critical role in regulating gene expression in different cell types (Duan et al. 2021; Winick-Ng et al. 2021; Schaeffer and Nollmann 2023; Tan et al. 2023) and in disease (Krumm and Duan 2019; Zheng and Xie 2019).
Most of our current understanding of 3D genome architecture centers around chromatin folding within individual chromosomes, that is, on intrachromosomal or cis contacts. These contacts give rise to a variety of hierarchical features at different genomic scales, including different types of DNA loops (i.e., cohesin-mediated looping and promoter-enhancer pairing), topologically associating domains (TADs; submegabase domains of preferential self-interaction) (Dixon et al. 2012), and A/B compartments (chromosome-wide segregation of active/inactive chromatin resulting from sparse intra-TAD interactions driven mainly by phase separation of heterochromatin) (Hildebrand and Dekker 2020). In contrast, interactions across different chromosomes (interchromosomal or trans contacts) are poorly understood.
Chromosome-wide trans genome architecture of nonholocentric chromosomes in eukaryotic species exhibits two nonmutually exclusive features: Rabl-like configuration (i.e., featuring centromere clustering, telomere clustering, and/or a telomere-to-centromere axis) and chromosome territories (Hoencamp et al. 2021). The latter is typical of mammalian chromosomes, which tend to occupy distinct domains of the interphase nucleus (Cremer and Cremer 2010). Although chromosome territories limit the possibility for trans contacts—compared with an alternative model of “spaghetti” DNA fibers (Longo and Roukos 2021)—they do not represent hard boundaries: Regions that overcome territorial topological restrictions engage with each other within mRNA and tRNA factories, polycomb domains, the nucleolus, nuclear speckles, and potentially other nuclear subcompartments (Fig. 1A; Bhat et al. 2021). Some of these trans contacts involve specific loci whose interactions are important to gene regulation in enhancer hubs (Monahan et al. 2019), transcription factories (Osborne et al. 2004, 2007; Papantonis et al. 2012), and splicing factories (Bertero et al. 2019). Despite these and a few other examples, whose discovery was serendipitous or informed by domain-specific prior knowledge (Takizawa et al. 2008; De Wit et al. 2013; Ito et al. 2016), the systematic discovery of functional trans interactions is currently very challenging.
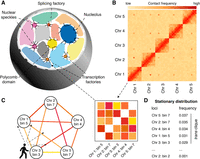
The trans-C algorithm. (A) Schematic of typical interchromosomal genome organization in mammals. Interchromosomal (trans) interactions mainly involve genomic domains that extrude from chromosome territories and engage with a variety of membrane-less structures involved in gene regulation. (B) A Hi-C matrix captures the contact frequency of loci in a genome-wide fashion. Besides intrachromosomal (cis) contacts, specific loci can exhibit strong interchromosomal (trans) contacts among themselves. (C) Trans-C employs a random walk algorithm that traverses the Hi-C contact graph choosing to move to a node (bin) probabilistically based on the strength of the edge (interaction). (D) The output is a list of loci ranked by how frequently they are visited during the random walk: More frequently visited loci interact more strongly in trans as a clique.
One reason for this difficulty is that in a typical Hi-C matrix the number of reads from trans contacts is two to four times smaller than that from cis contacts, depending on cell type and assay type. Furthermore, the number of possible pairs of loci that can interact in trans is also much larger than the number of possible pairs of loci that can interact in cis. Collectively, therefore, trans contact data are typically quite sparse. Most importantly, there is a lack of robust statistical and computational approaches to confidently identify reproducible trans contacts. In particular, available methods are limited to the identification of pairwise trans contacts (Cook et al. 2020) or large patterns of trans contacts across broad subnuclear structures (Joo et al. 2023). There remains an important knowledge gap in detecting smaller, specific sets of trans contacts (cliques) that could underlie important local regulations of DNA and RNA biochemistry.
In this paper, we overcome this limitation by providing a computational framework that systematically finds sets of jointly interacting loci from Hi-C data. The method, trans-C, takes as input a Hi-C contact map as well as, optionally, one or more seed loci and uses a random walk with restart algorithm to identify sets of trans-contacting loci. Trans-C provides a powerful way to uncover and measure various types of trans interactions, empowering both the discovery and hypothesis-driven studies of genome structure–function relationships.
Results
Trans-C randomly walks through the Hi-C graph
Our goal is to algorithmically identify, from a given set of Hi-C data, a collection of genomic loci that exhibit strong trans interactions with each other (i.e., a “clique”). We represent the Hi-C data as a matrix, referred to as a “contact map,” in which each axis corresponds to the complete genome, and entries in the matrix represent Hi-C contact counts (Fig. 1B). In practice, the genomic axes are discretized using fixed-width bins. The bin size is thus inversely proportional to the effective resolution of the contact map. The contact map can be thought of as the adjacency matrix of a corresponding Hi-C graph, in which nodes are genomic loci (bins) and edges are weighted by the corresponding contact counts (Fig. 1C). Our goal is thus to find dense subgraphs in this Hi-C graph.
The problem of dense subgraph discovery arises in many application domains and consequently has been very widely studied. Depending on the exact formulation and the notion of density, theoretical computer science has shown that the problem complexity ranges from easily solved in linear time via a max flow algorithm (Khuller and Saha 2009) to computationally intractable (NP-hard) (Charikar 2000). Common techniques to approximate the latter case, to which our specific problem belongs, are greedy approaches, which iteratively select the best option available at the moment without guaranteeing that this strategy will bring the global optimal result (Charikar 2000), and semirandom models, which account for model errors by incorporating both adversarial and random choices in instance generation (Bhaskara et al. 2010). Trans-C approaches the discovery task of finding strongly interacting loci in trans using a random walk with restart algorithm (Fig. 1C). This general approach has been applied successfully in domains as diverse as web searching (Gibson et al. 2005), protein remote homology detection (Weston et al. 2004), and gene functional prediction (Mostafavi et al. 2008). Prior to the random walk operation, trans-C performs three preprocessing steps on the provided Hi-C contact map. First, to control for sequencing and accessibility biases, Hi-C counts are ICE-normalized (Imakaev et al. 2012). Second, the resulting matrix is processed using a binomial model to estimate interaction P-values based on an empirical null model that accounts for potential biases arising from chromosomal territorialization (i.e., small, gene-rich chromosomes generally occupying the nuclear interior and interacting more with each other than with large, gene-poor chromosomes, and vice versa) (Lieberman-Aiden et al. 2009; Bertero et al. 2019). This step allows the algorithm to focus on interactions that stand out from the noise. Third, the negative log P-values are used as weights for the network edges and subsequently refined using a “donut filter” (Rao et al. 2014) to highlight points that are local maxima. The postprocessed Hi-C interaction matrix is finally represented as a weighted graph, in which each node corresponds to a bin and the weight on each edge corresponds to the negative log P-value computed in the previous step. Trans-C then carries out a random walk with restart algorithm, which exploits global patterns of connectivity on the graph. Each walk is initiated from a randomly selected “seed” locus and moves from a node to a neighboring one probabilistically based on the weight of the edge. A parameter α controls the probability that the walk will restart at a new, randomly selected seed locus. Mathematically, as an infinite number of walks are performed, the frequency with which each node is visited converges to a stationary distribution. This can be computed analytically using the Perron–Frobenius theorem. We use the stationary distribution to obtain a ranked list of trans-interacting bins (Fig. 1D), because the most frequently visited nodes are the ones that interact most strongly with the seed loci. Highly ranked genes are most likely to be functionally related with the seed loci, and therefore, a putative clique is obtained by extracting the top ranked loci.
Trans-C uncovers the clustering of var genes critical for P. falciparum immune evasion
Having developed trans-C, we set out to test its ability to uncover known sets of loci that interact together in trans in three different organisms. First, we focused on the protozoan Plasmodium falciparum, the parasite responsible for the most lethal form of malaria. The three-dimensional organization of the P. falciparum genome is strongly associated with gene expression (Ay et al. 2014), particularly for genes involved in pathogenesis, immune evasion, and master regulation of gene expression (Bunnik et al. 2018). Among these are the variant antigenic repertoire (var) genes, a family of 60 virulence genes responsible for the antigenic variation of the parasite and evasion of the host immune system. Only a single var gene is active at a given time, the other var genes being maintained in a perinuclear cluster of heterochromatic telomeres (Fig. 2A; Duffy et al. 2017). This cluster is an excellent test case to validate the ability of trans-C to uncover a group of biologically important genes that colocalize in 3D from Hi-C data.
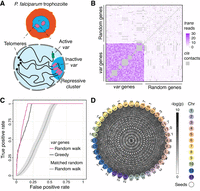
Trans-C identifies the var genes cluster in Plasmodium falciparum. (A) Schematic of P. falciparum in the trophozoite stage of its red blood cell life cycle, with a zoomed-in view of the nucleus highlighting its Rabl-like structure and the clustering of the var genes in a repressive heterochromatic cluster. (B) Contact heat map comparing trans contact counts among all 60 var genes versus 60 randomly selected 10 kb bins. Cis contacts are grayed out. (C) Performance evaluation of trans-C-mediated identification of var gene clustering. We plot the receiver operating characteristic (ROC) curve for the trophozoite life stage of P. falciparum. The var genes are uncovered by the random walk algorithm of trans-C with high area under the ROC curve (AUROC; 0.94). The cumulative distribution is statistically significant (P = 3 × 10−165) from a null model of 1000 random walks performed from seeds selected randomly but with an equal or greater collective interaction strength (matched random; the line reports the average and the shaded area the 95% confidence interval). We also report the performance of a simpler greedy heuristic. (D) Visualization of the var gene–associated trans clique identified by trans-C in P. falciparum trophozoite. Nodes are color-coded by chromosome and sequentially numbered based on their relative position along each chromosome (expressed in megabases). Edges are color-coded based on trans interaction significance (cis contacts are not plotted). The seed loci for the random walk are indicated by solid black lines around the nodes.
To this end, we examined Hi-C for two stages of the P. falciparum life cycle, trophozoite and schizont, both of which are characterized by trans contacts between var genes (Ay et al. 2014). To visually highlight the var cluster (Gardner et al. 2002), we extracted the bins containing var genes and also drew 60 bins at random from the full set of genomic loci. The submatrix of trans contacts formed by the concatenation of the two sets of bins showed a striking contrast between the var and non-var loci, as anticipated (Fig. 2B). Next, we selected three var genes at random to act as seed loci and examined whether trans-C (with α = 0.5) could automatically identify the remaining 57 var gene loci. For comparison, we used a method based on a greedy heuristic that iteratively selects the bins that interact most strongly with the selected loci (Supplemental Methods). For each approach, we plotted a receiver operating characteristic (ROC) curve, in which each element is a genomic bin, labeled as positive (var gene) or negative (other loci) (Fig. 2C; Supplemental Fig. S1A). In both Plasmodium life stages, trans-C quickly found the majority of the var genes by ranking their corresponding bins highly: Of the top 50 bins, 28 contained a var gene, and all 60 var genes were recovered within the top 280 bins. Trans-C outperformed the greedy heuristic baseline, with an area under the ROC curve (AUROC) of 0.94 compared with 0.88 for the trophozoite analyses (Fig. 2C), with similar findings in schizont (Supplemental Fig. S1A). This demonstrates that a random walk approach is more suited to the task of identifying trans cliques even in the context of a clear example.
As a negative control, we run trans-C starting from three seed loci that were randomly reselected until a trio could be identified so that its trans subnetwork showed the same or greater total interaction strength as the one for the three var genes previously used as seeds. We repeated this procedure, which henceforth will be referred to as “matched random control,” for a total of 1000 times in order to estimate empirical P-values for the var gene–associated subnetwork: This proved extremely significant (P = 3 × 10−165 and P = 3 × 10−171 for trophozoite and schizont, respectively) (Fig. 2C; Supplemental Fig. S1A). Visualization of the var gene–associated trans interaction subnetworks for the top 40 loci ranked by trans-C in both plasmodium life cycle stages showcased the intricacy of highly significant contacts (Fig. 2D; Supplemental Fig. S1B).
Identification of Greek islands regulating the expression of mouse olfactory receptor genes
To further validate trans-C, we turned to the mouse and its larger diploid nuclear genome. In mouse olfactory sensory neurons (mOSNs), chromatin regions associated to olfactory receptor gene clusters from 18 chromosomes make specific and robust interchromosomal contacts that increase in strength with differentiation (Lomvardas et al. 2006; Markenscoff-Papadimitriou et al. 2014; Monahan et al. 2017). These contacts are orchestrated by intergenic olfactory receptor enhancers that form a multichromosomal superenhancer driving the monoallelic and stochastic expression of a single mouse olfactory receptor gene (Fig. 3A; Monahan et al. 2019). The mOSN-specific trans contacts are arguably the strongest trans contacts in a mammalian genome known to date. The regions involved in such interactions were dubbed “Greek islands,” because they are sprinkled across the chromosomes as the tiny islands are in the Mediterranean Sea. Importantly, in horizontal basal cells (HBCs), the quiescent stem cell progenitors of mOSNs, these interchromosomal contacts are absent.
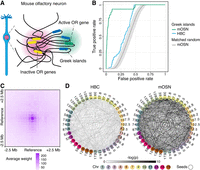
Trans-C identifies the Greek island cluster in mouse olfactory sensory neurons (mOSNs). (A) Schematic of trans contacts in a mOSN. The Greek islands form a multienhancer hub that is segregated from the inactive olfactory receptor (OR) genes. (B) Performance evaluation of trans-C-mediated identification of Greek island clustering. We plot the ROC curve for α = 0.5 in mOSNs versus their progenitors (horizontal basal cells [HBCs]). Trans-C correctly identifies Greek island clustering specifically in mOSNs in a way that is statistically significant (P = 6 × 10−194) versus a matched random seed null model (average and 95% confidence interval from 1000 runs). (C) Aggregated heatmap of trans contacts among the top 60 loci selected by trans-C in mOSNs. Each square in the grid represents an average 250 kb bin in a Hi-C matrix of 21 × 21 bins centered at each interacting pair of loci (reference). The exhibited spot-like structure highlights the highly specific nature of the interchromosomal interactions of the Greek islands. (D) Visualization of the Greek island-associated trans clique identified in mOSNs by trans-C, showcasing the increased significance of loci interactions after differentiation of HBCs, plotted as described for Figure 2D.
We applied trans-C to mOSN Hi-C data (Monahan et al. 2019), randomly selecting five Greek islands from the previously reported list of 63 to use as seeds in order to measure the ability of trans-C to uncover the remaining 58. Besides running a matched random control, as a biological negative control we used HBC Hi-C data. As expected, trans-C successfully found the Greek islands in mOSNs (AUROC = 0.93; P = 6 × 10−194) (Fig. 3B; Supplemental Table S1), although it failed to do so effectively in HBCs (AUROC = 0.71) (Supplemental Fig. S2A). At a false-positive rate of 10%, 95% of known Greek islands were identified in mOSNs, although we speculate that some of the false positives may actually represent previously unidentified Greek islands.
To visually verify whether trans-C detected specific interchromosomal contacts, we selected the top 60 predicted bins from the ranked stationary distribution (30% of which are known Greek islands). For each pair of loci from this set of 60, we extracted from the Hi-C data a 21-by-21 matrix centered at their interaction and then averaged these matrices (Fig. 3C). The resulting contact heatmap exhibited very strong punctuated signal in the middle, suggesting that the top 60 loci ranked by trans-C form specific interactions that are not driven by larger, nonspecific “neighborhood” features. Visualization of the Greek island–associated trans interaction subnetwork for the top 40 loci ranked by trans-C in mOSNs revealed a very dense network that greatly increases in significance when HBCs differentiate in mOSNs (Fig. 3D). Collectively, trans-C efficiently pinpoints trans cliques even in a complex eukaryotic genome.
We also used the Greek island data set in mOSNs to evaluate how strongly the behavior of trans-C depends on its primary parameter, the random walk restart probability α. We varied α between zero no restart to one (restart after every step) in small increments. We observed that the performance of trans-C was stable in the range [0.3, 0.7], whereas it deteriorated significantly in the two extremes when it approached zero or one (Supplemental Fig. S2B). This behavior is expected theoretically: When α is close to zero, the random walk restarts infrequently, and so, its stationary distribution becomes less dependent on the seeding bins and is mostly determined by the topology of the Hi-C graph. At the extreme, when α = 0 the walk is “memoryless” and entirely independent of the starting seed loci. On the other hand, when α is close to one there is little or no exploration along the graph. In this setting, the Hi-C data are essentially ignored, and consequently, no discoveries can be made. As an additional benchmarking, we evaluated the performance of trans-C with respect to the number of trans reads in the input Hi-C matrix. For this, we run trans-C with α = 0.5 using 100% of the trans contacts (about 436 million) versus decreasing subsamples down to about 87 million (Supplemental Fig. S2C). Trans-C maintained a comparable performance when 80% of the matrix was used, and an acceptable performance at 60% subsampling, whereas 40% and 20% of contacts proved insufficient, as could be anticipated. Lastly, we rebenchmarked trans-C against the greedy heuristic: Also in the context of Greek island discovery in mOSNs, our algorithm delivered a larger AUROC (0.93 vs. 0.92) (Supplemental Fig. S2D).
Dissecting the RBM20 splicing factory during cardiomyocyte differentiation
We next sought to explore the sensitivity of trans-C in a more challenging model in the human genome. We previously identified a network of gene loci that increase their association interchromosomally during cardiac development of human pluripotent stem cells (hPSCs) and are targets of the muscle-specific splicing factor RBM20 (Fig. 4A). Functional experiments indicated that the main RBM20 target, the large TTN pre-mRNA (which contains over 100 binding sites for RBM20), nucleates RBM20 foci. Secondary RBM20 targets interact in trans with TTN at RBM20 foci, which maximizes the efficiency of their alternative splicing (Bertero et al. 2019). We therefore dubbed the network a “trans-interacting chromatin domain” (TID) and the resulting structure a “splicing factory.” Of note, however, the cumulative interaction score of the TID calculated from shallow Hi-C data (about 90 million contacts) was only modestly enriched compared with a null model (P = 0.05). Thus, these interactions are less easily detected by Hi-C and are likely to be much more transient in nature compared with those involving the Greek islands.
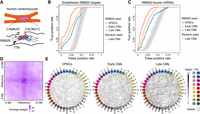
Trans-C identifies the RBM20 splicing factory in human cardiomyocytes (CMs). (A) Schematic of the RBM20 splicing factory, a muscle-specific interchromosomal structure organized by the TTN pre-mRNA. This pre-mRNA binds to more than 100 copies of RBM20 and nucleates foci that engage with other RBM20 targets to promote their alternative splicing (blue arrows). (B) Performance evaluation of trans-C in uncovering the RBM20 splicing factory in early (day 15) versus late (day 80) CMs differentiated from human pluripotent stem cells (hPSCs; also analyzed as “day 0” baseline control). Results for late CMs are statistically significant (P = 5 × 10−122) versus a matched random seed null model (average and 95% confidence interval from 1000 runs). Seed loci and ROC curves are based on a list of established RBM20 targets (Bertero et al. 2019). (C) Similar to B, but seed loci and ROC curves are based on loci directly bound by RBM20 as determined by eCLIP; P = 4 × 10−120. (D) Aggregated heatmap of trans contacts between the top 60 loci selected by trans-C in late CMs starting from eCLIP data. Each square in the grid represents an average 100 kb bin in a Hi-C matrix of 41 × 41 bins centered at each interacting pair of loci extracted from the Hi-C data (reference). The denser region in the middle reveals the specific nature of the trans interactions at the RBM20 splicing factory. (E) Visualization of the RBM20-associated trans clique identified by trans-C in late CMs starting from eCLIP data, showcasing the increased significance of loci interactions during hPSC differentiation and CM maturation, plotted as described for Figure 2D.
We set out to test whether trans-C would reidentify the RBM20 TID in an independent, more deeply sequenced Hi-C data set of hPSC differentiation into the cardiac lineage (about 3 billion read pairs per sample) (Zhang et al. 2019). Besides various progenitors and early hPSC-derived cardiomyocytes (hPSC-CMs), this data set also contains late hPSC-CMs obtained after 80 days of in vitro differentiation. Moreover, older hPSC-CMs were FACS-purified using an expression reporter for the mature marker ventricular myosin light chain 2 (MLC-2v; MYL2 gene). We first attempted to recover the trans network of 16 RBM20 target genes from our original report, using five of them (TTN, CACNA1C, CAMK2D, KCNIP2, CAMK2G) as seeds for trans-C. Figure 4B shows the ROC curve for day 0 (hPSCs), 15 (early CMs), and 80 (late CMs). The best performance was achieved using Hi-C data from day 80 (AUROC = 0.84, P = 5 × 10−122) (Supplemental Table S2); second was day 15 (AUROC = 0.78, P = 2 × 10−105) (Supplemental Fig. S3B); and last was day 0 (AUROC = 0.75, P = 6 × 10−101) (Supplemental Fig. S3A). The improvement in ROC area as differentiation advances is in line with the important role of RBM20 in cardiac maturation (Guo et al. 2012). RBM20 is not expressed at day 0, moderately expressed at day 15, and maximally expressed at day 80. We note, however, that the performance at day 0 was better than random, suggesting that some structure that brings the loci close together is present even in undifferentiated cells. Benchmarking of trans-C against a greedy heuristic demonstrated a strong increase in performance for late CMs (AUROC 0.84 vs. 0.72) (Supplemental Fig. S3C), highlighting the advantages of the approach particularly to find trans cliques that do not stand out strongly from the noise.
Encouraged by these results, we decided to use trans-C to expand our knowledge of the RBM20 TID. Our original list of 16 genes was not the result of an unbiased search but rather reflected our prior knowledge of RBM20 biology: These 16 genes were the known splicing targets of RBM20 in both human and rat hearts that were also upregulated in hPSC-CMs. As an alternative strategy to identify genes involved in the RBM20 TID in an unbiased fashion, we hypothesized that such genes would encode for transcripts most strongly bound by RBM20 and thus enriched within the splicing factory. To test this hypothesis, we leveraged a recent data set that measured RBM20 binding to RNAs using enhanced UV cross-linking and immunoprecipitation (eCLIP) (Van Nostrand et al. 2016).
We used RBM20 eCLIP data from hPSC-CMs (Fenix et al. 2021) and counted the number of peaks that fall in each genomic bin (mapping RNAs to the encoding DNA loci). We selected the five bins with the most eCLIP peaks, which contained the genes TTN, SLC8A1, OBSCN, NEAT1, and LBD3. Using these as seed loci, we ran trans-C with α = 0.5 on Hi-C matrices from differentiating hPSC-CMs (Zhang et al. 2019). Our goal was to test whether trans-C would uncover the remaining 202 bins with eCLIP peaks. We note that this experimental setup is very different from the previous one. Here, we used Hi-C data to find binding sites in an orthogonal eCLIP data set. Moreover, only a single seed locus, TTN, was shared between this analysis and the one reported in Figure 4B. The resulting ROC curves (Fig. 4C) show the same trend: the best performance was at day 80 (AUROC 0.82; P = 4 × 10−120) (Supplemental Table S3), the second at day 15 (AUROC 0.79; P = 1 × 10−106) (Supplemental Fig. S3E), and the last at day 0 (AUROC 0.72; P = 4 × 10−98) (Supplemental Fig. S3D), consistent with biological expectations.
Next, we performed a second performance recall analysis in the same trans subnetwork identified by trans-C from RBM20 eCLIP data, but in which we restricted the list of RBM20 targets to those whose RNA is bound by RBM20 on at least three sites and is differentially spliced in hPSC-CMs with RBM20 knocked out (Fenix et al. 2021). The resulting list of 45 high-confidence RBM20 targets was efficiently recovered in day 80 hPSC-CMs, with AUROC = 0.84 and a P-value of 2 × 10−125 (Supplemental Fig. S3F), a performance improvement compared with the full list of RBM20 bound loci.
Similarly to our observation for trans contacts between the Greek islands (Fig. 3C), the aggregated contact frequency heatmap for loci involved in the RBM20-associated trans interaction subnetworks identified from RBM20 eCLIP data showed a clear punctuated pattern, supporting the spatial specificity of these interactions (Fig. 4D). Visualization of this subnetwork showed that it is quite dense and that it clearly increases in significance when hPSC differentiate in CMs, and even more when CMs mature (Fig. 4E). Similar results were obtained for the subnetwork identified from established RBM20 targets (Supplemental Fig. S3G).
Because the ENCODE Project generated a large number of Hi-C matrices for the left ventricle (LV) (The ENCODE Project Consortium 2012), we used this model to both evaluate the reproducibility of trans-C and determine whether the RBM20 splicing factory could be identified in adult, fully mature cardiomyocytes. We ran trans-C starting from the same five seed bins prioritized using RBM20 eCLIP data, and compared the resulting list of ranked bins: The Pearson's correlation for analyses in the 10 biologically independent LV samples was very high (range 0.77–0.85) (Supplemental Fig. S3H), whereas negative control analyses in noncardiac samples showed a low correlation (range 0.50–0.70) (Supplemental Fig. S3H). The AUROC for recovering high-confidence RBM20-bound mRNAs in LV Hi-C data was high (range 0.73–0.79), further supporting the existence of a measurable RBM20-associated trans clique also in vivo.
In all, we conclude that trans-C captures even weak and/or unstable yet biologically meaningful trans subnetworks associated with RNA biogenesis.
Loci strongly bound by DNA-binding proteins often exhibit significant interactions in trans
The identification of dense subnetworks of trans Hi-C contacts that are enriched for RBM20 targets supports our hypothesis that RNA biogenesis influences 3D chromatin organization by bringing into proximity coregulated nucleic acids, so as to maximize the efficacy and specificity of their processing (Fig. 5A; Bertero 2021). We specifically propose that, as in the case of RBM20, “RNA factories” arise from the clustered binding of trans-acting factors to one or more core coregulated genes and/or their encoded transcripts. These, in turn, recruit accessory targets of the same factors. This hypothesis predicts the existence of both transcription factories specialized for certain transcription factors (TFs) and/or chromatin regulators, and other RNA factories specialized for various RNA-binding and regulatory proteins. We set out to test this hypothesis systematically using trans-C, as an example of its potential applicability to address biological questions.
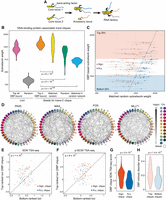
Trans-C identifies DNA-binding protein–associated trans cliques proximal to nuclear speckles. (A) Schematic of the mechanistic hypothesis for the formation of specialized RNA factories involving trans-interacting chromatin domains. Multiple copies of trans-acting regulatory factors (i.e., transcription or splicing factors) bind to core nucleic acids, aggregate to form new clusters and/or enrich pre-existing ones, and recruit accessory coregulated nucleic acids. RNA factories promote the efficacy and accuracy of RNA biogenesis processes (thicker black arrows). (B) Trans-C-identified subnetworks in lymphoblastoid cells built from loci characterized by strong binding of 110 DBPs have dense contacts. We plot the distribution of subnetwork weights for six types of sets of 40 loci: (1) loci with the highest number of ChIP-seq peaks for a given DBP (pink), (2) randomly drawn loci (gray), (3) top loci ranked by trans-C from a seed of five loci with the highest number of ChIP-seq peaks for a given DBP (orange), (4) top loci ranked by trans-C from a random seed of five loci whose starting subnetwork weight was matched to the seed of group 3 (yellow), (5) top loci ranked by trans-C from a seed of five randomly drawn loci (green), and (6) top loci ranked as for group 4 but starting from an interaction matrix that has been randomly shuffled (light blue). On average, sets seeded from loci most strongly bound by DBPs interact more strongly in trans than any of the other five types of sets of loci, including the stringent “matched random” control (P-values by Mann–Whitney U test). (C) For each DBP analyzed in B, we compare the weights of subnetworks obtained with trans-C from “top five DBP-bound” seeds (single data point) and “matched random” seeds (average of 1000 subnetworks ± SD). In red are comparisons with significantly different weights (P < 0.05 after FDR correction). Shaded areas highlight the top and bottom quartile of DBP-based subnetwork weights. (D) Visualization of selected significant DBP-associated trans cliques in lymphoblastoid cells, plotted as described for Figure 2D (PAX5 and MAX, strongest cliques; FOS and MLLT1, highest fold change of clique strength over average strength of cliques in the matched random null model). (E,F) Proximity to nuclear speckles of loci within trans-C-identified cliques, measured as the average SON and p-SC35 TSA-seq signal for the corresponding genomic regions. For each subnetwork, the signal is compared with that of an equal number of loci at the opposite end of the trans-C ranking. DBP-based cliques are overall significantly more proximal to both SON and p-SC35 than matched control sets (P-values by Mann–Whitney U test). Cliques that are significantly stronger compared with the null model in the analysis from panel C (Sign.) are in red, and nonsignificant ones (N.s.) are in blue. (G) The proximity to SON for significant versus nonsignificant cliques from panel C is significantly different by Mann–Whitney U test. (H) The strongest DBP-based subnetworks correspond to DBPs with a higher intrinsically disordered protein (IDP) score. We plot the IDP scores for DBPs resulting in the bottom and top quartiles of DBP-based subnetworks from panel C. The difference is statistically significant by Mann–Whitney U test.
First, we focused on DNA-binding proteins (DBPs), hypothesizing that the genes most strongly bound by a given DBP would be associated with strong TIDs. To test this notion, we used the most deeply sequenced Hi-C data set reported to date: an ultra-deep Hi-C map of human GM12878 lymphoblastoid cells (Harris et al. 2023). The ENCODE Project produced chromatin immunoprecipitation sequencing (ChIP-seq) data for 110 DBPs in this cell line (The ENCODE Project Consortium 2012), providing an ample resource to test our hypothesis in a systematic manner. For each ChIP-seq data set, we counted the number of peaks in each genomic bin.
First, we took the 40 bins with the most peaks for each DBP and calculated the weight of the subnetworks formed by these bins. The distribution of this subnetwork weight across all 110 DBPs is shown in pink in Figure 5B. For comparison, we randomly drew 1000 sets of 40 bins and plotted the distribution of their weight in gray. Clearly, the subnetworks of loci selected based on ChIP-seq peak density formed stronger interactions in trans than random sets of loci. This is a first important hint that many DBPs may be indeed involved in specific trans contact networks.
Second, for each DBP individually, we formed a seed by selecting the five bins with the most peaks from its corresponding ChIP-seq track, and we ran trans-C to identify a set of potential interactors in trans. We took the top 40 predicted bins for a given DBP and observed that these bins were enriched with ChIP-seq peaks not only for the DBP that spawned the seed but also for ChIP-seq peaks of other DBPs (Supplemental Fig. S4A). This is not surprising because many DBPs act in concert, and many loci contain proximal binding sites of several DBPs (Ibarra et al. 2020). When examining the weights of subnetworks formed by trans-C (“top five DBP-bound seeds,”) (Fig. 5B, orange), we noted that they were heavier on average than the subnetworks based on the ChIP-seq signal only (“top 40 DBP-bound loci”; pink). This observation validates that trans-C finds loci that interact even more strongly in trans with the seed bins than just the bins most bound by the respective DBP.
We also assessed how well trans-C can build dense subnetworks when it is seeded from biologically unrelated loci. To that end, we first drew 1000 times five random loci to use as seeds and ran trans-C (Fig. 5B, green). The subnetworks it built were significantly weaker than the DBP-based ones. This is likely because a randomly drawn seed set likely includes loci that are not interacting with one another, whereas the loci in the DBP-based seed tend to have strong interactions in trans. Thus, in order to establish a more stringent baseline, for each DBP we performed 1000 matched random controls with seeds of five random bins for each DBP (Fig. 5B, yellow). When using this matched random seed, the subnetworks that trans-C found were once again weaker on average than the ones it found using DBP-based seed (Mann–Whitney U test P = 0.006). At an individual level, the weight of subnetworks for 53% (58 out of 110) of the DBPs identified from the DBP-based seeds was significantly stronger than that from matched random seeds (P < 0.05) (Fig. 5C). Visualization of the two strongest subnetworks, associated to the TFs PAX5 and MAX, and the two most-significant subnetworks compared with their matched random controls, linked to the TF FOS and chromatin regulator MLLT1, demonstrated that these are interconnected with strong significance (Fig. 5D; Supplemental Fig. 4D).
Next, we performed an additional control in which matched random seeds were selected from a network that was previously randomly shuffled to remove all specific signals resulting from interchromosomal structure: As could be expected, the resulting cliques were on average the weakest recovered by trans-C (Fig. 5B, light blue), and individual comparisons for DBP-associated cliques were all significant compared with this type of control (Supplemental Fig. S4B). This confirms that interchromosomal genome architecture is far from random and that trans-C identifies signals much stronger than random noise. In all, these observations confirm the common sense conception that the interchromosomal interactions of biologically unrelated loci are mostly noise, while providing more rigorous support to the hypothesis that coregulated loci are often enriched for trans contacts (Bertero 2021).
To provide additional validation, we examined the strength of the DBP-associated cliques in an orthogonal data set based on split-pool recognition of interactions by tag extension (SPRITE) (Quinodoz et al. 2022), a proximity ligation-independent method to detect higher-order interactions within the nucleus. Specifically, we processed a SPRITE interaction matrix for GM12878 cells (Quinodoz et al. 2018) and evaluated whether the cliques’ trans-C identified using the Hi-C data exhibited strong interactions in this orthogonal SPRITE data set. All of the 110 DBP-based cliques showed much higher weight than a background of 1000 randomly drawn sets of loci (P = 6 × 10−48, Mann–Whitney U test) (Supplemental Fig. S5A). To use a more stringent background model, we statistically assessed whether the trans-C-derived subnetworks for each of the analyzed DBPs were stronger than their “matched random” controls in the orthogonal SPRITE data (Supplemental Fig. S5B). We observed that the majority (30 of 58) of the DBP-based subnetworks that were significantly stronger than their matched random controls in the Hi-C data were also significantly stronger than their matched random controls in the SPRITE data (Supplemental Fig. S5C). Notably, our top five most-significant cliques were all significant in the SPRITE data. These findings suggest that trans-C reliably identifies cliques that exhibit strong interactions in trans also when these are measured by proximity ligation-independent sequencing approaches.
Lastly, we evaluated whether the loci that are part of a trans-C clique are physically closer to one another. To that end, we used orthogonal imaging measurements with multiplexed error-robust fluorescence in situ hybridization (MERFISH) (Su et al. 2020). We computed the trans-interaction proximity matrix for loci in the IMR-90 fibroblast cell line and downloaded the corresponding Hi-C matrix along with the ChIP-seq tracks for 16 DBPs available in the ENCODE portal. Because the MERFISH study involved only 1041 loci, we devised a twofold experiment. First, we subsetted the Hi-C matrix to the loci surveyed in the MERFISH study and ran trans-C using the five loci most bound by a given DBP as a seed. We calculated the average trans-proximity in the MERFISH data set for the loci in the subnetworks’ trans-C found using the Hi-C data and compared them to 1000 randomly selected sets of loci. We found that the trans-C cliques exhibited significantly higher proximity in the orthogonal imaging data set (P = 3 × 10−11, Mann–Whitney U test) (Supplemental Fig. S6A). Second, we ran trans-C on the full Hi-C matrix with the five loci most bound by a given DBP as a seed to obtain a ranking of all loci, and then, we looked at the proximity of the 40 MERFISH loci that were ranked closest to the top of the trans-C ranking versus the proximity of the 40 MERFISH loci that were ranked closest to the bottom. We observed that for all DBPs the trans-C top-ranked MEFISH loci had significantly higher proximity than the bottom-ranked ones (P = 3 × 10−5, binomial test) (Supplemental Fig. S6B). These data extend the cross-validation of trans-C predictions of DBP-associated trans cliques from Hi-C data with orthogonal methods, including those based on imaging.
DNA-binding-protein-associated trans cliques are proximal to nuclear speckles
Our hypothesis is that DBP-associated cliques represent specialized RNA factories. To test this, we examined the nuclear localization of loci involved in trans-interacting subnetworks identified by trans-C. Specifically, we asked whether they are near nuclear speckles, membrane-less subnuclear organelles involved in various aspects of DNA and RNA metabolism (Galganski et al. 2017). To that end, we turned to data generated by tyramide signal amplification sequencing (TSA-seq) (Chen et al. 2018). TSA-seq is an experimental protocol that provides a “cytological ruler” for estimating mean chromosomal distances from nuclear landmarks genome-wide. We used the log2 fold change of TSA-seq signal compared with an input control from the lymphoblastoid K562 cell line (Chen et al. 2018). This measurement captures the distance to a target protein from loci genome-wide, with higher values corresponding to shorter distances and lower values to longer distances. First, for each DBP subnetwork we calculated the average TSA-seq signal strength when probing the SON protein, which plays a crucial role in the formation of nuclear speckles. Indeed, the SON TSA-seq score is proportional to the cytological distance of genes from nuclear speckles, and it can be even calibrated to estimate mean distance in micrometers (Chen et al. 2018). Next, as a control, for each DBP we took the 40 bins ranked lowest by trans-C but containing at least one ChIP-seq peak, and we compared their average SON TSA-seq score to that of the trans-C-identified subnetwork (Fig. 5E). We observed a statistically significant shift to higher values for the trans-C subnetworks, supporting the conclusion that these loci are closer to nuclear speckles. An analogous analysis leveraging on TSA-seq data for phosphorylated SC35 (p-SC35), a splicing factor that also marks nuclear speckles, led to similar results (Fig. 5F). We noticed that cliques that were significantly stronger than their matched random controls appeared to be particularly close to nuclear speckle markers, particularly SON (Fig. 5E). Indeed, the SON TSA-seq score was significantly higher for these cliques compared with the other cliques (Fig. 5G). Collectively, these observations suggest that several DBP-associated cliques identified by trans-C represent RNA factories located at nuclear speckles.
Examining the distribution of the trans-C subnetwork weights identified for different DBPs (Fig. 5B, orange), we noticed a bimodal distribution, indicating that some DBPs are associated with stronger TIDs. This bimodality did not correlate with differences in the expression level of the two groups of DBPs or in their preference to bind to loci in the A or B compartments (Supplemental Fig. S4C). Intrinsically disordered regions (IDRs) within proteins, which lack a defined tertiary structure and are thus prone to self-aggregation, are emerging as an important mediator of subcellular condensates involved in multiple aspects of cell function, including nuclear regulations (Wright and Dyson 2015; Hirose et al. 2023). We thus investigated the correlation between the intrinsic disorder in DBP structure and the strength of the trans-C subnetworks they are associated with. For this analysis, we took the DBPs whose seeds gave rise to the strongest and weakest subnetworks (Fig. 5C, top and bottom 25% along the y-axis; Supplemental Table S4). We calculated the average intrinsically disordered protein (IDP) score for each DBP in the two groups (Mészáros et al. 2018), and plotted them in Figure 5H. The difference between the two groups was statistically significant (Mann–Whitney U test P = 1.4 × 10−5), suggesting that the DBPs with more IDRs form stronger interactions in trans. In all, trans-C allowed us to identify a large set of DBP enriched for IDR regions that are involved in strong TIDs proximal to nuclear speckles and that may thus be important in efficient transcriptional regulation of their target genes.
Selected RNA-binding proteins are associated to significant nuclear speckle–proximal trans cliques
Encouraged by the results on DBP subnetworks, we also examined whether RNA-binding proteins (RBPs) are generally associated with TIDs. Only a few RBP binding profiles are available for GM12878. Thus, for this analysis, we turned to K562 cells, another human lymphoblastic line, for which ENCODE reports 139 eCLIP data sets and a deep Hi-C matrix. Similar to our DBP analysis, we counted the number of peaks in each genomic bin for each RBP (mapping RNAs to the encoding DNA loci). Then, for each RBP individually, we formed a seed by selecting the five bins with the most peaks and ran trans-C to identify a set of potential interactors in trans. To form a null model per RBP, we repeatedly drew 1000 contact frequency–matched random seeds. We report the average total weight of the matched random seeds compared with the RBP seed in Figure 6A. Most RBP subnetworks built by trans-C were comparatively as dense as those from the corresponding matched random control, lying broadly along the y = x line. Nevertheless, several outliers were notably denser. To assess this observation quantitatively, we performed a signed ranked test per RBP and FDR controlled the corresponding P-values. Thirteen proteins had corrected P-values lower than 0.05, which we considered as a significance threshold (Fig. 6A, red; for list, see Supplemental Table S5). Distinctly from DBPs, RBPs associated with significantly stronger trans-C subnetworks were not characterized by higher IDP scores (Supplemental Fig. S7A), indicating that other characteristics may explain their specific behavior in trans genome organization.
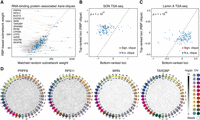
Trans-C identifies RNA-binding protein (RBP)–associated trans cliques proximal to nuclear speckles. (A) A subset of trans-C-identified subnetworks in lymphoblastoid cells built from loci characterized by strong binding of RBPs have denser contacts than the corresponding matched random null model. We plot the weight of a RBP-based subnetwork and the average weight of 1000 matched random seed subnetworks (error bars correspond to the SD) for 139 RBPs. In red and listed by name are those with significant P-values after FDR correction. (B,C) RBP-associated cliques identified by trans-C are significantly closer to nuclear speckles (stronger SON TSA-seq signal) and significantly further away from the nuclear lamina (weaker Lamin A TSA-seq signal) than matched control sets at the opposite end of the trans-C rankings. (D) Visualization of selected significant RBP-associated trans cliques in lymphoblastoid cells, plotted as described for Figure 2D.
We repeated the analyses of TSA-seq data and observed a very strong and significant global correlation between the trans-C subnetworks and both SON and p-SC35 signal (Fig. 6B; Supplemental Fig. S7B). This indicates that the trans-C eCLIP subnetworks are close in cytological distance to nuclear speckles. This same trend manifested for POL1RE TSA-seq signal, a measurement of proximity to transcription factories (Supplemental Fig. S7C). We observed the opposite result when we examined the Lamin A TSA-seq signal, indicating that the trans-C subnetworks are significantly further away from the nuclear lamina (Fig. 6C). This finding is in line with the notion that sites of active RNA biogenesis are localized in the euchromatic nucleoplasm and away from heterochromatin regions associated with the nuclear lamina.
Visualization of significant RBP-associated subnetworks showed that these are noticeably interconnected (Fig. 6D), more so than DBP-associated subnetworks of similar strength (Fig. 5D); on the other hand, the individual pairwise interactions were less significant, possibly owing to a more transient nature. In all, trans-C allowed us to identify a subset of RBPs associated with nuclear speckle–proximal and transcription factory–proximal TIDs that may contribute to gene regulation.
Discussion
Potential interactions between pairs of loci on different chromosomes occupy 90%–95% of the pairwise 3D DNA contact space (Supplemental Table S6) and a sizable fraction of experimentally measured interactions (Supplemental Table S7). Although in certain species whose nucleus is characterized by chromosome territories—such as humans and other mammals—a large fraction of trans contacts are likely nonspecific; illuminating an even small fraction of specific and functional interchromosomal interactions may provide important advances in our understanding of nuclear mechanisms such as transcription and splicing. In this context, trans-C is an important step toward refined analytical methods to probe the trans contact space for functional gene networks.
The study of trans contacts requires statistical methods designed for the specific task at hand. Approaches devised for the analysis of cis interactions control for some biases that are not applicable to trans ones, such as correction for the linear genomic distance between the interacting loci. To date, most analytical tools for Hi-C data have been limited to cis interactions (Lin et al. 2019). Recent network-based strategies to study interchromosomal interactions from bulk and single cell Hi-C (Kaufmann et al. 2015; Bulathsinghalage and Liu 2020; Joo et al. 2023) proposed probabilistic models that focus on identifying large patterns of trans contacts (i.e., those involved in nuclear speckles and nucleoli) rather than small sets of interactions linked to a specific process. Trans-C, in contrast, controls for chromosome territory biases to identify cliques that “stand out” from other trans interactions resulting from the random intermixing of neighboring chromatin domains. Given that the combinatorial number of possible sets is astronomical (4.7 × 10131 for sets of 40 loci in the human genome binned at 100 kb resolution), this problem cannot be solved directly. Trans-C addresses this challenge by applying a random walk algorithm to obtain a highly reproducible, approximate solution.
We first validate the ability of trans-C to detect known examples of functional trans contacts. We find that it outperforms a simple greedy heuristic even in the case of the small haploid genome of P. falciparum (22.9 Mb), which is characterized by remarkable trans contacts among var genes. In more complex and larger mammalian genomes, trans-C identifies with high precision not only the mOSN Greek islands but also the less striking example of trans contacts represented by the RBM20 splicing factory. Thus, trans-C may find applicability across nuclear genomes with different sizes and types of organization, as well as trans contacts of varying strength.
We demonstrate the broader utility of trans-C by using it to systematically search for trans cliques around loci most strongly bound by one of many DBPs or RBPs. These analyses support the existence of a large number of statistically significant TIDs readily measurable from Hi-C data, particularly in the case of intrinsically disordered DBPs. Orthogonal analyses of TSA-seq data confirm that such loci are proximal to nuclear speckles. The concept of “bookmarked” transcription factories, Pol II clusters that are specifically enriched for a set of DBPs and their target loci, was proposed over a decade ago (Cook 2010). However, examples of this phenomenon have been sparse (for review, see Bertero 2021). Our analysis of 110 DBPs provides an important piece of evidence to support this model for >50% of such DBPs, including leukemia-associated TFs (i.e., PAX5, MAX, and FOS) and chromatin regulators (i.e., MLLT1) (Zhou et al. 2018; Sigvardsson 2023). Nevertheless, a mechanistic dissection of these leads will be required to further validate this model.
The few RBPs associated with significant TIDs are involved in a wide variety of functions. Not only do we identify several factors involved in major and minor spliceosomes (PRPF8 and BUD13), but we also identify alternative splicing regulators (ZRANB2), a multifunctional RNA processing factor (TARDBP), a component of the RNA exosome complex (EXOSC10), a ribosomal protein (RPS11), and even a DNA helicase involved in homologous recombination (WRN). We speculate that these factors exemplify a wide range of chromatin structures involving both cis and trans interactions that regulate not only transcription but also other aspects of nucleic acid biology such as DNA replication and repair, or various aspects of RNA biogenesis. In line with this hypothesis, recent evidence published during the revision of our paper supports the notion that genome organization around nuclear speckles drives mRNA splicing efficiency (Bhat et al. 2024). Notably, several of the RBPs highlighted by our trans-C analysis are known to be mutated in severe human monogenic diseases: PRPF8 in retinitis pigmentosa (McKie et al. 2001), WRN in Werner syndrome (Yu et al. 1996), and TARDBP in amyotrophic lateral sclerosis (Sreedharan et al. 2008). Moreover, mutations in ZRANB2 have been linked to unfavorable prognosis in breast and liver cancer (Tanaka et al. 2020), whereas RPS11 has been shown to be a key player in poor outcomes of glioblastoma patients (Dolezal et al. 2018). Whether disorganization of trans genome architecture is implicated in the pathogenesis of these diseases is an interesting topic for future investigation.
Using trans-C, we confirmed the existence of significant RBM20-associated trans cliques in both hPSC-CMs from a different laboratory and in vivo samples of the human LV. These findings support the physiological relevance of muscle-specific interchromosomal splicing factories involving RBM20. We have previously shown that preventing TTN transcription disrupts RBM20 clustering, decreases the proximity of RBM20 targets to the TTN locus, and impairs their RBM20-dependent alternative splicing in trans (Bertero et al. 2019). TTN is the most commonly mutated gene in both familial and sporadic dilated cardiomyopathy (DCM) (Herman et al. 2012; Kayvanpour et al. 2017). Although RBM20 is mutated in only ∼2% of DCM patients, it leads to a particularly aggressive disease characterized by conduction system disorders (∼30%), malignant ventricular arrhythmia (∼44%), and a rapid progression to heart failure (Refaat et al. 2012; Kayvanpour et al. 2017). We and others recently showed that RBM20 mutations in the RS domain hotspot lead to nuclear mislocalization of RBM20 and severe changes in gene expression (Schneider et al. 2020; Fenix et al. 2021). We speculate that these or other mutations in RBM20, TTN, and/or other RBM20-associated targets may lead to disease in part through disruption of interchromosomal genome architecture. Trans-C will be a useful instrument in testing this hypothesis.
Although we validate and apply trans-C using seed loci selected from a priori hypotheses about interchromosomal genome architecture, either related to specific genes or to a general mechanism, trans-C can be run using all possible sets of seeds of a given size to conduct discovery. However, this approach is computationally challenging because the number of sets of possible seeds can become combinatorially very large. Moreover, the strongest cliques may not necessarily be the most biologically interesting, as showcased by our example for RBM20, which would not have stood out in an unbiased analysis of all hPSC-CM cliques.
It is important to point out that although trans-C identifies sets of loci that significantly interact with one another, this does not rule out the possibility that some of these interactions may be biologically unrelated. Indeed, in many cases, if we select seed bins at random, requiring only that they display trans interactions comparable in strength to those involving genes most strongly bound by DBPs or RBPs, we observe that trans-C sometimes identifies strong cliques. This observation is in line with the understanding that in mammals active chromatin tends to be situated at the periphery of chromosome territories (Di Pierro et al. 2016, 2017; Cheng et al. 2020; Su et al. 2020). Thus, although statistical assessment of trans-C results can allow the identification of cliques that are significantly stronger than matched random controls, a sizable portion of the signal is likely to nevertheless arise from compartmental interactions. Accordingly, predictions of novel cliques should be confirmed via orthogonal methods or experimentally validated for their biological significance, particularly if trans-C is applied to discovery research with no a priori hypothesis.
Another limitation to keep in mind is that the performance of trans-C analyses is strongly dependent on the sequencing depth of the Hi-C matrices. This could represent a bottleneck, because the generation of ultra-deep matrices requires not only substantial resources but also large enough cell numbers to capture a sufficient number of contacts for each locus. For rare samples, this may be infeasible even if the economic resources were available. When challenged by this situation, a compromise would be to reduce the resolution of genomic binning at the expense of increased noise and more complex biological interpretation of results.
Overall, our work focuses on poorly studied between-chromosome contacts and provides an efficient computational framework for identifying potentially biologically important sets of loci that interact in trans. We demonstrate the flexibility and sensitivity of trans-C and provide examples of how our approach can be used to identify candidate gene sets for subsequent hypothesis-driven studies. Application of trans-C to the growing number of Hi-C data sets from the ENCODE (The ENCODE Project Consortium 2012) and 4D Nucleome consortia (Reiff et al. 2022) will reveal novel cell-specific or disease state–specific trans networks. We also provide preliminary evidence that trans-C also allows exploration of SPRITE data (Quinodoz et al. 2018); minor adaptations of the approach will enable investigation of other proximity-ligation independent assays, such as GAM (Beagrie et al. 2017), and will collectively offer the potential to accurately characterize interchromosomal architecture at varying spatial resolutions.
Methods
Overview
The full mathematical formulation of trans-C is reported in the Supplemental Methods. In short, trans-C takes as input a Hi-C contact matrix H of interaction counts and an initial set S of loci of interest (“seed loci”); after processing, it outputs a set of loci U (containing S) that interact strongly together in trans. In practice, we model the Hi-C interaction matrix H as a weighted graph G = (V,E,W), in which nodes V correspond to the genomic loci (bins) of H, edges E between pairs of nodes correspond to interactions between their respective loci, and weights W on the edges reflect the strength of the interactions represented by the edges. For instance, the weight wij on edge eij between loci i and j corresponds to the Hi-C matrix entry hij. The goal of trans-C is to find a subset of loci that exhibit strong interchromosomal contacts. To solve this problem, which is computationally intractable to solve exactly, we employ a random walk with restart algorithm over the graph G. In essence, this reformulates the problem as a dense subgraph optimization.
Random walk with restart
The random walk traverses the graph by moving probabilistically from one node to another. The walk is initiated from a specified set S of seed loci. The goal of the random walk is to highlight the nodes that are strongly connected to those in S (Hristov et al. 2020). At each step, with a fixed probability α, the walk restarts from a randomly selected seed locus, and with probability 1 – α, the walk moves to a neighboring node picked probabilistically based upon the weights W. Specifically, if N(i) are the nodes that i interacts with, then the walk goes from node i to node j ∈ N(i) with probability proportional to wi,j. That is, for any node i, if at time t the walk is at i, then we calculate the probability pij that it will transition to node j at time t + 1 using only W and α. Hence, the random walk is fully described by a stochastic transition matrix P with entries pij. Importantly, this stochastic matrix P has certain mathematical properties (Supplemental Methods) that guarantee that, by the Perron–Frobenius theorem, the random walk converges. That is, the probability of the walk being at any given node at time t is constant as t → ∞. This probability π, known as the “stationary distribution” of the walk, can be analytically computed. Further, the probability πi reflects how well the node i is connected to the seed nodes because more strongly connected nodes are more frequently visited. The loci that have the largest probabilities are most frequently visited and, therefore, are more likely to be relevant because they are strongly connected to the seed loci. We use these probabilities as scores to rank all loci and include the top ℓ loci in U, where ℓ is a user-specified parameter. In this work, we use ℓ = 40 unless otherwise stated.
Data preprocessing
Prior to running the trans-C random walk algorithm, we perform three preprocessing steps on the Hi-C matrix to ensure that the weights W on the edges are not influenced by many of the biases common in Hi-C data.
First, we normalize the matrix using the iterative correction and eigenvalue decomposition (ICE) procedure (Imakaev et al. 2012; Servant et al. 2015). This procedure iteratively normalizes rows and columns of the matrix, equalizing their sum. We note that we carry out this procedure on the entire Hi-C matrix, including cis and trans contacts.
Second, we adjust the matrix entries to account for the fact that chromosomes tend to occupy chromosome territories; as a result, some pairs of chromosomes interact more frequently. We do this by using a binomial model to estimate interaction P-values based on an empirical null model that accounts for this territorialization (Supplemental Methods).
Third, we process each matrix entry using a “donut filter” as previously described (Rao et al. 2014). This step allows us to emphasize points that are local maxima in the contact map.
Matched random seed
We run trans-C with 1000 matched random seeds to generate a background model of cliques that are seeded at biologically unrelated loci that form a starting network of comparable strength to the loci of interest. This background model allows us to assess statistically (by Mann–Whitney U test) whether the clique trans-C found using the original seed is significantly stronger than the matched random background. Specifically, the procedure is as follows:
-
Given a seed S of b loci S = (s1,s2,…,sb), calculate the strength of that seed score(S) = Σi,j∈S wi,j.
-
Repeat 1000 times:
-
2.1 Draw sets of b random loci R = (r1,r2,…,rb) until score(R) ≥ score(S);
-
2.2 Use the set R as a seed to run trans-C to find a clique trans-C(R); and
-
2.3 Add trans-C(R) to the background list of cliques obtained from a “matched random seed.”
-
-
Assess statistically whether trans-C(S) is significantly stronger than the matched random background.
Clique visualization
To visualize the cliques, we use the Cytoscape software version 3.10.1 (Shannon et al. 2003).
Data sets
Validation experiments relied on Hi-C data from three organisms available publicly as either MCOOL or HIC files: P. falciparum trophozoite and schizont stages, binned at 10 kb resolution (available from the NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE126074) (Bunnik et al. 2018); mOSNs, binned at 250 kb resolution as in the original analyses (4DN Portal 4DNFI3M6726I) (Monahan et al. 2019); and human cardiomyocyte differentiation from embryonic stem cells (4DN Portal 4DNFIT5YVTLO, 4DNFIIOUG5RF, and 4DNFI8RH55DO) (Zhang et al. 2019). For this last data set, we used the cooltools package (Abdennur et al. 2024) to extract from each MCOOL file the interaction counts for its corresponding Hi-C matrix binned at 10 kb resolution. Then, we aggregated the Hi-C matrix to 100 kb resolution by summing the counts in each 10 consecutive bins of size 10 kb. RBM20 eCLIP data were previously reported (GEO GSE175886) (Fenix et al. 2021) and analyzed as described below using 100 kb bins. Human LV and unrelated tissue controls Hi-C were obtained from the ENCODE portal (ENCFF193CQL, ENCFF546TZN, ENCFF341WOY, ENCFF033WGK, ENCFF294GFP, ENCFF294GFP, ENCFF251VFA, ENCFF556RLR, ENCFF591MHA, ENCFF004YZQ) (The ENCODE Project Consortium 2012) and binned at 100 kb resolution.
Discovery analyses involved ChIP-seq data for 110 human DBPs in the GM12878 cell line and 139 eCLIP for RBPs in the K562 cell line from the ENCODE portal (for IDs, see Supplemental Tables S4, S5). We used the IDR thresholded peaks provided by ENCODE. We split the human linear genome in 100 kb bins and, for a given protein t, counted the number of peaks in each bin, producing a count vector Ct. For the DBP analysis in the GM12878 cell line, we used an ultra-deep sequenced Hi-C matrix (ENCODE ENCSR410MDC) (Harris et al. 2023), which contains 3.7 billion trans contacts. We performed our analysis at 100 kb resolution, which results in nonzero counts for 85% of all pairwise trans contacts. For the RBP analysis in the K562 cell line, we used an intact Hi-C matrix (ENCODE ENCFF621AIY), which has 360 million trans contacts and was binned at 100 kb resolution.
For the SPRITE analysis, we downloaded the processed SPRITE interaction matrix in GM12878 cells (4DN Portal 4DNFIUOOYQC3) and normalized it using the steps described above as done for the Hi-C matrices, binning at 100 kb resolution. For the imaging analysis, we downloaded the computed (x,y,z) coordinates (files: genomic-scale.tsv and genomic-scale-with transcription-and-nuclearbodies.tsv available at https://zenodo.org/records/3928890) of 1041 loci studied by MERFISH (Su et al. 2020), and we used the scripts provided by the authors to compute the trans-interaction proximity matrix. Because the study was done in the IMR-90 fibroblast cell line, we used a corresponding Hi-C matrix (ENCODE ENCFF281ILS) and ChIP-seq data (ENCODE ENCFF483ERE, ENCFF459DPT, ENCFF470FUH, ENCFF770ISZ, ENCFF585XWV, ENCFF567GON, ENCFF124ORZ, ENCFF170WDS, ENCFF718BQI, ENCFF566MPI, ENCFF890WEE, ENCFF150MNG, ENCFF453XKM, ENCFF786CKM, ENCFF448ZOJ, ENCFF699YDJ). For the nuclear speckles analysis, we used TSA-seq data in K562 cells (4DN Portal 4DNFI2WK5IVI, 4DNFI1WULK53, 4DNFII37TNR5, 4DNFIXWDLHDL).
Software availability
The trans-C code and the custom scripts used for data processing and figure preparation are available at GitHub (https://github.com/Noble-Lab/trans-C) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Irene Farabella for critical advice on MERFISH data analyses and Łukasz Truszkowski for insightful discussions and manuscript proofreading. The study has received funding from the European Research Council (ERC) under the European Union's Horizon Europe research and innovation programme (grant agreement no. 101076026; project acronym TRANS-3; A.B.). Views and opinions expressed are, however, those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. We also acknowledge financial support from the National Institutes of Health (award UM1HG011531; W.S.N.) and the Giovanni Armenise-Harvard Foundation (Career Development Award 2021; A.B.).
Author contributions: B.H.H. performed all of the analyses and wrote the first draft of the manuscript. W.S.N. supervised the analyses, edited the manuscript, and obtained funding. A.B. conceptualized the study, cosupervised the analyses, assembled the final figures, edited the manuscript, and obtained funding.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278327.123.
- Received August 8, 2023.
- Accepted August 30, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








