
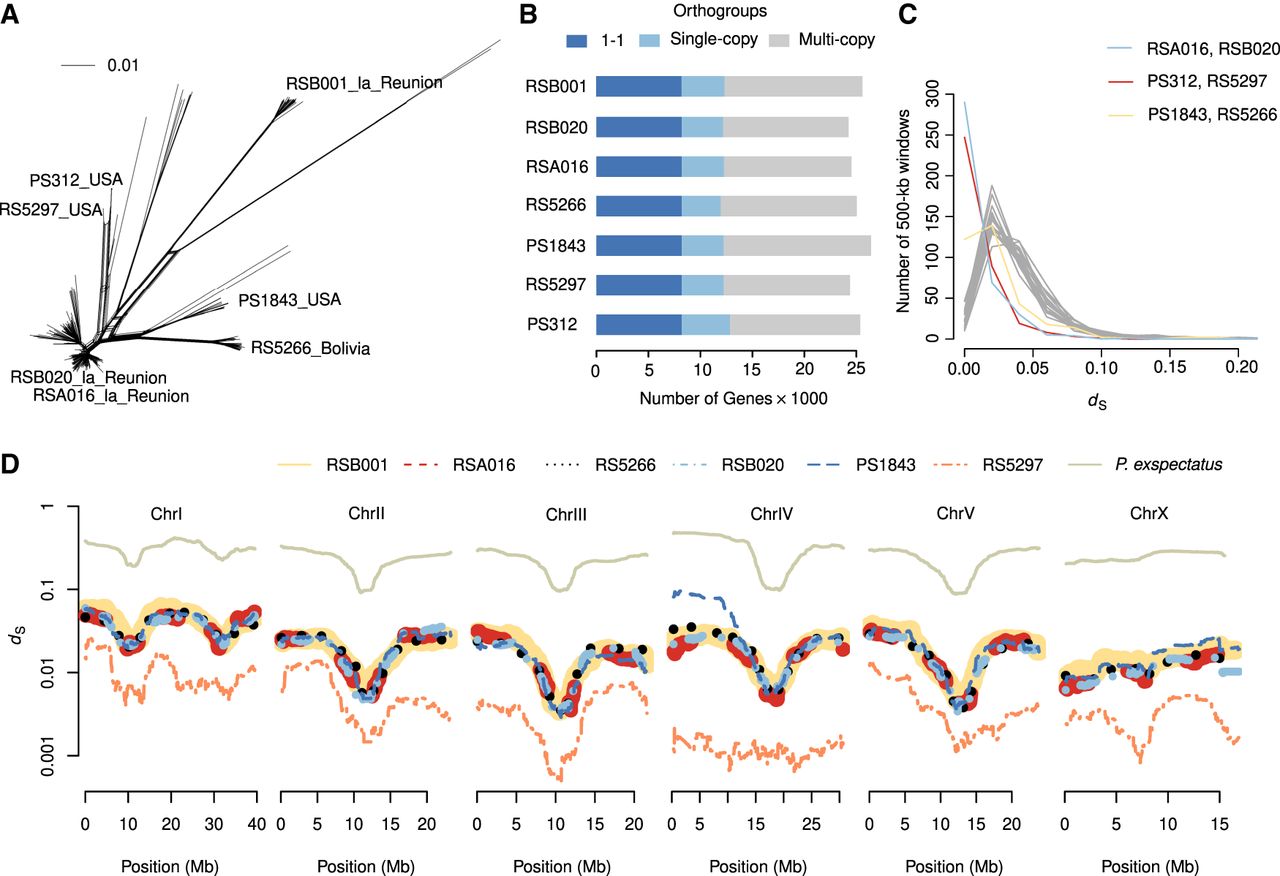
Multiple reference genomes facilitate comparisons at various timescales. (A) The evolutionary distances between 323 P. pacificus strains are displayed as a phylogenetic network. The distances are inferred from 600,000 variable sites that have been called from whole-genome sequencing data (Huson and Bryant 2006; Rödelsperger et al. 2017). (B) Orthologous clustering revealed 8214 genes with one–one orthologs across all seven genomes. Almost half of the genes are assigned to orthogroups with multiple genes per strain. (C) The plot shows the distribution of 500-kb blocks as a function of the mean dS values for every pairwise comparison. The peaks at dS = 0.00 identify recently shared haplotypes between the three most closely related strain pairs. (D) The average dS values are plotted across the megabase position of the chromosome-scale assembly of the reference strain PS312.








