
Chromatin interaction maps identify oncogenic targets of enhancer duplications in cancer
- Yueqiang Song1,6,
- Fuyuan Li1,6,
- Shangzi Wang1,6,
- Yuntong Wang1,7,
- Cong Lai1,
- Lian Chen1,
- Ning Jiang1,
- Jin Li1,
- Xingdong Chen1,2,3,
- Swneke D. Bailey4,5 and
- Xiaoyang Zhang1,8
- 1State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Fudan University, Shanghai 200438, China;
- 2Human Phenome Institute, Fudan University, Shanghai 200438, China;
- 3Fudan University Taizhou Institute of Health Sciences, Taizhou, Jiangsu 225312, China;
- 4Cancer Research Program, Research Institute of the McGill University Health Centre, Montreal, Québec H4A 3J1, Canada;
- 5Departments of Surgery and Human Genetics, McGill University, Montreal, Québec H4A 3J1, Canada
-
↵6 These authors contributed equally to this work.
Abstract
As a major type of structural variants, tandem duplication plays a critical role in tumorigenesis by increasing oncogene dosage. Recent work has revealed that noncoding enhancers are also affected by duplications leading to the activation of oncogenes that are inside or outside of the duplicated regions. However, the prevalence of enhancer duplication and the identity of their target genes remains largely unknown in the cancer genome. Here, by analyzing whole-genome sequencing data in a non-gene-centric manner, we identify 881 duplication hotspots in 13 major cancer types, most of which do not contain protein-coding genes. We show that the hotspots are enriched with distal enhancer elements and are highly lineage-specific. We develop a HiChIP-based methodology that navigates enhancer–promoter contact maps to prioritize the target genes for the duplication hotspots harboring enhancer elements. The methodology identifies many novel enhancer duplication events activating oncogenes such as ESR1, FOXA1, GATA3, GATA6, TP63, and VEGFA, as well as potentially novel oncogenes such as GRHL2, IRF2BP2, and CREB3L1. In particular, we identify a duplication hotspot on Chromosome 10p15 harboring a cluster of enhancers, which skips over two genes, through a long-range chromatin interaction, to activate an oncogenic isoform of the NET1 gene to promote migration of gastric cancer cells. Focusing on tandem duplications, our study substantially extends the catalog of noncoding driver alterations in multiple cancer types, revealing attractive targets for functional characterization and therapeutic intervention.
The focus of cancer genome analyses has expanded from protein-coding genes to the entire genome, which has started to shed light on noncoding regions that account for the majority of the human genome. Noncoding regions harbor regulatory elements such as enhancers, promoters, and insulators that control the expression of protein-coding genes. Recent studies have shown that somatic genomic alterations, such as point mutations, indels, and structural variants, affecting these regulatory elements may cause dysregulation of oncogenes or tumor suppressor genes to promote tumorigenesis (Cuykendall et al. 2017; Zhang and Meyerson 2020; Elliott and Larsson 2021). As a major type of structural variants, duplication of oncogenes is a key cancer driver event causing oncogene overexpression (Stratton et al. 2009; Zack et al. 2013). In addition to the oncogenes themselves, we and others have recently shown that noncoding enhancers are also critical subjects of duplications in cancer. For instance, distal enhancers of oncogenes such as KLF5, AR, SOX2, MYC, and EGFR are selectively duplicated with or without the oncogenes in diverse cancer types, which activates their expression through up to 800 kb long-distance enhancer–promoter interactions (Zhang et al. 2016, 2018; Takeda et al. 2018; Morton et al. 2019; Wu et al. 2019; Kim et al. 2020; Liu et al. 2021). In addition, genes within the duplicated regions are not necessarily the actual oncogenic targets of the duplications, as exemplified by duplications of enhancers within the SNCAIP gene causing activation of a distal oncogene PRDM6 in medulloblastoma (Northcott et al. 2017). These findings demonstrated the critical roles of enhancer elements and their associated enhancer–promoter interactions in the oncogenic duplication events.
Despite these individual examples, it remains largely unknown, at the genome-wide scale, which enhancers are selectively duplicated in which cancer types and what their target genes are. With the advance of technologies to map structural alterations as well as enhancer–gene interactions, it is now becoming feasible to address these questions. In particular, using whole-genome sequencing, the Pan-Cancer Atlas of Whole Genomes (PCAWG) project has provided a comprehensive catalog of high-confidence somatic structural variants including duplications in a large number of cancer samples (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). Furthermore, the development of high-resolution 3D genome technologies such as HiChIP, PLAC-seq, Micro-C, and Micro-Capture-C has enabled fine mapping of enhancer–promoter contacts (Hsieh et al. 2015; Fang et al. 2016; Mumbach et al. 2016; Hua et al. 2021). Finally, statistical models such as the activity-by-contact (ABC) have been developed to weight the contribution of a given enhancer to its target gene (Fulco et al. 2019; Nasser et al. 2021). Here, we integrated these types of information to systematically investigate enhancer duplications in cancer. We aimed to reveal the prevalence of cancer-type-specific enhancer duplications in the cancer genome, to identify the potential target genes for duplication hotspots harboring enhancer elements in diverse cancer types, and to functionally validate some of the target genes subject to enhancer duplications.
Results
A catalog of duplication hotspots in the cancer genome
We first aimed to identify recurrently duplicated regions in the genome in an unbiased manner. The PCAWG project has identified 288,417 somatic structural variants, including duplications, deletions, inversions, and translocations, in 2439 samples across 37 cancer types (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). The project released structural variants that were stringently called by at least two independent algorithms. To focus on the focal events, we selected duplications that are <1 Mb in length, which are found in 1944 samples from 35 cancer types (Supplemental Table S1) and account for over 70% of the identified duplications (Supplemental Fig. S1A). For cancer types that have more than 30 samples harboring duplications (Supplemental Table S1), we performed hierarchical clustering based on their duplication profiles—duplication frequency of 5 kb bins tiling the entire genome. This analysis revealed that the duplications exhibit cancer-type-specific distributions, since cancer types bearing biological similarities tend to cluster together (Fig. 1A). For instance, adenocarcinomas of the stomach (STAD) and esophagus (ESAD) clustered together, as did adenocarcinomas of the uterus (UCEC) and ovary (OV), while squamous cancers of the lung (LUSC) and head and neck (HNSC) were found in a separate cluster.
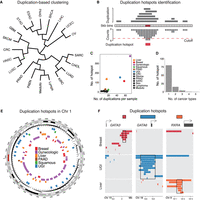
Identification of duplication hotspots in the cancer genome. (A) Hierarchical clustering of cancer types based on the duplication frequencies of continuous 5 kb bins in the genome. The full names of the cancer types are listed in Supplemental Table S1. (B) Schematic illustrating the methodology applied to identify duplication hotspots in each cancer type. Based on a Poisson distribution, 5 kb bins that are duplicated more often than expected were identified (FDR < 0.05). The significant 5 kb bins that are adjacent to each other or one 5 kb bin apart were then stitched together as a duplication hotspot. (C) Presented are the number of duplication events observed per sample (x-axis) and the number of identified duplication hotspots (y-axis) in each cancer type. (D) The number of the identified duplication hotspots that are unique to a single cancer type or shared by multiple cancer types. (E) The distribution of the identified duplication hotspots from multiple cancer types in Chromosome 1. (F) The distribution of the duplication events observed at the GATA3, GATA6, and RXRA loci in breast, upper gastrointestinal (UGI), and liver cancers.
We then sought to assess if these duplications are recurrently present in specific regions—duplication hotspots. For the analysis, we combined the cancer types that are biologically similar to each other based on the clustering results as well as previous literature (Berger et al. 2018; Campbell et al. 2018), which resulted in 17 major cancer types for the subsequent analysis (Supplemental Table S1). Based on the Poisson distribution of the duplication occurrence of continuous 5 kb bins across the genome, we identified bins that are significantly duplicated in each of the major cancer types (Supplemental Fig. S1B). Significant bins that are adjacent or only one 5 kb bin apart were then stitched together for calling the duplication hotspot regions (as illustrated in Fig. 1B). The analysis identified in total 881 duplication hotspots (median size = 45 kb) in 13 major cancer types (Supplemental Table S2; Supplemental Fig. S1C), 94.3% of which were found in breast, upper gastrointestinal (UGI), liver, gynecologic, pancreatic (PAAD), and squamous cancers (Fig. 1C). Overall, the number of the duplication hotspots is correlated with the duplication frequency across the examined cancer types (Fig. 1C). We also examined TCGA SNP-array copy number data, which showed that the duplication hotspots are indeed enriched with focal amplification events in the relevant cancer types (Supplemental Fig. S1D). The duplication hotspots are highly cancer-type-specific, with 79.5% of the hotspots being found in only one cancer type (Fig. 1D). The cancer-type specificity was exemplified by the distribution of the duplication hotspots found across the Chromosome 1 (Fig. 1E) and at the GATA3, GATA6, and RXRA loci (Fig. 1F).
Enrichment of cancer-type-specific distal enhancers in duplication hotspots
Next, we sought to annotate the function of the duplication hotspot regions. Known amplified oncogenes such as ERBB2, EGFR, MDM2, and CDK4 were found within the duplication hotspots; however, 61.9% of the duplication hotspots do not harbor full-length protein-coding genes (Fig. 2A). Even for the gene-harboring hotspots, gene bodies occupy only 30.0% of their space. All these observations suggest the involvement of other functional elements in the duplication hotspots. Indeed, using TCGA ATAC-seq chromatin accessibility data (Corces et al. 2018), we found that both the gene-harboring and non-gene-duplication hotspots are significantly enriched with ATAC sites distal to gene promoters (Fig. 2B). In contrast, promoter ATAC sites are only enriched in the gene-harboring hotspots (Supplemental Fig. S2A). We also examined the ENCODE SCREEN data set that has defined promoters, enhancers, and CTCF-only regions across the human genome using ChIP-seq data derived from hundreds of biosamples (The ENCODE Project Consortium et al. 2020). We showed that the majority of the distal ATAC sites found in the duplication hotspots are indeed annotated as distal enhancers in this data set (Fig. 2C).
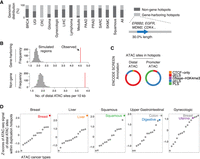
Enrichment of distal enhancers in the duplication hotspots. (A) The percentage of the identified duplication hotspots harboring intact protein-coding genes (from the start codon to the stop codon) in each cancer type. For gene-harboring hotspots, the harbored genes only account for 30.0% length of the hotspots wherein the genes are found. (B) The number of distal ATAC sites per 10 kb within the duplication hotspots or randomly shuffled regions matching the lengths of the hotspots. (C) The percentage of the distal and promoter ATAC sites residing in different categories of regulatory elements annotated in the ENCODE SCREEN data set. (dELS) distal enhancer-like signature, (pELS) proximal enhancer-like signature, (PLS) promoter-like signature. (D) Z-scores of the averaged ATAC-seq signal of distal ATAC sites within duplication hotspots (sum of ATAC signal for all distal ATAC sites within the hotspots divided by the number of ATAC-seq profiled samples) across different cancer types.
We then examined if the distal ATAC sites in the duplication hotspots exhibit cancer-type-specific activity. We focused on breast, UGI, liver, gynecologic, and squamous cancers, each of which has at least 20 duplication hotspots identified and also has available ATAC-seq data (Corces et al. 2018). We found that distal ATAC sites within the duplication hotspots exhibit stronger chromatin accessibility in the matched cancer types from which the hotspots were identified (Fig. 2D), suggesting cancer-type-specific regulatory activity at these hotspots. HOMER motif analysis (Heinz et al. 2010) showed that these distal ATAC sites are enriched with recognition sites of oncogenic transcription factors such as AP-1, KLF5, and FOXA1 that often exhibit lineage-specific binding at enhancers in the genome (Supplemental Fig. S2B; Eferl and Wagner 2003; Lupien et al. 2008; Liu et al. 2020). Taken together, these results suggest that the duplication hotspots are enriched with cancer-type-specific putative distal enhancer elements.
A methodology to prioritize target genes of the duplication hotspots
We next aimed to identify the target genes of the putative enhancers found in the duplication hotspots. As the identified duplication hotspots range from 5 to 1905 kb in length, they often contain multiple putative enhancer elements that form complex enhancer–promoter pairs, making it difficult to identify the real targets. Recent studies have revealed that the effect of a given enhancer on its target gene depends on the enhancer's own activity, as well as its contact intensity with the gene promoter, a model named ABC (Fulco et al. 2019). The H3K27ac HiChIP assay combines H3K27ac ChIP-seq (enhancer activity) and Hi-C (contact intensity) (Mumbach et al. 2017), enabling us to capture the two types of information from a single experiment. Indeed, previous work has shown the H3K27ac HiChIP data capture sufficient ABC information that is comparable to that obtained from the combination of Hi-C, ATAC-seq, and H3K27ac ChIP assays (Fulco et al. 2019). Based on the ABC model, we analyzed HiChIP results that were previously generated in cancer cell lines corresponding to the relevant cancer types (Supplemental Table S3) to define the enhancer–gene relationships for the duplication hotspots from both the enhancer and the gene's perspectives.
For enhancers in each of the duplication hotspots, we used HiChIP paired-end tags (PETs) to measure the amount of enhancer activity delivered to each gene's promoter via significant HiChIP loops (hotspot-delivered enhancer activity, Ein). Then, for each of the candidate genes, we followed the ABC model and measured the relative contribution of the hotspot-delivered enhancer activity as percentage of activity delivered from all enhancers (inside and outside of the hotspot) interacting with the gene's promoter (Ein/(Ein + Eout)) (Fig. 3A). We multiplied these two factors to prioritize target genes for the enhancers in the duplication hotspots (Fig. 3A), a methodology that we referred to as ranking of target genes based on enhancer–promoter contacts (RENC). For genes that have multiple promoters, the promoter with the highest RENC score is used to represent the gene. We refrain from combining scores from different promoters of the same gene because, while some genes may have promoters that drive isoforms with redundant or synergistic functions, others may produce isoforms with distinct or even opposing functions. A gene's RENC score is calculated per duplication hotspot. Therefore, factors such as duplication size, number of enhancers per hotspot, and GC content of the enhancers will be the same for the analysis of each hotspot. In addition, because we only consider enhancers interacting with gene promoters but not gene bodies, gene sizes do not affect the analysis. The calculation of the RENC score, including the formula used, is described in detail in the Method section. We also provided the number of PETs connecting gene promoters and enhancers within the duplication hotspot (Supplemental Table S2).
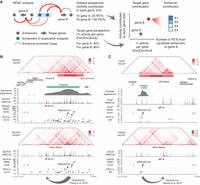
A methodology to identify the target genes activated by the duplicated enhancers. (A) Schematic illustrating the HiChIP-based RENC methodology that we applied to rank the target genes for the enhancers within the duplication hotspots. (B) Presented tracks include H3K27ac HiChIP signal, positions of duplication hotspots, duplication events observed in squamous cancer, H3K27ac ChIP-seq signal, RENC scores prioritizing the KLF5 promoter that is more likely to be activated by enhancers within the left duplication hotspot presented in the window, and the number of PETs contributed by each interacting enhancer to KLF5. (Upper) Data from BICR31 cells with the KLF5 enhancer amplification; (bottom) data from BICR16 cells without amplification at the KLF5 locus. Two of the previously published functional enhancers for KLF5 were indicated (Zhang et al. 2018). (C) Presented tracks include H3K27ac HiChIP signal, positions of duplication hotspots, duplication events observed in prostate cancer, H3K27ac ChIP-seq signal, RENC scores prioritizing the AR promoter that is more likely to be activated by enhancers within the duplication hotspot, and the number of PETs contributed by each interacting enhancer to AR. (Upper) Data from VCaP cells with AR and its enhancers amplified; (bottom) data from LNCaP cells that are near diploid at the AR locus. A previously published functional enhancer for AR was indicated (Takeda et al. 2018).
Although the cancer cell line models we used may not harbor the duplications observed in real tumors, we and others have shown that duplications, except for the ones that span boundaries of topologically associated domains (TADs), do not disrupt existing enhancer–promoter contacts or create de novo ones (Beroukhim et al. 2016; Weischenfeldt et al. 2017; Zhang et al. 2018). We found that only 7.0% of the identified duplication hotspots span previously annotated TAD boundaries (Akdemir et al. 2020), including the previously reported IGF2 duplication in colorectal cancer (Supplemental Fig. S3A; Weischenfeldt et al. 2017). Although the copy number status of the enhancers may affect the calculation of the enhancer contribution in the RENC analysis, the ranking of the candidate genes will unlikely be affected, because the analysis is done per duplication hotspot and the candidate genes are compared to each other in the same context (a pseudo example is presented in Supplemental Fig. S3B). We, therefore, reasoned that cell lines of the corresponding cancer types are sufficient to prioritize target genes for a large proportion of the enhancer duplication events.
To test this, we applied the RENC methodology to assess previously characterized enhancer duplications. For instance, we have previously identified multiple enhancers amplified in the 13q22 noncoding region in squamous cancer, which activates the expression of the KLF5 oncogene (Zhang et al. 2018). We analyzed HiChIP results in the head and neck squamous cancer cell lines BICR31 harboring amplification at the locus and BICR16 without the amplification based on the CCLE copy number data (Barretina et al. 2012; Ghandi et al. 2019). The RENC methodology identified the KLF5 promoter as the top candidate target for the duplication region in both cell lines regardless of their copy number status (Fig. 3B). Some of the driver enhancers for KLF5, previously identified from CRISPR-interference (CRISPRi) assays, also show strong interactions with the KLF5 promoter (Fig. 3B).
In addition, we applied RENC analysis to another enhancer duplication event that was previously identified in the Xq12 noncoding region in prostate cancer (Takeda et al. 2018; Viswanathan et al. 2018). RENC identified AR as the top candidate target gene for the duplication hotspot in prostate cancer cell lines VCaP that is amplified with AR and the hotspot region as well as LNCaP that is near diploid at the locus (Fig. 3C). A previously reported driver enhancer of AR also show a strong interaction with the AR promoter in both the cell lines. All these data showed that the RENC methodology prioritizes target genes of the duplication hotspots regardless of the copy number status in the cell lines used, further supporting its robustness.
Large-scale target gene identification for duplication hotspots
We then proceeded to apply RENC to comprehensively identify target genes for the remaining duplication hotspots found in our study. We focused on breast, UGI, squamous, prostate, and skin (melanoma) cancers, as HiChIP data are available in their representative cell lines (Supplemental Table S3). In total, RENC-prioritized target genes for 58.1% of the duplication hotspots in the five cancer types, of which 61.5% are not harbored within the hotspots per se (Fig. 4A–D; Supplemental Fig. S3C,D), suggesting long-distance gene regulation. The GC content of promoters of the RENC-prioritized genes is not significantly different from the other genes (Supplemental Fig. S3E). We also assessed if the RENC-prioritized genes are determined by the hotspot-delivered enhancer activity to each gene promoter (Ein) or their contribution to each gene promoter relative to all the linked enhancers (Ein/(Ein + Eout)), the two key components of the RENC formula. We found that the Ein only, the Ein/(Ein + Eout) only, or their combination, can independently prioritize the same target gene promoter for 51.4% of duplication hotspots. Target gene promoters for 38.0% and 8.4% of the hotspots were determined by only Ein or Ein/(Ein + Eout), respectively (Supplemental Fig. S4A).
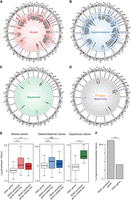
Comprehensive identification of target genes for the duplicated enhancers. (A–D) circlize plots presenting the genomic positions of the identified duplication hotspots, their duplication occurrences (scale: 0 to ≥10), and the associated target genes based on the RENC analysis in breast cancer (A), UGI cancer (B), squamous cancer (C), melanoma, and prostate cancer (D). For UGI cancer, due to the space limitation, presented are hotspots supported by duplications from more than five samples (the remaining hotspots were presented in Supplemental Fig. S3D). The colored genes indicate the previously annotated oncogenes (Chakravarty et al. 2017; Liu et al. 2017; Sondka et al. 2018). (E) Expression fold change (log2-transformed) of RENC-prioritized target genes for the duplication hotspots in samples with the associated duplications versus the ones without duplications or deletions of the hotspot regions in breast, UGI, and squamous cancers. RENC-prioritized genes inside dup: RENC-prioritized target genes that are within the duplication hotspots; RENC-prioritized genes outside dup: RENC-prioritized target genes that are outside of the duplication hotspots; Other genes: genes that are not prioritized for the duplication hotspots based on RENC analysis, which are used as negative controls. For the “other genes,” expression fold changes between samples with duplications of any hotspot versus the ones without duplications or deletions of these hotspots were calculated. P-values were derived from two-sided Wilcoxon tests: (***) P < 0.001, (NS) not significant. (F) The percentage of RENC-prioritized target genes that encode transcription factors, as compared to that of all the other protein-coding genes. P-value was derived from a Fisher's exact test: (***) P < 0.001.
Next, we analyzed the expression of the target genes using PCAWG RNA-seq data (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). Overall, the target genes exhibit higher expression in samples of breast, UGI, and squamous cancers with the duplication events, regardless of whether they are within the duplication hotspots (Fig. 4E), suggesting that enhancer duplications may activate the expression of the identified target genes. A clear trend is not observed in melanoma samples (Supplemental Fig. S4B), which is likely due to the low number of RENC-prioritized genes identified. RNA-seq data are not available for PCAWG prostate cancer samples.
The list of the predicted target genes exhibits high relevance in tumorigenesis. Indeed, some of the target genes are known oncogenes, but have not been associated with enhancer duplications. For instance, we found the cell cycle gene BCL3, the TP53 E3 ligase gene MDM2, the angiogenesis-promoting gene VEGFA, the translation elongation gene EEF1A1, the receptor tyrosine kinase gene ERBB3, and the Rho-signaling activator gene JUP are targeted by enhancer duplications (Fig. 4A–D; Supplemental Fig. S3D). Furthermore, transcription factor genes (Lambert et al. 2018) account for 22.2% of the identified target genes, showing a significant overrepresentation (Fig. 4F). In particular, we found duplications of enhancers targeting genes encoding lineage-specific oncogenic transcription factors, such as ESR1, FOXA1, and GATA3 in breast cancer, GATA6, ELF3, and EHF in UGI cancers, SOX2, KLF5, and TP63 in squamous cancers, and MITF in melanoma (Fig. 4A–D). In addition, we identified potentially novel oncogenic transcription factor genes targeted by enhancer duplications, including GRHL2, IER2, and IRF2BP2 in breast cancer and ZNF217, CREB3L1, and IRF2BP2 in UGI cancers (Fig. 4A–D; Supplemental Fig. S3D). The potential oncogenic features of these transcription factors require future investigations.
Distinct patterns of enhancer duplications
Using the RENC method, we revealed different patterns of enhancer duplications activating the expression of the target genes. For instance, we found that duplication hotspots linked to promoters of oncogenes like MYC, ESR1, TP63, and GATA3 in diverse cancer types contain only their associated enhancers but not the target genes (Fig. 5A; Supplemental Fig. S4C). We applied CRISPRi to repress the enhancers with relatively strong activity, based on the number of PETs connected to the RENC-prioritized genes, in the duplication hotspots linked to MYC, ESR1, and TP63, which resulted in significant reductions in their expression in the M14 (melanoma), MCF-7 (breast cancer), and BICR16 (squamous cancer) cell lines, respectively (Fig. 5B). In contrast, duplication hotspots associated with GATA6 and VEGFA and the potentially novel oncogenes IRF2BP2 and CREB3L1 harbor both the enhancers and the genes themselves (Fig. 5C; Supplemental Fig. S4D). CRISPRi-mediated repression of the enhancers with relatively strong activity in the duplication hotspots linked to IRF2BP2, GATA6, and VEGFA resulted in significant reductions in their expression in the gastric cancer cell line AGS (Fig. 5D). Among the six tested enhancers, the ESR1, TP63, and IRF2BP2 enhancers are amplified in the tested cancer cell lines, based on SNP-array copy number data from CCLE, while the GATA6 and VEGFA enhancers are not amplified and copy number of the MYC enhancer is unknown in the corresponding cell lines. However, as illustrated by the KLF5 and AR examples (Fig. 3B,C) and the pseudo example presented in Supplemental Figure S3B, we expect the RENC analysis to identify the target genes for the hotspots regardless of the copy number status in the tested cell lines.
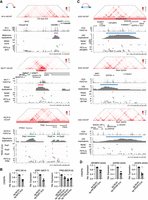
Different patterns of enhancer duplications activating cancer-related genes. (A) Duplications of noncoding enhancers activating promoters of the oncogenes MYC in melanoma, ESR1 in breast cancer, and TP63 in squamous cancer. Presented tracks include H3K27ac HiChIP signal, H3K27ac ChIP-seq signal, positions of the identified duplication hotspots, duplication events observed in the corresponding cancer types, RENC scores prioritizing gene promoters that are more likely to be activated by enhancers within the duplication hotspot, and the number of PETs of each significant interaction between the enhancers and the RENC-prioritized gene promoter. The SNP-array-based copy number (CN) status of the hotspot in the corresponding cancer cell lines is indicated (the SNP-array data are not available for the M14 cell line). Note that the RENC analysis prioritized the short isoform of TP63 (deltaNP63) as the target gene for the duplication hotspot presented in squamous cancer, so that the RENC score of the deltaNP63 promoter is used to represent the TP63 gene. (B) CRISPRi of the indicated enhancers for MYC, ESR1, and TP63 caused significant reductions in their expression, as measured by RT-qPCR, in the M14, MCF-7, and BICR16 cell lines, respectively (n = 3 biological replicates). P-values were derived from two-sided t-tests: (*) P < 0.05, (**) P < 0.01. (C) Duplications of noncoding enhancers together with their target genes such as IRF2BP2, GATA6, and VEGFA in UGI cancer. Similar to A, presented are H3K27ac HiChIP signal, H3K27ac ChIP-seq signal, positions of the duplication hotspots, the duplication occurrence within the highlighted windows, RENC scores prioritizing gene promoters that are more likely to be activated by enhancers within the hotspot, and the number of PETs of each significant interaction between the enhancers and the RENC-prioritized gene promoter. SNP-array-based copy number status of the hotspot in AGS cells is indicated. (D) CRISPRi of the indicated enhancers for IRF2BP2, GATA6, and VEGFA caused significant reductions in their expression, as measured by RT-qPCR, in AGS cells (n = 3 biological replicates). P-values were derived from two-sided t-tests: (*) P < 0.05, (**) P < 0.01.
Plotting the RENC scores of RENC-prioritized genes and other genes nearby the duplication hotspots showed that, although the examined regions have different scales of RENC scores, RENC analysis enables the separation and prioritization of the target genes (Supplemental Fig. S5). We then tested the effects of CRISPRi on nearby expressed genes of the six validated target genes, which are genes ranked second to the prioritized ones (ARMT1 for the ESR1 locus, CLDN1 for the TP63 locus, TARBP1 for the IRF2BP2 locus, ESCO1 for the GATA6 locus, and MAD2L1BP for the VEGFA locus) or adjacent to the duplication hotspot if no additional genes are linked to the hotspot via enhancer–promoter loops (FAM49B for the MYC locus) (Supplemental Fig. S6). Among the tested genes, only ARMT1 showed a significant reduction after CRISPRi of the corresponding enhancer, supporting the robustness of the RENC analysis in prioritizing gene targets.
Among the validated target genes, MYC is a known oncogene driving cancer cell proliferation (Dang 2013), while ESR1 and GATA6 are also required for proliferation of the corresponding cell lines based on the DepMap CRISPR knockout screen data (CRISPR Chronos gene effect score <−0.5, based on the DepMap 23Q4 release) (Dempster et al. 2019). We then performed cell proliferation assays following CRISPRi of the validated enhancers of the three target genes, which show that these enhancers are necessary for the proliferation of the corresponding cell lines (Supplemental Fig. S7). These data support the notion that functional enhancers are important components of oncogenic duplications.
A novel duplication hotspot and its oncogenic target
We focused on an UGI cancer-specific duplication hotspot at Chromosome 10p15 that harbors a cluster of strong enhancer elements based on the H3K27ac ChIP-seq signal (Fig. 6A). These enhancers are duplicated in seven UGI cancer samples (4.7%). Located at the center of the duplication hotspot, these enhancers are adjacent to the CALML3 and CALML5 genes that encode two calcium-binding proteins. However, the RENC analysis predicted that these enhancers skip over CALML3 and CALML5 to activate the neuroepithelial cell transforming 1 (NET1) gene at the far end of the duplication hotspot. The NET1 gene contains two isoforms driven by two distinct promoters, of which only the short one (NET1-short) was predicted to be the target of the enhancers (Fig. 6A).
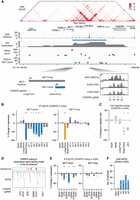
Functional dissection of a duplication hotspot at the NET1 locus. (A) Presented tracks are H3K27ac HiChIP and H3K27ac ChIP-seq signal from the AGS cells, the position of the identified duplication hotspot, and the duplication events observed in UGI cancer. The RENC methodology prioritized NET1-short as the target gene isoform of the enhancers within the duplication hotspot, and also identified a cluster of four individual enhancers as the main driver for NET1-short transcriptional activation. TCGA ATAC-seq signal at the highlighted region containing the e1–e4 enhancers is presented. (B) RT-qPCR results showing expression changes of NET1-short and NET1-long isoforms after CRISPRi of the enhancers e1–e4 in AGS cells (n = 3 biological replicates). sgRNAs (NC#1 and NC#2) with no recognition sites in the genome serve as negative controls. sgRNAs targeting the promoters of NET1-short and NET1-long serve as positive controls. P-values were derived from two-sided t-tests: (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (C) Cell migration rate changes of AGS cells after CRISPRi of the NET1-short promoter, the NET1-long promoter, or the e3 enhancer (n = 3 biological replicates). P-values were derived from two-sided t-tests: (*) P < 0.05, (**) P < 0.01. (D, top) phastCons score (0:1 range) representing the conservation level of the DNA sequences in the e3 enhancer; (middle) distribution of JASPAR transcription factor-binding motifs identified in the e3 enhancer; (bottom) CRISPR cutting positions and the disrupted DNA motifs. (E) RT-qPCR results showing expression changes of NET1-short and NET1-long isoforms after CRISPR cutting of the annotated motifs (n = 3 biological replicates). P-values were derived from two-sided t-tests: (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (F) SOX9 ChIP-qPCR results showing the binding of SOX9 at the enhancers e1–e4 (n = 3 biological replicates) in AGS cells. NR1 and NR2 are two negative control regions. The ChIP-qPCR signal was normalized to the sonicated genomic input and then normalized to the averaged signal of NR1 and NR2.
To validate these predictions, we applied CRISPRi assays to individually repress these enhancers, followed by RT-qPCR assays to measure the expression changes of the two isoforms of NET1, as well as CALML3 and CALML5, in the gastric cancer cell lines AGS and SNU719 (Fig. 6B; Supplemental Fig. S8A,B). Repression of the four tested enhancers e1–e4 caused a range of reductions in the expression of NET1-short, with the e3 enhancer exhibiting the strongest effects in both tested cell lines (74.8% and 63.6% reductions in AGS and SNU719, respectively) (Fig. 6B; Supplemental Fig. S8B). In contrast, repressing these enhancers in AGS and SNU719 cells had little effect on the expression of CALML3 and CALML5 that are located between NET1 and the enhancers (Supplemental Fig. S8A,B). Luciferase reporter assays validated the regulatory activity of the strongest enhancer e3, which is doubled when the enhancer is duplicated on the reporter construct (Supplemental Fig. S8C).
Previous studies have shown that the NET1 gene promotes cell migration of gastric cancer cells (Murray et al. 2008), a key step for tumor metastasis; however, it was unknown which isoform of NET1 plays the role. We showed that repression of e3, the strongest enhancer regulating the NET1-short isoform, caused significant reductions in cell migration of AGS cells, which is comparable to the effects observed from repression of the NET1-short promoter (Fig. 6C). In contrast, repression of the NET1-long promoter had little effects on cell migration of AGS cells (Fig. 6C). These results showed that enhancer-activation of NET1-short is a potentially oncogenic event in gastric cancer.
Focusing on the e3 enhancer, we next sought to identify the bound oncogenic transcription factors. Motif analysis of the e3 enhancer identified DNA sequences recognized by multiple transcription factor families, some of which are distributed in regions that are highly conserved across diverse species (Fig. 6D; Supplemental Fig. S8D). We applied CRISPR cutting to disrupt the DNA motifs, if applicable, and assessed their effects on NET1 expression in AGS cells. We observed significant reductions in the NET1-short expression after disruption of DNA motifs recognized by transcription factor families ETS, SOX, STAT, and TCF7, with disruption of the SOX motif showing the strongest effect (on average 49.5% reduction) (Fig. 6E). In contrast, disruptions of these motifs have little effects on NET1-long expression, which is consistent with our finding that the e3 enhancer only regulates the NET1-short isoform. Among the SOX family members, SOX9 is a known oncogenic transcription factor in gastric cancer (Santos et al. 2016; Chen et al. 2023). We performed SOX9 ChIP-qPCR in AGS and SNU719 cells, which validated the binding of SOX9 at the enhancers e1–e4, with the strongest binding observed at e3 (Fig. 6F; Supplemental Fig. S8E). Together, these data identified an oncogenic isoform of the NET1 gene that is transcriptionally activated by SOX9-bound enhancers within a duplication hotspot in gastric cancer.
Discussion
Our understanding of tumorigenesis requires comprehensive annotation of the cancer genome, as genomic alterations of noncoding regulatory elements are emerging as critical oncogenic mechanisms. Previous copy number analyses were mainly gene-centric and were based on SNP-array data that is relatively low-resolution and misses structural information. Our study used the PCAWG high-resolution whole-genome sequencing data to identify duplication hotspots in an unbiased manner. We found that the majority of the duplication hotspots do not harbor protein-coding genes but, instead, are enriched with potential enhancer elements. Consistent with the cancer-type-specific distributions of the duplication events, their associated enhancers also exhibit activity specific to the corresponding cancer types. Although individual examples of enhancer duplications have been previously reported, this study reveals the prevalence of these events, which suggests that these enhancers may serve as the selection force to shape the duplication profiles during tumorigenesis and implicates their potential value in tumor diagnosis.
One key step of studying noncoding alterations is to identify their target genes. Although 3D genomics assays have enabled mapping of enhancer–promoter interactions throughout the genome, the duplication events are often associated with multiple enhancers and complex enhancer–gene links, which makes it challenging to tease out the real targets. Built on the newly developed ABC model (Fulco et al. 2019; Nasser et al. 2021), we proposed a methodology that utilizes HiChIP results to prioritize target genes for the duplicated enhancers. The method takes into account the activity delivered to each of the candidate genes from the duplicated enhancers (enhancer perspective, Ein), as well as their relative contribution as compared to the rest of the enhancers for each gene (gene perspective, Ein/(Ein + Eout)). Overall, Ein plays a larger role in prioritizing the target genes than Ein/(Ein + Eout); however, the latter serves as an important additional parameter to separate genes with similar Ein values. With more comprehensive functional validation of duplicated enhancers and new insights into the oncogenic features of the target genes, we anticipate further optimization of the methodology in the future.
In addition to previously reported individual enhancer duplication events, the analysis prioritized novel target genes for over a hundred duplication hotspots found in five different cancer types, largely extending the category of functional enhancer duplications. We expect that further characterization of these events will reveal novel oncogenic mechanisms in these cancer types, as exemplified by our finding of the oncogenic isoform of NET1 and its predominant enhancers. While our work uses PCAWG data that are mostly derived from primary tumors, future efforts comparing these data versus those derived from metastatic tumors such as the Hartwig Medical Foundation data (Priestley et al. 2019) may reveal enhancer duplication events that promote tumor metastasis.
Among the identified targets, we found a strong enrichment of transcription factor genes. These include known oncogenes such as ESR1, ELF3, FOXA1, GATA3, GATA6, and TP63, as well as potentially novel oncogenic transcription factor genes such as IRF2BP2, CREB3L1, and GRHL2. This is likely because that, different from other oncogenes such as the RTK/RAS/RAF members that tend to be activated by amino acid changes in their enzymatic domains, oncogenic transcription factor genes are more often activated by their aberrantly high abundance (Bhagwat and Vakoc 2015). Noteworthy, the duplicated enhancers are also enriched with motifs recognized by several of the oncogenic transcription factors themselves, suggesting positive feedback loops activating their expression. As most of the transcription factors lack enzymatic domains, it has been difficult to identify small molecules to inhibit them. Future work identifying chromatin regulators bound at their duplicated enhancers may reveal novel drug targets. Additionally, the PROTAC technology and its derivatives (Sakamoto et al. 2001) may also provide additional avenues for degrading these oncogenic transcription factors.
Our study has several limitations. First, we used HiChIP data from one or two cell lines of each cancer type, which may have missed enhancers that are only present in certain subtypes and not represented by these cell lines. Second, it has been reported that duplications spanning TAD boundaries may cause changes in chromatin architecture and the formation of novel enhancer–promoter interactions. Although only a small proportion of the duplication hotspots span TAD boundaries, our analysis may have missed oncogenes that are activated by such mechanisms. Third, the RENC methodology prioritizes a single target gene for each duplication hotspot. However, it is possible that large duplication events, especially the ones harboring many enhancers, may affect multiple genes. Addressing these limits requires future efforts such as obtaining enhancer–promoter interaction maps from more cell models and primary tumor samples, as well as updating the current methodology with new knowledge about gene regulation.
In summary, combining genomics, epigenomics, and 3D genomics approaches, our study demonstrated the prevalent involvement of enhancers in DNA duplications and identified their target genes, which expands our understanding of this type of structural alteration in cancer and nominates novel, potential therapeutic targets for future investigation.
Methods
Cell culture
MCF-7, AGS, and SNU719 cells were obtained from ATCC. BICR16 cells were obtained from Sigma. MCF-7 cells were grown in DMEM media supplemented with 10% fetal bovine serum and 1% of penicillin–streptomycin, while the remaining cell lines were grown in RPMI-1640 media with the same supplements. Cells were tested negative for mycoplasma using the Beyotime Mycoplasma PCR detection kit. Cell line identities were verified by short tandem repeat analysis.
CRISPRi
For CRISPRi assays, we used a previously generated lentiviral vector stably expressing the dCas9–KRAB–MeCP2 fusion (Liu et al. 2021). Cells infected with the lentiviral vector were selected with blasticidin (10 µg/mL) for at least 5 days. Enhancer-targeting sgRNAs were designed to recognize regions close to the centers of ATAC-seq peaks and were cloned into the LentiGuide-Puro vector (Addgene 52963). The dCas9–KRAB–MeCP2 cells were then infected with LentiGuide-Puro carrying either enhancer-targeting sgRNAs or previously published negative control sgRNAs that have no recognition sites in the genome (Liu et al. 2021). The sgRNA-infected cells were selected with puromycin (2 µg/mL) for 5 days before RNA extraction. For RT-qPCR, the extracted RNA was first converted into cDNA with NEB LunaScript SuperMix kit and then processed with real-time PCR using the NEB Luna Universal qPCR Master Mix. The qPCR signal was normalized to the internal reference gene HPRT1 or CTCF. The sgRNA oligos and RT-qPCR primers were listed in Supplemental Table S4.
Cell proliferation and migration assay
For cell proliferation assays, the infected cells were selected for 3 days and then seeded at the same number (AGS: 0.02 million; M14 and MCF-7: 0.03 million) per well in a 6-well plate and counted after 7 days, using the Countess 2 automated cell counter. For cell migration assays, 0.5 million infected and selected AGS cells were seeded per well in a 6-well plate and waited till they form confluent monolayers in the wells. A 200 µL tip was used to create a scratch across each well. PBS was used to wash away the debris and then replaced with fresh cell culture media. Cell images were captured using an EVOS imaging system at hours 0 and 48 after cell scratch to monitor cell migration. The distances between the sides of the scratches were quantified using the ImageJ software. We took six to eight images of each well to calculate the mean of the migrated distance. Cell migration rate was normalized to that of cells treated with negative control sgRNAs.
Luciferase reporter assay
The enhancer regions (e2 and e3) were cloned upstream of the minimal promoter of the pGL3-Promoter luciferase reporter construct using the MluI and XhoI restriction enzyme cutting sites. To duplicate the enhancer, an additional e2 or e3 region was cloned into the KpnI and MluI sites. The enhancer luciferase constructs were cotransfected with a control Renilla luciferase construct into AGS and SNU719 cells. The luciferase signal, measured 2 days posttransfection, was first normalized to the Renilla luciferase signal and then to the signal from the empty control.
PCAWG duplication analysis
The list of structural variants, including duplications, deletions, inversions, and translocations, was downloaded from the PCAWG project (https://docs.icgc-argo.org/docs/data-access/icgc-25k-data?; The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). We focused on duplications that are <1 Mb in length for the subsequent analysis. We used continuous 5 kb bins to tile the entire genome and counted their duplication occurrence in each cancer type using BEDTools (Quinlan and Hall 2010). Based on the duplication frequency, we then calculated the Euclidean distance among cancer types with more than 30 samples and performed hierarchical clustering. Cancer types that are biologically similar to each other based on the clustering results as well as previous literature were combined for duplication hotspot identification.
To identify duplication hotspots, P-values were calculated using a Poisson distribution based on the average duplication occurrence across the continuous 5 kb bins. Significantly duplicated bins that are adjacent or only one 5 kb bin apart from each other were stitched together to form the duplication hotspots in each cancer type. For comparing duplication hotspots in different cancer types, a minimum of 50% overlap is required for a hotspot to be categorized as “shared” in another cancer type. If a hotspot region harbors an entire protein-coding gene (from the start codon till the stop codon), then the region will be called as a gene-harboring hotspot, otherwise a nongene hotspot. The Circlize tool (Gu et al. 2014) was used to present the distribution of the identified duplication hotspots.
Enrichment analysis of regulatory elements
We downloaded the Pan-cancer chromatin accessible sites identified by ATAC-seq in 410 samples from 23 cancer types included in the TCGA project (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG) (Corces et al. 2018). We converted the DNA coordinates of the ATAC sites from hg38 to hg19 using the liftOver tool (Kent et al. 2002), and then divided the ATAC sites into promoter and distal sites based on their overlap with gene promoters (±2.5 kb of TSS). Using the BEDTools shuffle function (Quinlan and Hall 2010), we performed 1000 times of simulations to generate randomly distributed duplication hotspots within genomic regions that are duplicated at least once, which serves as the background for the enrichment analysis. We then calculated and compared the density of promoter and distal ATAC sites (number of ATAC sites every 10 kb) within the identified versus the simulated duplication hotspots. For cancer-type specificity analysis, we used the normalized ATAC-seq insertion counts data (ATAC signal) released by TCGA (Corces et al. 2018). In particular, for the duplication hotspots found in each cancer type, we calculated Z-scores based on the averaged ATAC signal of distal ATAC sites (sum of ATAC signal of distal ATAC sites within the hotspots divided by the number of ATAC samples) across different cancer types. HOMER motif analyses (Heinz et al. 2010) were performed to identify transcription factor motifs enriched at distal ATAC sites within the duplication hotspots.
HiChIP analysis
H3K27ac HiChIP sequencing reads were aligned to hg19 using the HiC-Pro pipeline (Servant et al. 2015). For presenting HiChIP signal, we used the AllValidPairs output from HiC-Pro that lists all valid PETs. We then mapped the PETs to continuous 5 kb bins within the regions of interest and presented the interactions as heatmaps using the gTrack package (Hadi et al. 2020). For calling significant chromatin loops, we used the hichipper pipeline (Lareau and Aryee 2018). In particular, we first used TCGA ATAC sites identified in the corresponding cancer types as predetermined peaks for loop calling by hichipper (i.e., breast cancer ATAC sites for MCF-7 and ZR751 HiChIP data; stomach cancer ATAC sites for AGS HiChIP data; squamous cancer ATAC sites for BICR16 and BICR31 HiChIP data; prostate cancer ATAC sites for LNCaP and VCaP HiChIP data; melanoma ATAC sites for M14 HiChIP data). We then selected the significant loops (PETs ≥ 3 and FDR < 0.05) of which only one anchor overlaps with the transcription start site (TSS) of a RefSeq protein-coding gene, which we referred to as enhancer–promoter loops. The TSS-overlapping HiChIP anchors will be treated as promoters for the corresponding genes and the remaining HiChIP anchors will be treated as enhancers.
Ranking of target genes based on enhancer–promoter contacts
Built on the ABC model (Fulco et al. 2019; Nasser et al. 2021), we developed a HiChIP-based methodology to rank candidate target genes for duplication hotspots, using the following formula: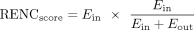 For a given duplication hotspot, Ein is the sum of the PETs from all significant HiChIP loops (PETs ≥ 3 and FDR < 0.05) connecting the enhancers within the hotspot
to a candidate gene's promoter (EPin):
For a given duplication hotspot, Ein is the sum of the PETs from all significant HiChIP loops (PETs ≥ 3 and FDR < 0.05) connecting the enhancers within the hotspot
to a candidate gene's promoter (EPin): 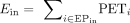 . Eout is the sum of the PETs from all significant HiChIP loops connecting the enhancers outside the hotspot to the candidate gene's
promoter (EPout):
. Eout is the sum of the PETs from all significant HiChIP loops connecting the enhancers outside the hotspot to the candidate gene's
promoter (EPout):  . Therefore, Ein represents the absolute enhancer activity delivered from the hotspot to each gene promoter.
. Therefore, Ein represents the absolute enhancer activity delivered from the hotspot to each gene promoter. 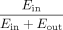 represents the relative contribution of hotspot-delivered enhancer activity as a percentage of activity delivered from all
its enhancers (including the enhancers inside or outside the duplication hotspots). The multiplication of the two factors:
the absolute enhancer activity delivered (PETs) and their relative contribution to each gene promoter (%), is used for ranking
the target genes for each hotspot.
represents the relative contribution of hotspot-delivered enhancer activity as a percentage of activity delivered from all
its enhancers (including the enhancers inside or outside the duplication hotspots). The multiplication of the two factors:
the absolute enhancer activity delivered (PETs) and their relative contribution to each gene promoter (%), is used for ranking
the target genes for each hotspot.
For genes that have multiple promoters, each of the promoters will be treated separately and the promoter with the highest RENC score will be used to represent the gene. Genes not linked to the duplication hotspot through a significant enhancer–promoter HiChIP loop are given a RENC score of zero. For cancer types that have HiChIP data available from more than one cell line, we selected the prioritized gene promoters that are shared. As the RENC score is calculated per duplication hotspot, genes linked to different hotspots are not compared to each other.
When plotting the RENC scores of individual genes linked to each duplication hotspot, we also indicated the copy number status of the hotspot in the corresponding cancer cell line. Based on the SNP-array data released by the CCLE project (Barretina et al. 2012; Ghandi et al. 2019), we annotated hotspot-overlapping regions with segment values (log2(Copy number/2)) > 0.5 as amplified, the ones with segment values <−0.5 as deleted, and the rest as near diploid.
Gene expression analysis
We downloaded the PCAWG RNA-seq results (normalized gene-level FPKM upper quantile normalized values) from the UCSC Xena Data Hubs (https://xenabrowser.net/hub/), and selected the samples that have both RNA-seq and WGS structural variants data available for gene expression analysis. For each RENC-prioritized gene of the duplication hotspots, we identified samples in the corresponding cancer type with duplications overlapping with the hotspot and the ones without duplications or deletions at the hotspot region. Between the two groups, we calculated log2 expression fold changes of the RENC-prioritized genes. As controls, we used genes not prioritized for any of the duplication hotspots and calculated their log2 expression fold changes between samples with duplications of any hotspot region versus the ones without duplications or deletions of these hotspot regions.
Public data usage
This study used PCAWG consensus SV calls downloaded from https://docs.icgc-argo.org/docs/data-access/icgc-25k-data?, PCAWG RNA-seq data downloaded from https://xenabrowser.net/hub/, TCGA SNP-array-based copy number data downloaded from https://gdc.cancer.gov/about-data/publications/pancanatlas, TCGA ATAC-seq data downloaded from https://gdc.cancer.gov/about-data/publications/ATACseq-AWG, and publicly available ChIP-seq and HiChIP data listed in Supplemental Table S3.
Reference genome
This study used the hg19 reference because the released PCAWG processed data were in this reference. We do not expect using hg38 or other more recent reference genomes would significantly affect the conclusions, because this study focused on genes and regulatory elements that are well preserved among these reference genomes.
Software availability
Custom code used in this study is included in the Supplemental Code and is accessible at GitHub (https://github.com/yqsongGitHub/RENC).
Competing interest statement
The authors declare no competing intersts.
Acknowledgments
We thank Jiacheng Dai and Yiqiao Li for technical support and useful discussions. S.D.B. is the recipient of a Chercheur-boursier Junior 1 award from the Fonds de Racherche du Québec—Santé (FRQS).
Author contributions: Y.S. and S.W. performed the bioinformatics analysis; F.L., Y.W., C.L., and L.C. performed the wet-laboratory experiments; S.D.B. and X.Z. designed the study; N.J., J.L., X.C., S.D.B., and X.Z. supervised the study; Y.S., F.L., S.W., X.C., S.D.B., and X.Z. wrote the manuscript with input from other coauthors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278418.123.
- Received August 18, 2023.
- Accepted September 18, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








