
Multiple Pristionchus pacificus genomes reveal distinct evolutionary dynamics between de novo candidates and duplicated genes
Abstract
The birth of new genes is a major molecular innovation driving phenotypic diversity across all domains of life. Although repurposing of existing protein-coding material by duplication is considered the main process of new gene formation, recent studies have discovered thousands of transcriptionally active sequences as a rich source of new genes. However, differential loss rates have to be assumed to reconcile the high birth rates of these incipient de novo genes with the dominance of ancient gene families in individual genomes. Here, we test this rapid turnover hypothesis in the context of the nematode model organism Pristionchus pacificus. We extended the existing species-level phylogenomic framework by sequencing the genomes of six divergent P. pacificus strains. We used these data to study the evolutionary dynamics of different age classes and categories of origin at a population level. Contrasting de novo candidates with new families that arose by duplication and divergence from known genes, we find that de novo candidates are typically shorter, show less expression, and are overrepresented on the sex chromosome. Although the contribution of de novo candidates increases toward young age classes, multiple comparisons within the same age class showed significantly higher attrition in de novo candidates than in known genes. Similarly, young genes remain under weak evolutionary constraints with de novo candidates representing the fastest evolving subcategory. Altogether, this study provides empirical evidence for the rapid turnover hypothesis and highlights the importance of the evolutionary timescale when quantifying the contribution of different mechanisms toward new gene formation.
Genetic studies across multiple animal phyla showed that phenotypic innovations are frequently associated with the evolution of new genes (Mayer et al. 2015; Santos et al. 2017). New genes can arise from gene duplication events or emerge de novo from previously noncoding sequences (Chen et al. 2013; Van Oss and Carvunis 2019). Distinguishing products of gene duplications from de novo genes is often not straightforward, as extensive sequence divergence can prohibit the detection of homologous sequences. Consequently, depending on the taxonomic resolution of available genomic data, up to one-third of genes in a given organism are classified as orphan genes that do not have recognizable homologs in other phylogenetic lineages (Toll-Riera et al. 2009; Tautz and Domazet-Lošo 2011). Although the formation of new genes from previously noncoding sequences was initially thought to be unlikely (Jacob 1977), over the past decades a growing number of studies have identified multiple instances of de novo genes across various taxonomic lineages (Heinen et al. 2009; Knowles and McLysaght 2009; Carvunis et al. 2012; Zhao et al. 2014; Vakirlis et al. 2018; Zhang et al. 2019a). With them, the number of identified de novo genes varied greatly with their definition. The resulting numbers ranged from thousands of species-specific open reading frames with transcriptional activity in yeast to a single gene with evidence of purifying selection and a well-supported noncoding status of the ancestral sequence in Drosophila (Zile et al. 2020; Li et al. 2021). Recent studies using deep transcriptomics and ribosome-profiling identified thousands of species-specific sequences with evidence of transcription and translation that form the raw material for incipient de novo gene formation (Ruiz-Orera et al. 2014; Neme and Tautz 2016; Schmitz et al. 2018; Blevins et al. 2021; Li et al. 2021). The numbers of products of pervasive transcription and translation by far exceed the numbers of duplications found in individual comparisons (Pegueroles et al. 2013; Baskaran and Rödelsperger 2015). However, the fact that most genes in a given genome are members of known gene families implies that a higher loss rate counteracts the higher birth rate of de novo genes (Palmieri et al. 2014; Schmitz et al. 2018). We have recently incorporated these contradicting observations into a hypothetical model for the lifetime dynamics of duplicated and de novo genes (Rödelsperger et al. 2019). One major prediction of this rapid turnover model is that the contribution of both categories of origin will depend on the evolutionary distance of the comparison. In this study, we explicitly test this model using genomic data of the nematode Pristionchus pacificus. P. pacificus is a free-living nematode that shared a common ancestor with Caenorhabditis elegans 60–90 million years ago (Cutter 2008; Prabh et al. 2018). P. pacificus was initially introduced as a model system for comparative biology (Hong and Sommer 2006). Both P. pacificus and C. elegans are androdioecious species, in which populations are mostly composed of hermaphrodites that can self-fertilize. Males are produced at a low frequency and allow for recombination between distinct lineages. Ongoing field trips and sampling efforts have resulted in more than 50 described Pristionchus species and several hundred natural isolates of P. pacificus (McGaughran et al. 2016; Kanzaki et al. 2021). The deep taxonomic sampling together with small genome sizes (∼200 Mb) makes this clade of nematodes an ideal model system for phylogenomic studies (Rödelsperger 2018). Previously, we exploited this deep taxonomic sampling by selecting eight Pristionchus and two outgroup species to establish a phylogenomic framework to study the evolutionary dynamics and origin of orphan genes (Prabh et al. 2018; Prabh and Rödelsperger 2019). Thereby, the underlying ladder-like phylogeny allowed us to contrast evolutionary processes in various age classes defined at the species level. In the current study, we extend this to the intraspecies level. To this end, we generated genome assemblies for six divergent strains of P. pacificus. Three of these strains were found on La Réunion Island in the Indian Ocean, which represents a hotspot of P. pacificus biodiversity (McGaughran et al. 2016). P. pacificus strains from different clades can be successfully mated in the laboratory, but population genomic analysis found little evidence for admixture in the wild (Morgan et al. 2017). In addition, the ability to use pheromone profiles to drive other strains into dispersal strategies in the presence of food suggested intraspecific competition (Mayer et al. 2015). This is further supported by the recent finding of predation across divergent strains (Lightfoot et al. 2021). Thus, these strains display certain characteristics of different species without being reproductively isolated (McGaughran et al. 2016). The resulting combination of species and population-level phylogenomic data allows us to perform evolutionary comparisons at various timescales and to provide empirical support for the rapid turnover hypothesis.
Results
Multiple reference genomes facilitate comparisons at various timescales
To gain insights into the intraspecific changes in gene repertoires of P. pacificus, we complemented the existing genome of the P. pacificus reference strain PS312 by generating draft genome assemblies of six additional isolates (Fig. 1A). Initial genome assemblies were generated based on Illumina sequencing of paired-end and mate-pair libraries as described previously (Prabh et al. 2018). The assembly sizes range between 151 and 174 Mb with N50 values of several hundred kilobases (Table 1). Completeness analysis based on the benchmarking of universal single-copy orthologs (BUSCO) yields values between 86% and 92% (Simão et al. 2015). These completeness values are close to 93%, which was obtained for the chromosome-scale P. pacificus reference assembly (Rödelsperger et al. 2017), and are comparable to many other nematode genome assemblies (Supplemental Fig. S1). Evidence-based gene annotations were generated by integrating the community-curated annotations for the reference strain PS312 and strain-specific transcriptome data (Rödelsperger 2021). This resulted in 24,400–26,757 protein-coding gene models with BUSCO completeness values between 87% and 92% (Table 1). Although the quality of these computationally generated gene annotations is comparable to many other nematode genomes (Supplemental Fig. S1), further improvements of genome assemblies and gene annotations are needed to reach the 98% BUSCO completeness of the community-curated P. pacificus reference annotations (Athanasouli et al. 2020). To maximize comparability, we include the PS312 genome from Prabh et al. (2018) in our analysis, which was generated and reannotated using the same methodology. The chromosome-scale assembly is only used for plotting chromosomal distributions of genomic features (Rödelsperger et al. 2017). We applied the OrthoFinder software to detect orthologous groups from the seven P. pacificus strains together with data from 13 different nematode species (Emms and Kelly 2015). This combined all-against-all BLASTP searches with a Markov clustering algorithm (Enright et al. 2002) and identified 37,228 orthogroups in total. Although 8214 orthogroups had a single-copy gene in all of the seven P. pacificus strains (Fig. 1B), around 12,000 orthogroups could not be resolved to the level of one–one orthologs based on protein similarities alone. However, pairwise gene order alignments could identify up to 20,294 (83%) gene pairs as syntenic orthologs (Supplemental Fig. S2). For most of the analyses, we used the OrthoFinder results. The pairwise syntenic orthologs were only used for studying the mechanisms of gene loss. To better quantify genetic distances between the strains, we screened for shared ancestry by calculating mean synonymous site diversity, dS, in 500-kb windows (Fig. 1C). Among all pairwise comparisons, only three pairs showed a high number of recently shared haplotypes with dS < 0.01 (Fig. 1C). The strains RSB020 and RSA016 are from La Réunion Island, which harbors diverse populations that occasionally hybridize (McGaughran et al. 2016). In contrast, the strains PS312 and RS5297 are isolated soil samples from the United States. Similarly, the strains PS1843 and RS5266 were sampled from the United States and Bolivia, respectively, and show smaller amounts of shared haplotypes. Please note that the strain RSB001 is also from La Réunion Island but shows no evidence of admixture with either RSB020 or RSA016, which further supports a process of incipient speciation between different P. pacificus clades. The distribution of dS values across the chromosomes shows reduced diversity at the chromosome centers (Fig. 1D), which is consistent with previous analysis (Rödelsperger et al. 2017). Please note that the P. pacificus Chromosome I is known to have two center-like regions. The strain PS1843 shows a pronounced increase in dS on the left arm of Chromosome IV. This could be associated with an atypical crossover pattern that was recently identified by recombination mapping in hybrids and is suggestive of intrachromosomal structural variations (Rillo-Bohn et al. 2021). Altogether, these analyses show a wide range of genetic distances across P. pacificus strains and different haplotypes, facilitating evolutionary comparisons at various timescales.
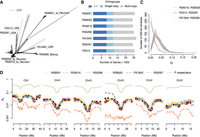
Multiple reference genomes facilitate comparisons at various timescales. (A) The evolutionary distances between 323 P. pacificus strains are displayed as a phylogenetic network. The distances are inferred from 600,000 variable sites that have been called from whole-genome sequencing data (Huson and Bryant 2006; Rödelsperger et al. 2017). (B) Orthologous clustering revealed 8214 genes with one–one orthologs across all seven genomes. Almost half of the genes are assigned to orthogroups with multiple genes per strain. (C) The plot shows the distribution of 500-kb blocks as a function of the mean dS values for every pairwise comparison. The peaks at dS = 0.00 identify recently shared haplotypes between the three most closely related strain pairs. (D) The average dS values are plotted across the megabase position of the chromosome-scale assembly of the reference strain PS312.
Features of seven P. pacificus genomes
Duplications, divergence, and de novo formation shape genome evolution
To investigate the dynamics of gene family evolution, we assigned orthogroups into age classes based on the species’ presence/absence pattern (Fig. 2A). Under the most parsimonious scenario, these patterns assign the origin of an orthogroup to a specific branch in the species tree. We consider an orthogroup to be present in P. pacificus if an ortholog exists in at least one of the strains. Adding the six P. pacificus strains to this analysis identifies 3396 orthogroups that lack an ortholog in the reference strain PS312 but have been preserved in at least one of the six additional strains (Supplemental Fig. S3). To contrast evolutionary patterns with regard to the mechanism of origin, we defined one orthogroup category of known gene families based on the presence of protein domains in more than half of the members of a given orthogroup. Known orthogroups of the older age classes represent, to a large extent, highly conserved single-copy genes. On the contrary, known orthogroups of a younger age derived most likely from a duplication and divergence mechanism; that is, if rapidly evolving members of large superfamilies diverge beyond a certain level, they will appear as a new gene family. Note that there are also duplications within orthogroups that give rise to multiple paralogs. These duplicates are not the subject of the current study, as we focus on the emergence and loss of new gene families. Next, we defined a category of orthogroups representing de novo candidates based on the absence of BLASTP hits in a set of outgroup species that are defined by the age class (see Methods). This category includes potential de novo genes but also orphan genes that have no detectable sequence similarity in a set of outgroup species (Vakirlis et al. 2020; Weisman et al. 2020). All remaining orthogroups were grouped into a mixed category that represents fast-evolving members of known gene families or divergent duplicates of older de novo candidates (Fig. 2B). Although older age classes are dominated by known orthogroups, there is a gradual increase in the contribution of de novo candidates toward younger age classes. However, such de novo candidates may not be as evolutionary stable as new genes originating from duplication events. This would explain their decreasing contribution to older age classes (Fig. 2B). We tested the robustness of this trend by varying the definition of known genes and changing the inflation parameter that controls the granularity of orthogroups in the OrthoFinder pipeline. This shifted the ratio between known gene families and de novo candidates (Supplemental Fig. S4). However, irrespective of the parameter settings, the contribution of de novo candidates increases in the younger age classes, whereas the contribution of the known category increases toward older age classes. In summary, the separation of orthogroups into age classes and categories of origin allowed us to quantify the contribution of both categories across different timescales. This showed a gradual decrease in the contribution of de novo candidates with age and supports that de novo candidates are born at a high rate but are evolutionarily less stable than gene families that arose by duplication and divergence.
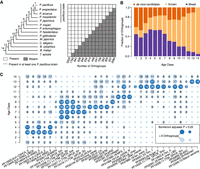
Duplications and new gene formation shape genome evolution. (A) The presence/absence patterns of species in a given orthogroup were used to assign orthogroups to 14 distinct age classes. The age classes define a branch in the schematic phylogeny (Rödelsperger et al. 2018; Smythe et al. 2019), at which a given orthogroup evolved. (B) Orthogroups were classified as known if more than half of the genes had recognizable protein domains and classified as de novo candidates if there was no BLASTP hit against sequences from a set of outgroup species. The remaining orthogroups were classified as mixed. Although older age classes are dominated by members of known gene families, younger age classes are biased toward de novo candidates. (C) The circles represent the number of orthogroups with a given protein domain, and the color code indicates significant enrichments within an age class.
Characterization of core features reflects the molecular and functional dynamics of new gene formation
To further characterize genes from both categories of origin, we investigated some core features like protein length, copy number, and expression level and compared these features between age classes and categories of origin. The comparison of protein lengths reveals that protein length increases with age and that known genes are significantly larger than de novo candidates (Supplemental Fig. S5). Specifically, de novo candidates of age class 1 form the smallest proteins, with a median length of 96 amino acids. These results suggest that the initial products of de novo formation are relatively small peptides that may become larger over time. Next, we explored whether de novo candidates or known genes have different duplication rates, which would manifest in higher copy numbers of genes per orthogroup per genome. This showed that known genes tend to have higher duplication rates, with age classes 8–11 showing the strongest differences (Supplemental Fig. S5). Comparison of expression levels in mixed-stage cultures of all P. pacificus strains (Rödelsperger et al. 2018) indicates that >80% of genes across both categories of origin and all age classes show at least some expression evidence (Supplemental Fig. S5). However, around half of the de novo candidates and known genes in age classes 1–3 are only weakly expressed (0 < FPKM ≤ 1), whereas most genes of the older age classes are robustly expressed (FPKM > 1) (Supplemental Fig. S5). This suggests that new genes of both categories initially have weak or restricted expression but that expression may become stronger or broader over time. Contrasting de novo candidates with known genes, we find that the latter category generally shows higher fractions of robustly expressed genes (Supplemental Fig. S5). Finally, although the absence of homologs impedes any inference of gene function for de novo candidates, we characterized the large category of known orthogroups at different age classes by performing protein domain overrepresentation analysis (Fig. 2C). This revealed stable expansions of specific gene families in the evolutionary lineage leading to P. pacificus. For example, collagens show a specific expansion at the branch leading to the diplogastrid family, which includes the genera Pristionchus, Parapristionchus, and Micoletzkya. Several P. pacificus collagens have been recently shown to be part of a highly conserved network controlling nematode development (Sun et al. 2021). Taken together, these results reflect the dynamic evolution of new genes and reveal evidence that duplications of specific gene families served key functions in the evolution of the P. pacificus lineage.
Young de novo candidates are nonuniformly distributed across chromosomes and show an X Chromosome bias
The chromosomes of P. pacificus and C. elegans are holocentric and have centromeric function distributed along each chromosome (Albertson and Thomson 1993; Rillo-Bohn et al. 2021). Even though they are not monocentric, they have center-like regions that are distinguished from chromosomal arms by lower recombination frequency and genetic diversity, lower repeat content, and higher gene density (The C. elegans Sequencing Consortium 1998; Rödelsperger et al. 2017). The P. pacificus Chromosome I has two such center-like regions (Fig. 1D), which likely represents a fusion of ancestral linkage groups, also called Nigon elements (Tandonnet et al. 2019). Although for most autosomes of C. elegans and P. pacificus, these center-like regions coincide with regions of low recombination rate, this pattern is not observed for P. pacificus Chromosome I, which only shows a single recombination-poor region in the middle of the chromosome (Rockman and Kruglyak 2009; Rödelsperger et al. 2017; Rillo-Bohn et al. 2021). To investigate the distribution of different gene classes across the P. pacificus genome, we plotted the number of known genes and de novo candidates in 500-kb windows across the chromosomes (Fig. 3A,B) and tested for positional bias between the center-like regions and chromosome arms, as well as overrepresentation of gene classes across chromosomes (Fig. 3C,D; Supplemental Fig. S6). Here, we defined chromosome centers based on genetic diversity rather than recombination rate, because the pattern of recombination likely represents a derived feature that was caused by the chromosome fusion. Evidence from hybridization experiments showed that recombination patterns can change very rapidly, and comparative analysis across Pristionchus species suggests that the ancestor of P. pacificus and Pristionchus exspectatus had seven chromosomes (K Yoshida, C Rödelsperger, W Röseler, et al., in prep.). Thus, by defining the chromosome centers based on genetic diversity, we hope to capture the evolutionary signal in the ancestral Nigon elements. This showed that de novo candidates of age classes 1 and 3 are significantly depleted from chromosome centers (Wilcoxon test, Bonferroni-corrected P < 0.05) (Fig. 3C; Supplemental Fig. S6). Furthermore, we find that although Chromosome IV is overrepresented in old gene classes of the known category, Chromosome X is enriched in de novo candidates of age classes 1 and 3 (Wilcoxon test, Bonferroni-corrected P < 0.05) (Fig. 3D; Supplemental Fig. S6). This is consistent with previous findings of an enrichment of novel genes on the X Chromosome of Drosophila (Betrán et al. 2002; Levine et al. 2006). Thus, de novo candidates are not uniformly distributed across chromosomes and are particularly enriched on P. pacificus Chromosome X.
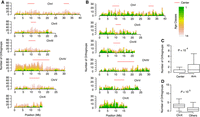
New gene classes are nonuniformly distributed across chromosomes. (A) The number of known orthogroups is plotted in nonoverlapping 500-kb windows across the P. pacificus genome. Center-like regions were defined based on genetic diversity (Rödelsperger et al. 2017) and are indicated by red lines. (B) The plot shows the distribution of de novo candidates across the P. pacificus chromosomes. (C) The box plot shows the number of de novo candidates per 500-kb window. De novo candidates of age class 1 are significantly depleted from chromosome centers. (D) De novo candidates of age class 1 are significantly enriched on P. pacificus Chromosome X.
De novo candidates evolve more rapidly than recent duplicates of known gene families
The observation of a gradual decrease in the contribution of de novo candidates with gene age is in line with our previously postulated rapid turnover model, which proposed distinct evolutionary trajectories posited by new gene families that arose by either duplication or de novo formation (Rödelsperger et al. 2019). However, because different age classes imply various time spans for selection to act on, these gene classes and categories of gene origin should be compared at a similar timescale. To do this, we focused on recent patterns comparing P. pacificus and its sister species P. exspectatus, as well as detecting trends in divergent strains of P. pacificus. First, we measured evolutionary constraint in one–one orthologs between P. pacificus (PS312) and P. exspectatus as the ratio of nonsynonymous and synonymous changes, dN/dS, for different age classes and categories of origin (Yang 1997). This shows substantial evidence for purifying selection in most genes of each class, which implies strong support for biological function (Prabh and Rödelsperger 2016). Moreover, older genes are more constrained than younger ones for both categories of origin (Fig. 4A). This is consistent with previous phylogenomic analyses of yeasts and rice (Stein et al. 2018; Vakirlis et al. 2018). Contrasting de novo candidates with duplicated genes shows a significant trend for weaker constraints in de novo candidates (Wilcoxon test, Benjamini and Hochberg–corrected P < 0.05). To include age class 1 (P. pacificus–specific genes) in this analysis, we compared dN/dS ratios of one–one orthologs between the P. pacificus reference strain PS312 and the strain RSB001 (Fig. 4B). Although the shorter separation time between both strains implies reduced removal of deleterious alleles leading to higher dN/dS ratios, basically the same trends are revealed as for the comparison with P. exspectatus (Fig. 4A). Thus, de novo candidates evolve more rapidly than new gene families that arose by duplication and divergence.
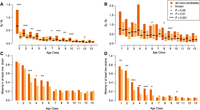
De novo candidates are less constrained and more frequently lost than recent duplicates. (A) The box plot shows the median and interquartile ranges of dN/dS ratios with regard to P. exspectatus orthologs for different age groups. The comparison between de novo candidates and known orthogroups shows evidence that de novo candidates are less constrained than recent duplicates of known gene families. (B) Estimation of evolutionary constraint between the P. pacificus strains PS312 and RSB001 shows a trend toward significantly higher dN/dS ratios of de novo candidates for multiple age classes. (C) The plot shows for each age class the fraction of orthogroups, where an ortholog is missing in at least one P. pacificus strain. Comparison between de novo candidates and known categories shows that de novo candidates are significantly more likely to be lost in three age classes. (D) De novo candidates show an even stronger trend toward higher probabilities of being lost than recently duplicated members of known gene families, if we consider only orthogroups with predicted losses in at least two P. pacificus strains.
De novo candidates are more frequently lost than duplicated gene families
Next, we wanted to test whether the birth rate of the de novo candidates (Fig. 2B) is counteracted by a high loss rate. Therefore, we computed the ratio of orthogroups with a missing ortholog in at least one of the seven P. pacificus strains (Fig. 4C). We must keep in mind that missing orthologs in age class 1 could represent either a loss or a very recent gene birth event that never got fixed. The results confirm our previous findings that young genes tend to be lost much more frequently (Prabh et al. 2018). Specifically, although old age classes show a baseline frequency of ∼20% orthogroups with a missing ortholog in at least one P. pacificus strain, young genes are up to four times more likely to be lost (Fig. 4C). Contrasting the de novo candidate and known category shows a consistent trend for younger age classes that de novo candidates tend to be lost more frequently than known orthogroups. This difference is significant for age classes 3, 4, and 5 (binomial test, Benjamini and Hochberg–corrected P < 0.05). Because missing orthologs could be due to problems during the genome assembly or gene annotation process, we repeated the analysis by screening for missing orthologs in at least two P. pacificus strains. In total, we identified 902 orthogroups with missing orthologs in two strains. Sixty percent of these losses are shared between the three most closely related strain pairs (Fig. 1C). This strong phylogenetic signature supports that the pattern of missing orthologs is not completely stochastic. Overall, the analysis of double losses reveals highly consistent trends with regard to the analysis of single losses (Fig. 4C,D). The frequency of orthogroups with gene losses drops from ∼70% for the youngest age class to ∼10% for older age classes. In addition, age classes 1–6 and 10 show a significantly higher frequency of predicted gene losses in de novo candidates. In summary, these analyses support that de novo candidates are more frequently lost than young duplicates of known gene families.
Structural variations and nonsense mutations are the major source of gene loss
To study the nature of gene loss in P. pacificus, we compared syntenic regions between the different P. pacificus strains. We manually classified 221 missing orthologs that fall into syntenic regions. This was performed by visual inspection of syntenic alignments, TBLASTN searches, and screening for assembly gaps in the genome browser. Among these cases, we find that structural variations, such as deletions and partial translocations, form the most abundant category leading to a missing ortholog (N = 99) (Fig. 5A). In a comparison of different C. elegans strains, it was shown that structural variations affect more nucleotides than single-nucleotide polymorphisms and were predicted to change the function of 2694 genes (Kim et al. 2019). In the case of the Pristionchus-specific gene PS312000018427, the gene seems to be completely deleted from the intergenic region between the two syntenic anchor genes in the strain RSB020 (Fig. 5B). Only an N-terminal fragment of 23 amino acids could be mapped to a different contig of the RSB020 assembly. Another portion of missing orthologs showed assembly gaps in the region between the two closest syntenic anchor genes (N = 77) (Fig. 5A). This indicates that the analysis of gene loss is still hampered by technical problems, and these cases could potentially be false calls. However, a conclusive statement can only be made if the syntenic region would be completely assembled. We found 17 clear annotation errors in which the apparently missing orthologs showed complete TBLASTN alignments, but the gene was still not annotated. This could happen if some genic feature (e.g., minimum intron length) was outside the allowed range or if a gene was classified as a potential isoform and removed because of overlaps with other gene models. Twenty-eight of the candidate loci showed premature stop codons. For example, the Pristionchus-specific gene PS312000020222 encodes a small peptide of 78 amino acids, and orthologs are present in all strain genomes except PS1843. In this strain, it has acquired a premature stop codon at amino acid position 23 (Fig. 5C). In this particular example, one of the downstream amino acids was substituted into a methionine. This could still result in a shortened peptide of 41 amino acids, yet this would fall below our minimal protein length threshold of 60 amino acids. Although the manual inspection of candidates showed that a substantial fraction could be incorrect calls, the majority of candidates could be validated, and there is no bias across different categories of origin (χ2-test, P > 0.05) (Fig. 5A). Thus, we would conclude that missing orthologs may be useful to investigate and compare signatures of gene loss across larger gene sets, but missing orthologs should not automatically be considered as gene-loss events and individual cases need to be carefully validated. Among the confirmed cases, we found that structural variations and nonsense mutations seem to be the major causes of gene loss in P. pacificus.
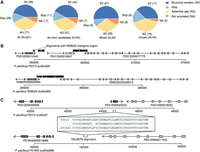
Nonsense mutations and structural variations are the major source of gene loss. (A) The pie charts show the classification of missing orthologs after manual inspection. De novo candidates were defined as genes that are specific to the pacificus clade. (B) The plots show syntenic regions surrounding the Pristionchus-specific gene PS312000018047 in two P. pacificus strains. The x-axis indicates the nucleotide position; exons are shown as rectangles; and syntenic orthologs are color-coded. The presence of two intergenic blocks between the syntenic anchor genes without homology suggests that at least two structural variations occurred that have led to the loss of PS312000018047. (C) The plot shows syntenic regions surrounding the gene PS312000020222 in two P. pacificus strains. The gene is Pristionchus specific and is present in all P. pacificus genomes except in the PS1843 assembly, where it has acquired a premature stop codon.
Loss of sex-biased genes preferentially affects de novo candidates
The previous sections showed how the evolutionary history of a gene determines its likelihood of being lost. However, other factors also contribute to gene loss. In both genera, Pristionchus and Caenorhabditis, it has been shown that hermaphroditism evolved multiple times independently from gonochoristic ancestors and coincided with a subsequent loss of male-specific genes (Rödelsperger et al. 2018; Yin et al. 2018). To test if this gene loss as a consequence of a sexual evolution follows the general dynamics of de novo candidates being lost at a higher rate, we reanalyzed sex-specific transcriptomes of the gonochoristic species Pristionchus arcanus (Rödelsperger et al. 2018). Differential gene expression between male and female transcriptomes (three biological replicates) yielded 2354 (6%) female-biased and 8845 (23%) male-biased genes (Benjamini and Hochberg–adjusted P < 0.05, greater than twofold expression change). This corresponded to 3963 male-biased and 1686 female-biased orthogroups. Protein domain overrepresentation analyses identified multiple male-enriched domains (Fig. 6A) such as motile sperm protein (PF00635), phosphatases (PF00102), metallophosphatases (PF00149), DUF236 (PF03057), and SH2 (PF00017), all of which were recently found to be enriched in sperm-related regions in P. pacificus (Rödelsperger et al. 2021). Female-biased genes are significantly enriched in the yolk protein Vitellogenin (PF01347) and multiple peptidases (Fig. 6A). Next, we investigated the age distribution of sex-biased orthogroups by comparing the most abundant absence–presence patterns. The three most abundant patterns among male-biased orthogroups with putative losses in P. pacificus define pacificus clade-specific orthogroups (Fig. 6B). Specifically, the most frequent pattern represents 228 (6%) gene families that likely originated in the ancestor of P. pacificus, P. exspectatus, and P. arcanus and are now missing in at least one P. pacificus strain (Fig. 6B). For the same pattern, there are only 62 (4%) instances among the female-biased orthogroups (Fig. 6C). Consistent with previous analysis, we find a significant trend toward a higher loss of male-biased genes in the hermaphroditic P. pacificus (binomial test, P < 0.05) but not in the gonochoristic species P. exspectatus (Fig. 6D). Moreover, the difference in the number of male-biased orthogroups that are putatively lost in at least one (28%) versus all (7%) strains implies that the process of male-specific gene degeneration is still ongoing. Furthermore, relative to the overall composition of sex-biased orthogroups, male- and female-biased orthogroups with predicted losses in either P. pacificus or P. exspectatus show a significantly different composition with de novo candidates being preferentially lost (χ2-test, P < 0.001 in all comparisons) (Fig. 6E,F). In summary, there is a trend toward a loss of male-specific genes in the hermaphroditic P. pacificus. However, de novo candidates in general are preferentially affected by gene loss irrespective of the type of expression bias and reproductive mode.
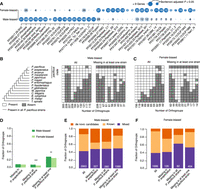
Loss of male-biased genes after the evolution of hermaphroditism preferentially affects de novo candidates. (A) Protein domain overrepresentation analysis reveals multiple protein domains that are enriched in orthogroups with sex-biased expression in P. arcanus. (B,C) The heatmaps next to the schematic phylogeny show the 10 most abundant presence/absence patterns for orthogroups with sex-biased expression. (D) Gene loss in the hermaphroditic P. pacificus preferentially affects male-biased genes but not in the gonochoristic P. exspectatus. (E,F) De novo candidates are overrepresented among predicted losses of male-biased (E) and female-biased (F) orthogroups in either P. pacificus or P. exspectatus.
Discussion
For a gene, what is past is prologue, and thus its age and mechanism of origin may be predictive of its evolutionary stability and impact its longevity, respectively. We previously postulated that in contrast to de novo genes, new duplicates might be engendered with a higher evolutionary stability at inception as their protein sequences are less likely to be toxic and they may readily interact with other proteins (Rödelsperger et al. 2019). This facilitates faster integration into cellular networks, resulting in biological functions that are essential at an organismic level (Keeling et al. 2019). Moreover, processes like pervasive transcription and translation generate far more gene-like sequences than gene duplication. Thus, to explain the dominance of members of known gene families in individual genomes, one has to assume differential turnover rates between both categories of new genes. Although numerous studies have investigated patterns of gene loss showing high turnover rates for young genes (Palmieri et al. 2014; Prabh et al. 2018), this study explicitly tests the rapid turnover hypothesis by comparing de novo candidates with orthogroups that arose by gene duplication and divergence.
The category of de novo candidates includes potential de novo genes and also rapidly evolving orphan genes of unknown origin. This category could equally be called taxonomically restricted genes or orphan genes as they are defined by the absence of protein similarity in a set of outgroup species. In this study, we decided to use the term de novo candidates to be more in line with the rapid turnover hypothesis. We use genome sequencing of multiple P. pacificus isolates to compare all these different categories at the same evolutionary timescale. Our results are in complete agreement with the rapid turnover hypothesis, revealing an increasing contribution of de novo candidates at young age classes and showing that de novo candidates are significantly more often lost than new orthogroups that arose by duplication and divergence. Thus, our study reveals strong evidence for the distinct evolutionary dynamics between de novo candidates and duplicated genes.
A major implication of this work is that the question, which is the dominant process generating new genes, has to be discussed in the context of evolutionary stability. Certainly, pervasive transcription and translation provide the raw material out of which new functions may evolve (Ruiz-Orera et al. 2014; Neme and Tautz 2016; Blevins et al. 2021; Li et al. 2021). Still, a large fraction of these sequences might be neutrally evolving. This is reflected in their elevated dN/dS ratios and a higher probability of being lost. Pervasive transcription is particularly abundant in sperm-related tissues, which has led to the “out of testis” hypothesis (Betrán et al. 2002; Dai et al. 2006; Levine et al. 2006; Vinckenbosch et al. 2006; Zhang et al. 2010). However, it is still not entirely clear whether the expression of new genes in testis reflects the activity of fast-evolving genes with biological functions or might just be the result of a permissive epigenetic state (Soumillon et al. 2013; Witt et al. 2019; Rödelsperger et al. 2021). To emphasize the rare chance of a new sequence being evolutionary stable, the survival of a de novo gene has been previously termed a “frozen accident” (Schmitz et al. 2018). Consequently, the outcomes of studies testing divergence versus de novo processes will depend on the evolutionary distance between the species and may not be generalizable (Vakirlis et al. 2020). Finally, to characterize the evolutionary dynamics of different gene classes, deep taxonomic sampling is absolutely invaluable (Vakirlis et al. 2016; Prabh et al. 2018). We would therefore like to highlight the utility of the Pristionchus system with an extensive phylogenomic data set, generated by a single laboratory and annotated by the same computational pipelines, to study genome evolution at a cross-species and intraspecies level. Our concerted attempt to contrast phenotypic and molecular evolution of diverging strains on both sides of a speciation boundary marked by a major life-history change, that is, shift to a hermaphroditic form, has captured ongoing changes such as accelerated loss of male-specific young genes and variation in selective constraint at the chromosome level among different strains. Thus, our phylogenomic analysis provides an excellent opportunity to unravel the evolutionary processes acting on individual genes, gene families, chromosomes, and genomes. Such a scale of analysis is only possible in a few model systems.
Future work may further use the phylogenomic data of the Pristionchus system to gain a more systematic understanding of the molecular mechanisms driving the emergence of new genes. Through detailed manual inspections of individual candidate genes, we had previously identified multiple mechanisms driving the evolution of new genes in P. pacificus, including de novo formation, divergence, ORF switching, and chimeric origin (Prabh and Rödelsperger 2019). Until now, we have been using rather conservative definitions of genes requiring multiple exons and a minimal protein length of 60 amino acids. Consistently, most genes in our analysis have expression evidence and show signs of purifying selection. However, similar analyses could be performed on a more broadly defined set of biologically active sequences. In combination with recently developed approaches to study the potential origin of orphan genes (Vakirlis et al. 2020; Weisman et al. 2020), such efforts could be used to further dissect the heterogeneous sets of de novo candidates and to clarify to what extent they represent de novo genes or fast-evolving members of other existing genes. However, to ultimately confirm the de novo origin, enabling mutations have to be identified (Vakirlis et al. 2018), which is difficult as noncoding sequences in outgroup species typically degrade very fast. In our previous analysis, we could only identify two instances of de novo genes in P. pacificus with homology with noncoding regions in an outgroup species (Prabh and Rödelsperger 2019). Such a small number is consistent with a recent analysis in Drosophila and C. elegans (Zhang et al. 2019b; Zile et al. 2020). Thus, it will be unlikely that many more candidates of young age classes will be ultimately confirmed as de novo genes. However, the phylogenomic data from multiple divergent P. pacificus strains will offer the opportunity to study the mechanisms of gene origin at a much finer evolutionary timescale. Finally, the different P. pacificus genomes will also be a fundamental resource for future studies elucidating the genetic basis of various traits (Moreno et al. 2016; Namdeo et al. 2018). Given the high levels of genetic diversity between different P. pacificus isolates, the availability of reference genomes for selected clades will improve the analysis of whole-genome sequencing data from mutants and other natural isolates, as structural variations leading to loss and gains of new genes can be more reliably detected (Mayer et al. 2015; Falcke et al. 2018).
Methods
DNA extraction, sequencing, assembly, and scaffolding
All P. pacificus strains were grown on nematode growth medium (NGM) plates before DNA extraction. We rinsed the plates with M9 buffer and collected worm pellets by slow centrifugation at 1300 rpm for 3 min at 4°C. DNA extraction was performed as described previously (Rödelsperger et al. 2017). PCR-free libraries were generated with a TruSeq DNA PCR-Free Library Prep Kit following the manufacturer's protocol, and sequencing was performed on Illumina MiSeq. Initial assemblies were constructed with the DISCOVAR de novo assembler (version r52488). We checked for Escherichia coli contamination by BLASTN against in-house and NCBI E. coli genomes and removed contaminated contigs after manual inspection. Finally, scaffolding was performed with SSPACE_Basic_v2.0 (Boetzer et al. 2011) using four mate-pair libraries of sizes 1.5, 3, 5, and 8 kb (that were generated with a Nextera Mate Pair Sample Preparation Kit). The BUSCO software (version 3.1.0 with the odb9 nematode data set) was run in genome mode to assess the completeness of the genome assemblies and for the comparison with phylogenomic data of 54 other nematode genomes (Rödelsperger 2021).
Gene annotation
Evidence-based gene annotations were generated using the PPCAC pipeline (version 1.0) (Rödelsperger 2021). In summary, the highly curated reference gene annotations (El Paco gene annotation, version 3) of the P. pacificus reference strain PS312 (Athanasouli et al. 2020) and strain-specific transcriptomes for all seven strains (Rödelsperger et al. 2018) were mapped to the respective genome assemblies using the exonerate alignment program (version 2.2.0) (Slater and Birney 2005). Next, one representative gene model per 100-bp window was chosen by selecting the gene model with the longest open reading frame (minimum protein length 60 amino acids, at least three exons). All other alternative gene models within a 100-bp window were discarded. We previously noticed that coding capacity on the antisense strand of protein-coding genes could lead to gene annotation artifacts, resulting in species-specific orphan genes (Athanasouli et al. 2020). Therefore, we performed all-against-all BLASTP searches (e-value < 10−5) and overlap analysis (at least 1-bp exonic overlap) to remove strain-specific proteins residing on the antisense strand of conserved genes. To maximize comparability, we have used the alternative assembly of the PS312 strain (version Pinocchio) in our analysis (Prabh et al. 2018). This assembly was generated using the same methodology and was also reannotated using the PPCAC pipeline. To visualize genomic features across the chromosome-scale assembly of the reference strain PS312 (Rödelsperger et al. 2017), the PPCAC gene annotations were mapped back to the chromosome-scale El Paco assembly by the exonerate protein2genome (options: ‐‐bestn 1 ‐‐dnawordlen 20 ‐‐maxintron 20000) program. The BUSCO software (version 3.1.0 with the odb9 nematode data set) was run in protein mode to assess the completeness of the gene annotations and compared with phylogenomic data of 54 other nematodes (Rödelsperger 2021).
Orthologous clustering, age classes, and categories of origin
To define orthologous clusters, protein files for the seven P. pacificus strains were combined with the reannotated diplogastrid genome data from Prabh et al. (2018), and protein sets from C. elegans, Bursaphelenchelus xylophilus (Kikuchi et al. 2011), Brugia malayi (Foster et al. 2020), and Trichinella spiralis (WormBase ParaSite version WBPS14) (Mitreva et al. 2011; Bolt et al. 2018). Subsequently, the program OrthoFinder (version 2.5.2, default mode) was run, resulting in 37,228 orthogroups (Emms and Kelly 2019). Protein domains were annotated by using the hmmsearch program (version 3.3, -E 0.001) and searching against the Pfam database (version 3.1b2) (Potter et al. 2018; Mistry et al. 2021). If at least half of the proteins were annotated with a given protein domain, the corresponding orthogroup was categorized as known. To categorize orthogroups as de novo candidates, we defined for each age class a set of ingroup species/strains and a set of outgroup species. An orthogroup was categorized as de novo candidate if there were no BLASTP hits (e-value < 10−5) between ingroup sequences (sequences within an orthogroup) and any sequences from an outgroup species. Apart from the nematode species with deeper rooting ancestry than was defined by an age class, we included protein sequences from Drosophila melanogaster (Ensembl Metazoa release 40) and Mus musculus (Ensembl release 93) as additional outgroups. Any orthogroup that was not classified as known or de novo candidate was categorized as “mixed.” We also ran OrthoFinder with different MCL inflation parameters and varied the threshold for the fraction of genes with Pfam hits to assess the robustness of patterns between known and de novo candidates.
Synteny analysis and investigation of the nature of gene loss
Syntenic gene order alignments between the reference strain PS312 and all other P. pacificus strains were generated by the software CYNTENATOR (options -filter 0 -gap -2) (Rödelsperger and Dieterich 2010). This software computes local alignments of sequences of genes using BLASTP bit-scores to measure similarities between genes. Although gaps and mismatches of unrelated gene pairs will lower the local alignment score, aligned gene pairs with BLASTP matches will increase the alignment score and thus define syntenic orthologs. The syntenic orthologs were only used as anchor genes for investigating the nature of gene loss. For all other analyses, the OrthoFinder results were taken. Candidate loci for manual inspections were mostly taken from orthogroups that included an ortholog in the reference strain PS312 but were missing in one other strain. Only candidates in CYNTENATOR alignments were further studied by performing a TBLASTN search against the genome assembly (e-value < 10) and assessing the alignments for completeness. Cases were classified as “stops” if a nonsense mutation was found in conserved regions. In case of partial alignments, the candidate region between the two closest syntenic anchors was screened for assembly gaps. A partial alignment was scored as “structural variation” if the candidate region was fully assembled or as “assembly gap” if such a gap was found. Complete TBLASTN hits were classified as “not annotated.”
Selection analysis
To perform selection analysis, pairwise one–one orthologs were extracted from the orthogroups. Protein alignments were computed by the program MUSCLE (version 3.8.31) and converted into codon alignments with the help of the PAL2NAL program (version v14) (Edgar 2004; Suyama et al. 2006). The program CodeML of the PAML package (version 4.9) was then run on the codon alignments, and dN and dS values were extracted from the result files (Yang 1997). To generate maps for the chromosomal distributions of dS values, individual data points were smoothed by computing a running mean with an offset of 200 data points.
Analysis of RNA-seq data
To measure expression values of age classes and categories of origin, raw reads of mixed-stage transcriptomes were aligned against the genome assemblies with the STAR aligner (version 2.5.4b) (Dobin et al. 2013; Rödelsperger et al. 2018). The number of reads were counted using the featureCount function of the Rsubread package and subsequently converted into FPKM values (Liao et al. 2014). For the analysis of sex-biased gene expression, raw reads for three biological replicates of P. arcanus male and female transcriptomes (Rödelsperger et al. 2018) were aligned against the P. arcanus reference assembly using the STAR aligner. Genes with significant differential expression between the sexes were identified by the DESeq2 package in R (version 4.0.0) with a Benjamini and Hochberg–adjusted P-value cutoff of 0.05 and an absolute log2 expression fold change of one (Love et al. 2014). An orthogroup was considered as sex biased if at least one gene was captured as significantly differentially expressed between the sexes. De novo candidates for this data set were defined based on the ingroup consisting of all P. pacificus strains, P. exspectatus, P. arcanus, Pristionchus maxplancki, and Pristionchus japonicus.
Statistical analysis
To test for overrepresentation of protein domains in a given set of orthogroups or set of genes, a Fisher's exact test with Bonferroni correction was applied. To test for significant differences among fractions (e.g., fraction of missing orthologs) between two categories of orthogroups, two reciprocal binomial tests were performed by estimating the probability parameter p in the first category and testing against the values (x : = number of successes, n : = number of trials) of the second category. From both reciprocal tests, the maximum P-value was used and corrected with Benjamini and Hochberg correction for multiple testing. Differences in dN/dS ratios were assessed using Wilcoxon tests with Benjamini and Hochberg correction. Differences between the contributions of categories of origin were tested using a χ2-test. All tests were performed using the corresponding functions in R (version 4.0.0) (R Core Team 2020).
Data access
All raw sequencing reads and assemblies from this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena) under accession numbers PRJEB26358, PRJEB26361, and PRJEB48368. The P. pacificus strain genome data are also accessible at http://www.pristionchus.org/download/.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Eduardo Moreno for providing PCR free sequencing data for the P. pacificus strain RSB001. We also thank Ralf Sommer for long-term support and the whole Sommer laboratory for helpful discussions. Finally, we highly appreciate the comments from three anonymous reviewers. This work was funded by the Max Planck Society.
Author contributions: N.P. and C.R. were both responsible for the following: conceptualization, resources, investigation, data curation, formal analysis, visualization, writing the original draft, reviewing and editing, project administration, and funding acquisition.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276431.121.
- Received November 24, 2021.
- Accepted May 20, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








