
Tissue-specific reprogramming of host tRNA transcriptome by the microbiome
- 1Guangdong Provincial Key Laboratory of Insect Developmental Biology and Applied Technology, Institute of Insect Science and Technology, School of Life Sciences, South China Normal University, Guangzhou 510631, China;
- 2Department of Molecular Genetics and Cell Biology, The University of Chicago, Chicago, Illinois 60637, USA;
- 3Department of Biochemistry and Molecular Biology, The University of Chicago, Chicago, Illinois 60637, USA
-
↵4 These authors contributed equally to this work.
Abstract
Transfer RNAs (tRNAs) are essential for translation, and tRNA expression and modifications are regulated by many factors. However, the interplay between the microbiome and host tRNA profiles through host-microbiome interactions has not been explored. In this study, we investigated host-microbiome interactions via the tRNA profiling of four tissue types from germ-free and specific pathogen-free mice. Our analyses reveal that cytosolic and mitochondrial tRNA expression and tRNA modifications in the host are reprogrammed in a tissue-specific and microbiome-dependent manner. In terms of tRNA expression, the intestines and brains are more sensitive to the influence of the microbiome than the livers and kidneys. In terms of tRNA modifications, cytosolic tRNAs show more obvious changes in the livers and kidneys in the presence of the microbiome. Our findings reveal a previously unexplored relationship among the microbiome, tRNA abundance, and epitranscriptome in a mammalian host.
Transfer RNA (tRNA) expression and modifications are crucial parameters in the regulation of protein synthesis (El Yacoubi et al. 2012; Pan 2018; Schimmel 2018; de Crécy-Lagard et al. 2019; Thornlow et al. 2020). Human tRNA expression is tissue-specific and plays crucial roles in tumor development and progression (Dittmar et al. 2006; Pavon-Eternod et al. 2009; Gingold et al. 2014; Goodarzi et al. 2015). As the most extensively modified type of RNA in the epitranscriptome, nuclear-encoded tRNAs contain 13–14 post-transcriptional modifications on average (Clark et al. 2016; Pan 2018; Pereira et al. 2018), and mitochondrial-encoded tRNAs contain 2–9 modifications (Suzuki and Suzuki 2014; Suzuki et al. 2020). tRNA modifications are critical for all core aspects of tRNA functions, such as controlling tRNA stability, decoding, charging efficiency and fidelity, and generating tRNA fragments that regulate stress responses and development (Whipple et al. 2011; Zaborske et al. 2014; Clark et al. 2016; Chanfreau 2017; de Crécy-Lagard et al. 2019; Pinkard et al. 2020). Dynamic RNA modifications, such as N6-methyladenosine (m6A), have important biological functions (Liu et al. 2015; Kan et al. 2017; Louloupi et al. 2018; Gkatza et al. 2019; Yang et al. 2019). Epitranscriptomic modifications can be induced by multiple internal and external factors, including the presence of the microbiome in a mammalian host. Increasing evidence suggests that the gut microbiota and its metabolites are associated with the interactions among the host diet, genome, RNA expression, and RNA modifications (Pang et al. 2014; Pan et al. 2018; Cuevas-Sierra et al. 2019; Miro-Blanch and Yanes 2019). Moreover, the microbial composition is responsible for reversible changes in host DNA modifications and gene expression (Qin et al. 2018; Kim et al. 2019; Louwies et al. 2020). We have recently demonstrated that the presence of the microbiome can markedly influence m6A modifications in the mouse messenger RNA (mRNA) transcriptome in a tissue-specific manner (Wang et al. 2019). We have also revealed taxon- and diet-specific variations in tRNA abundance and modifications among the gut microbial communities of mice (Schwartz et al. 2018). However, the associations among the presence of the microbiome, tRNA expression, and tRNA modifications across mammalian host tissue types remain to be studied.
tRNA modifications impair complementary DNA (cDNA) synthesis, which interferes with the efficiency and accuracy of the high-throughput sequencing of tRNA (Zheng et al. 2015; Schwartz et al. 2018). The technical difficulty of tRNA sequencing (tRNA-seq) has been partially overcome via the recent development of sequencing strategies combining demethylase enzymes to remove Watson-Crick face methylations (e.g., N1-methyladenosine [m1A], N1-methylguanosine [m1G], N2,N2-dimethylguanosine [m22G], N1-inosine [m1I], and N3-methylcytosine [m3C]) and thermophilic reverse transcriptases (TGIRTs) for processive cDNA synthesis (Zheng et al. 2015; Clark et al. 2016; Schwartz et al. 2018). Therefore, our demethylase tRNA-seq approach (referred to as DM-tRNA-seq) facilitates the comprehensive elucidation of microbiome effects on host tRNAs by systematically quantifying tRNA expression and tRNA modifications under different physiological conditions.
Whether the microbiome of mice can reprogram the host tRNA transcriptome is an open question in the field, and the molecular basis of tRNA expression and tRNA modifications associated with translation in the mammalian decoding system remains to be investigated. In the present study, we applied DM-tRNA-seq and obtained the cytosolic and mitochondrial tRNA expression and modification profiles of four tissue types (brain, intestine, kidney, and liver) in germ-free (GF) and specific pathogen-free (SPF) mice. This study aimed to investigate host-microbiome interactions via the tRNA profiling of different tissue types from GF and SPF mice and to further verify the potential of DM-tRNA-seq for studying the epitranscriptome and developmental biology.
Results
Expression-related analyses at the isodecoder level
Figure 1 shows the experimental workflow of tRNA-seq applied in this study. We constructed a total of 48 tRNA libraries from 24 tissue samples (brain, intestine, kidney, and liver samples from three GF mice and three SPF mice), each with (+DM) or without (−DM) demethylase treatment (Supplemental Table S1). After quality control, each of the clean tRNA-seq data was mapped to modified cytosolic (21 amino acid isotypes, 49 isoacceptors, 216 isodecoders) and mitochondrial reference tRNA data sets (20 amino acid isotypes, 22 isoacceptors). On average, each tRNA library produced 6.0 and 2.8 million mapped reads for cytosolic and mitochondrial tRNAs, respectively (Supplemental Table S1).
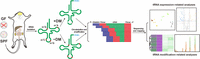
Workflow of this study. tRNAs were isolated from the total RNAs extracted from four tissue types (brain, intestine, kidney, and liver) in specific pathogen-free (SPF) and germ-free (GF) mice. Each tRNA sample was treated with (+DM) or without demethylase (−DM), respectively. After reverse transcription, circularized cDNAs were amplified and sequenced on an Illumina HiSeq system. The sequencing data were used for downstream analyses after mapping to the constructed cytosolic and mitochondrial reference tRNA data sets. (m1A) N1-methyladenosine, (m1G) N1-methylguanosine, (m3C) N3-methylcytosine.
tRNA expression–related analyses were performed based mainly on the tRNA expression normalized and measured by fragments per kilobase per million (FPKM) values for the −DM libraries. We first evaluated the expression of 216 cytosolic tRNAs (Fig. 2A) and 22 mitochondrial tRNAs (Fig. 2B) via principal component analysis (PCA). In these PCAs, all four tissues could be distinctly separated from each other, and the GF mice generally showed higher divergences within biological replicates than the SPF mice (Fig. 2A,B). According to the PCA of cytosolic tRNAs, PC1 and PC2 explained 50.6% of the total variance (Fig. 2A; Supplemental Fig. S1A). The SPF replicates of the brain, intestine, and liver tissues all showed considerable differences from the GF replicates, and these differences were significantly greater than those among the corresponding replicates (Fig. 2A). All 216 cytosolic tRNAs contributed to both PC1 and PC2 to some extent (Supplemental Fig. S1B–D). According to the PCA of mitochondrial tRNAs, 67.2% of the total variance was represented by PC1 and PC2 (Fig. 2B; Supplemental Fig. S1E). This PCA showed that the SPF replicates (especially those of the brain and liver tissues) could not be distinctly separated from the GF replicates (Fig. 2B). Similarly, the 22 mitochondrial tRNAs unevenly contributed to both PC1 and PC2 (Supplemental Fig. S1F–H). tRNATyr and tRNAGln explained nearly 30% variance of PC2 in total, which was significantly higher than the other mitochondrial tRNAs (Supplemental Fig. S1H). Overall, the PCA of cytosolic tRNAs showed that the microbiome has a large effect on tRNA expression of the intestines, brains, and livers, but not in the kidneys; in contrast, the effect of the microbiome on mitochondrial tRNA expression is relatively stronger in the intestines and kidneys rather than the livers or brains. We then performed a correlation analysis of tRNA expression for the three intestinal biological replicates of the GF mice. We found that, although high variability was observed among the replicates in the PCAs (Fig. 2A,B), the calculated FPKM values still showed strong linear correlations (R > 0.96, P < 0.0001) (Supplemental Fig. S2).
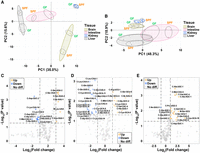
tRNA expression–related analyses at the isodecoder level. (A,B) Principal component analyses (PCAs); (C–E) Volcano plots based on the fragments per kilobase per million (FPKM) values of 216 cytosolic and 22 mitochondrial tRNAs from three tissues (n = 3, SPF vs. GF mice). (A) PCA based on the FPKM values of 216 cytosolic tRNA isodecoders. (B) PCA based on the FPKM values of 22 mitochondrial tRNA isoacceptors. (C) Volcano plot for the brains. (D) Volcano plot for the intestines. (E) Volcano plot for the livers. For C–E, P < 0.05 and log2 (fold change) > 1 were set as the thresholds of significance, and the names of the significant cytosolic tRNA isodecoders follow the names in the Genomic tRNA Database (GtRNAdb, http://gtrnadb.ucsc.edu/genomes/eukaryota/Mmusc10/). (C) Cytosolic tRNA, (M) mitochondrial tRNA.
Next, we analyzed the tRNA expression differences in each tissue between the GF and SPF mice (Fig. 2C–E; Supplemental Fig. S3). Both the brains and intestines were found to exhibit significant numbers of up- and down-regulated tRNAs (Fig. 2C,D); the livers primarily exhibited up-regulated tRNAs when the microbiome was present (Fig. 2E), whereas the kidneys showed very limited changes (Supplemental Fig. S3). Overall, we found that the influence of the microbiome on tRNA expression was not only tissue-specific but also derived from isodecoder-level differences.
Expression-related analyses at the amino acid isotype level
We further analyzed tRNA expression differences at the amino acid isotype level (Fig. 3A–D). Overall, the results indicated that the tRNA isotype patterns and the influence of the microbiome on these patterns among the four tissues were obviously different. The brains, intestines, and kidneys all contained numerous tRNA isotypes that were significantly different between the GF and SPF mice, and the colonization of the microbiome generally significantly increased the expression of tRNA isotypes in the intestines and kidneys (Fig. 3B,C). The variability (standard deviations, SDs) of tRNA expression between the GF and SPF mice in the brains and kidneys was significantly lower than that in the intestines and livers, which contributed greatly to the numbers of significant tRNAs (Fig. 3A,C). Mitochondrial tRNAPhe showed the highest expression ranges beyond the other 40 tRNA amino acid isotypes in the four tissues. The colonization of the microbiome generally significantly increased mitochondrial tRNAMet levels and reduced mitochondrial tRNAPhe levels in three mouse tissues (P < 0.05 for all) (Fig. 3A–C), except for the liver data (Fig. 3D).
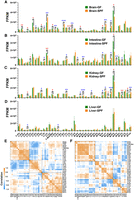
tRNA expression–related analyses at the amino acid isotype level. tRNA expression was measured by the FPKM values of 21 cytosolic and 20 mitochondrial tRNA isotypes. (A–D) tRNA expression in four tissues (n = 3 for both GF and SPF mice). (E,F) tRNA coexpression correlation plots for the GF and SPF mice obtained by combining the data from the four tissue types (brain, intestine, kidney, and liver; each n = 3). (A) tRNA expression in the brains. (B) tRNA expression in the intestines. (C) tRNA expression in the kidneys. (D) tRNA expression in the livers. (E) tRNA coexpression correlation plot for the GF mice. (F) tRNA coexpression correlation plot for the SPF mice. Significant differences were determined with the unpaired Student's t-test. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. For A–D, red asterisks indicate the tRNA isotypes significantly down-regulated in the SPF mice compared with the GF mice, whereas blue asterisks indicate the tRNA isotypes significantly up-regulated in the SPF mice compared with the GF mice. In E and F, the squares indicate hierarchical clusters.
We then analyzed tRNA coexpression correlations at the amino acid isotype level (Fig. 3E,F; Supplemental Fig. S4). Similarly, we found that there were large differences in tRNA coexpression patterns among the tissues and that the microbiome influenced these patterns to some extent (Fig. 3E,F; Supplemental Fig. S4). Moreover, we noted that the presence of the microbiome distinctly enhanced the complexity of the clustering results for tRNA coexpression patterns, especially in the brains and livers (Supplemental Fig. S4A–D). When we combined the data from the four tissues, the 41 tRNA amino acid isotypes were roughly divided into three hierarchical clusters in the GF mice and four hierarchical clusters in the SPF mice. The largest clusters included more than half of the mitochondrial tRNA isotypes (Fig. 3E,F). The correlation of tRNA coexpression in the livers, which showed only two large unambiguous hierarchical clusters (Supplemental Fig. S4G,H), was much simpler than that in the other three tissues (Supplemental Fig. S4A–F).
Expression-related analyses at the isoacceptor level
The compositions of the tRNA isoacceptors provided a representation at the anticodon level for revealing both tissue-specific and microbiome-dependent differences (Fig. 4). Overall, the clustering analyses divided both the 49 cytosolic isoacceptors (Fig. 4A) and the 22 mitochondrial isoacceptors (Fig. 4B) into four unequal-sized clusters based mainly on their proportions among total cytosolic or mitochondrial tRNAs. Among these isoacceptors, cytosolic tRNAGln (Fig. 4A) and mitochondrial tRNAPhe (Fig. 4B) were the most prominent isoacceptors due to their high proportions among cytosolic or mitochondrial tRNAs. Similarly, the influence of the microbiome on the changes in tRNA proportions depended on the tissue types and isoacceptor types. For example, the proportion of cytosolic tRNALeu-CAA distinctly increased in the brains and decreased in the intestines, whereas no significant changes were seen in the kidneys and livers when the microbiome was present (Fig. 4A). Overall, the tRNA compositions at the tRNA isoacceptor level showed similar results as those from the PCAs, that is, the cytosolic tRNA compositions of the intestines, brains, and livers, as well as the mitochondrial tRNA compositions of the intestines and kidneys, were more sensitive to the influence of the microbiome. By comparing the fold changes (FCs) of the tRNA isoacceptor proportions between the GF and SPF mice, we found that with the colonization of the microbiome: mitochondrial tRNALeu-UAA increased (FC = 2.91) and cytosolic tRNAAsp decreased (FC = 0.37) most significantly in the brains; cytosolic tRNAiMet-CAU increased (FC = 2.00) and tRNASer-GGA decreased (FC = 0.22) most significantly in the intestines; mitochondrial tRNAMet increased (FC = 1.69) and tRNAThr decreased (FC = 0.61) most significantly in the kidneys; mitochondrial tRNASer-GCU increased (FC = 6.71) and cytosolic tRNASer-GGA decreased (FC = 0.18) most significantly in the livers.
tRNA composition heat maps at the isoacceptor level. tRNA proportions were calculated based on the FPKM values of 49 cytosolic and 22 mitochondrial tRNA isoacceptors among total cytosolic or mitochondrial isoacceptors, respectively. (A) Heat map of cytosolic tRNAs. (B) Heat map of mitochondrial tRNAs.
Modification-related analyses between +DM and −DM libraries
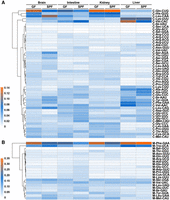
tRNA modification–related analyses were performed based mainly on the mutation fractions of Watson-Crick face methylations within each tRNA isoacceptor or amino acid isotype at single-base resolution (Fig. 5; Supplemental Figs. S5, S6). We first compared the differences in tRNA methylations between the +DM and −DM samples to confirm the effect of demethylase treatment in this study. Compared with the −DM libraries, the mutation fractions of most types of tRNA methylations (including known cytosolic m1A58, m1G37, m3C32, and m3C47 and mitochondrial m1A/G9 according to the standard tRNA nomenclature) in the +DM libraries were significantly reduced (Fig. 5A–F); an exception was observed for known cytosolic m1I37 in the tRNAAla amino acid isotype (Fig. 5C), where demethylase converts m1I to inosine, and different bases are further incorporated by TGIRT during reverse transcription. To provide more details about the changes in mapping coverage and mutations, we selected four representative tRNA amino acid isotypes/isoacceptors to visualize the effect of demethylase treatment between the −DM and +DM libraries of the brains (Supplemental Figs. S5, S6). Consistent with related studies (Zheng et al. 2015; Clark et al. 2016), all common types of tRNA methylations except for cytosolic m22G26 were sensitive to demethylase treatment and exhibited less reverse transcriptase (RT) stops and smaller mutation fractions (Supplemental Figs. S5, S6). Among mitochondrial tRNAs, bulky tRNA modifications such as 2-methylthio-N6-isopentenyladenosine (ms2i6A) within tRNAPhe (Supplemental Fig. S6), tRNATrp, and tRNATyr were insensitive to demethylase treatment and could cause serious RT stops and induce many deletions during cDNA synthesis (Supplemental Fig. S6).
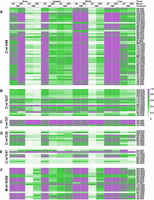
Mutation heat maps of common methylation types within cytosolic or mitochondrial tRNAs according to the standard tRNA nomenclature. The modification level of each type of tRNA methylation was semiquantified based on the corresponding mutation fraction, and presented data are from four tissues (n = 3 for both GF and SPF mice). (A) N1-methyladenosine of position 58 (m1A58) within cytosolic tRNAs. (B) N1-methylguanosine of position 37 (m1G37) within cytosolic tRNAs. (C) N1-inosine of position 37 (m1I37) within cytosolic tRNAs. (D) N3-methylcytosine of position 32 (m3C32) within cytosolic tRNAs. (E) N3-methylcytosine of position 47 (m3C47) within cytosolic tRNAs. (F) N1-methyladenosine or N1-methylguanosine of position 9 (m1A/G9) within mitochondrial tRNAs.
Modification-related analyses among tissues, and between GF and SPF mice
We further identified the tissue-specificity and microbiome-dependence of host tRNA modifications. Among the −DM libraries of the four tissues, the kidneys and livers of both the GF and SPF mice displayed very similar high mutation fractions among multiple methylations (Fig. 5); in contrast, the intestines of the SPF mice showed universally low mutation fractions among these modifications (Fig. 5), whereas the brains of the GF mice showed lower mutation fractions only for cytosolic m1A58 (Fig. 5A). We visualized the mutation and stop fractions across the cytosolic isoacceptor tRNALeu-CAG to show the above-mentioned differences among the tissues (Fig. 6). In the mutation plots (Fig. 6A,C,E,G), known methylations can be identified easily. With the colonization of the microbiome, the mutation fraction of m1A58 was slightly increased in the brains (Fig. 6A), whereas those of m3C47 and m1A58 were decreased in the intestines (Fig. 6C). The stop patterns of m22G26, m1G37, and m3C47 of this tRNA were different between the GF and SPF mice in all four tissues (Fig. 6B,D,F,H). In the brains, m22G26 and m3C47 decreased in the SPF mice (Fig. 6B); in the intestines, all three modifications decreased in the SPF mice (Fig. 6D); in the kidneys, no obvious difference in the stop fraction was seen between the GF and SPF mice (Fig. 6F); whereas in the livers, m22G26 increased and m1G37 decreased in the SPF mice (Fig. 6H).
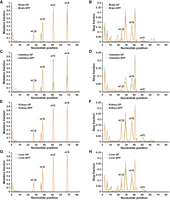
Mutation and stop plots for the cytosolic tRNALeu-CAG isoacceptor in four tissues (n = 3 for both GF and SPF mice) from the −DM libraries. (A) Mutation plot for the brains. (B) Stop plot for the brains. (C) Mutation plot for the intestines. (D) Stop plot for the intestines. (E) Mutation plot for the kidneys. (F) Stop plot for the kidneys. (G) Mutation plot for the livers. (H) Stop plot for the livers. The stop fractions at and after m1A58 are not shown. The x-axis corresponds to the nucleotide position along the tRNA. Watson-Crick face methylations that are sensitive to demethylase treatment can be easily identified from the mutation plots. (m22G) N2,N2-dimethylguanosine.
Compared to the modification-related analyses of the −DM libraries (Fig. 6), we noted that the host tRNA methylations in the +DM libraries were more comparable among the tissues and between the GF and SPF mice in our study because the patterns of mutation fractions showed more distinct tissue-specificity and microbiome-dependence. In the +DM libraries, the results indicated that the livers and intestines showed higher mutation fractions for multiple methylations than the kidneys and brains (Fig. 5). The colonization of the microbiome also strongly influenced the semiquantified results of tRNA methylations. In the brains and intestines, the mutation fractions only significantly increased for mitochondrial m1A/G9 (Fig. 5F), whereas the kidneys and livers showed different mutation patterns for multiple cytosolic methylations between the GF and SPF mice. Furthermore, we found that the mutation fractions of mitochondrial m1A/G9 (Fig. 5F) were more easily influenced by the presence of the microbiome than other cytosolic tRNA methylation types in all four tissues (Fig. 5A–E).
Discussion
The elucidation of the roles and effects of the microbiome in mammalian hosts has become an increasingly important issue in a range of biological fields. The response between microbiome-host interactions and the host epitranscriptome currently remains poorly studied. In this study, we explored this unresolved question by performing expression-related analyses of different tRNA levels and modification-related analyses of known methylations across mammalian tRNAs. Overall, the results obtained from the expression-related analyses revealed the associations between the microbiome and host tRNA genes in different tissues, and modification-related analyses provided semiquantified results regarding tissue-specific and microbiome-dependent differences. Altogether, our results showed that there were very distinct tRNA abundance and modification patterns among the tissues and that the presence of the microbiome resulted in the tissue-specific reprogramming of host tRNA expression and modifications. It is still unclear whether certain microbes are associated with variations in tRNA expression, tRNA modifications, mRNA translation, or protein expression. These questions will become the foci of interest in the field and remain to be investigated in future studies.
The impacts of the microbiome on host tRNA expression and modification
Among the four tissue types, host tRNA expression in the brains and intestines was more sensitive to the influence of the microbiome than that in the kidneys and livers in nearly all analyses conducted in this study. These findings are consistent with our previous findings from mRNA analyses that the m6A contents in the brains and intestines show more noticeable differences between the GF and SPF mice than those in the livers (Wang et al. 2019). Recent advances have shown that cellular gene expression patterns in the microbiota-gut-brain axis can be interfered with by the microbiome through epigenetic modifications (Canani et al. 2012; Fofanova et al. 2016; Hoban et al. 2017). These findings could be explained by the direct influence of the activities or metabolites produced by the gut microbiota, which represents one of the densest microbial communities in mammals, with growing evidence showing its contributions to maintaining homeostasis and various physiological processes (Sandhu et al. 2017).
In our study, we noted that the microbiome could reprogram not only the cytosolic but also the mitochondrial tRNA profiles of the host. The changes in mitochondrial tRNA expression in the kidneys and intestines were particularly noteworthy when the microbiome was present (Figs. 2B, 3B,C). On the other hand, mitochondrial m1A/G9 (Fig. 5F) was more sensitive to the microbiome than all other cytosolic types of tRNA methylations (Fig. 5A–E). All of these results suggest that the presence of the microbiome could influence mitochondrial translation activity and energy metabolism. Colonization of the gut microbiota in the GF mice was shown to cause rapid weight gain or increase obesity (Bäckhed et al. 2004; Heiss and Olofsson 2018). This phenomenon may be linked to changes in mitochondrial tRNA expression and associated mitochondrial translational regulation. The relatively high variance of mitochondrial tRNA expression among biological replicates from the same tissues should be noted and taken into consideration. One possible explanation could be related to PCR bias during library construction, which will be optimized in our future work.
The accuracy of expression-related analyses is closely related to the correct construction of a cytosolic tRNA reference database
The construction of tRNA reference data sets is an important step that can profoundly influence the accuracy of expression-related analyses. Although the cytosolic tRNA reference data set of Mus musculus was based mainly on the high confidence set from the Genomic tRNA Database (GtRNAdb), there remains substantial room for further improvements in data set construction. First, the extremely short lengths of tRNAs limit the traceability of their sources on chromosomes. One hundred ninety of the 401 cytosolic tRNAs from the high confidence set were merged to reduce redundancy from the beginning due to their entirely identical sequences. For example, tRNAGlu-CTC-1 isodecoder contained nine entirely identical tRNA sequences derived from five chromosomes. However, it is still unknown whether the influence of the microbiome on different sources of tRNAs is consistent and linearly correlated. Additionally, because the primary filter of the cytosolic tRNA reference data set of M. musculus contained 36,415 tRNA pseudogenes with low general tRNA model scores predicted by the tRNAscan-SE search server, whether these pseudogenes are involved in determining the composition of mature tRNA abundance remains to be studied.
The noncanonical roles of tRNAs beyond translation may be the main reason for their large expression differences in the presence of the microbiome
It is widely accepted that the most canonical role of tRNAs is to transfer activated amino acids from aminoacyl-tRNA synthetases to ribosomes for protein synthesis (Katz et al. 2016). Translational control by tRNA abundance has been inferred to play roles via codon usage (Dittmar et al. 2006). Therefore, we attempted to investigate the correlations between the codon usage of coding sequences (CDSs) and the expression of corresponding tRNA isoacceptors in the GF and SPF mice to determine whether the presence of the microbiome can influence the dynamic balance between mRNAs and tRNAs, in conditions such as breast cancer (Pavon-Eternod et al. 2009). The liver was selected as the representative tissue type for this analysis because its tRNAs in the SPF mice showed a universally up-regulated trend in the volcano plot (Fig. 2E). We first calculated the SPF/GF ratio for each tRNA isoacceptor type. By using the transcriptome data from our previous mRNA study (Wang et al. 2019), we extracted the expression of all 15,901 CDSs from the livers of the GF and SPF mice. We then calculated the total codon usage and obtained the SPF/GF ratio for each codon type. Finally, we established the connections between codons and anticodons considering the wobble base pairing that occurs between position 34 of the tRNA anticodon and the third base of the mRNA codon. We failed to find a linear or near-linear correlation between the codons and anticodons even if a series of efforts had been made to optimize the analysis (Supplemental Fig. S7). The potential positive association between codon usage and tRNA expression could only be found for a few specific codon types within overexpressed genes (Pavon-Eternod et al. 2009). It is worth noting that the influence of the microbiome only slightly altered the codon usage of total CDSs for any codon type in the livers (with SPF/GF ratios ranging from 0.98 to 1.03), whereas the expression of tRNA isoacceptors was sensitive to the microbiome and showed much higher variations (with SPF/GF ratios ranging from 0.57 to 2.23) (Supplemental Fig. S7). The noncanonical roles of tRNAs beyond translation include synthetic, regulatory, and information functions (Rogers et al. 2012; Colussi et al. 2014; Raina and Ibba 2014; Katz et al. 2016; Pan 2018). In this analysis, the variations in tRNA expression were much greater than the requirements of codon usage, indicating that the colonization of the microbiome might markedly influence the noncanonical roles of tRNAs beyond the transfer of amino acids. Additionally, the tissue-specificity of tRNA coexpression patterns (Supplemental Fig. S4) may shed light on the dominant noncanonical function of tRNAs in further studies.
Methods
Sample collection and tRNA isolation
GF and SPF mice (4-wk-old C57BL/6NTac male mice, both n = 3) were purchased from Taconic Biosciences. A total of 24 tissue samples (brains, intestines, kidneys, and livers) from the six mice (Supplemental Table S1) were dissected and freshly frozen on dry ice and then stored at −80°C. These tissues were homogenized with glass beads using a tissue homogenizer, and their total RNA was isolated using TRIzol reagent (Ambion) following the manufacturer's protocol. The RNA samples were dissolved in RNase-free water, and the concentration and purity of the total RNAs were spectrophotometrically measured at 260 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Total RNA was first deacylated with Tris-HCl (pH = 9.0) at 37°C for 30 mins and then purified in a 10% urea denaturing gel according to standard procedures. The tRNA bands were cut out under UV shadowing, and the isolated total tRNAs (∼3 µg, 60 pmol) were dissolved in RNase-free water.
DM-tRNA-seq and regular tRNA-seq
We performed DM-tRNA-seq as previously reported in human (Zheng et al. 2015; Clark et al. 2016) and microbial tRNA studies (Schwartz et al. 2018). Briefly, ∼1.5 µg of total tRNA from each sample were treated with a 3× molar ratio of wild-type AlkB (180 pmol) and a 4× molar ratio of the D135S mutant (240 pmol) to remove Watson-Crick face methylations on tRNAs, which can impede the processivity of reverse transcription, while another 1.5 µg of total tRNA extracted from the same sample were left untreated. Thereafter, we performed reverse transcription from 150-ng tRNA samples of the −DM and +DM libraries with TGIRT (InGex). We purified and eluted cDNAs from 10% urea denaturing gels by using linear acrylamide (Thermo Fisher Scientific AM9520) as the carrier. The purified cDNAs were circularized with CircLigase II (Epicentre) at 60°C overnight following the manufacturer's protocol and then extracted with phenol-chloroform-isoamyl alcohol (25:24:1), followed by ethanol precipitation. For library construction, the circularized cDNAs were amplified with Phusion-HF (NEB) by PCR amplification for 12 cycles after ligation with Illumina universal adapter sequence (5′-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATC-3′) and RNA adapter sequence (5′-CAAGCAGAAGACGGCATACGAGAT[Index]GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′). The PCR products were purified with AMPure XP beads (Beckman Coulter) and sequenced on an Illumina HiSeq system.
Construction of tRNA reference data sets
For cytosolic tRNAs, we obtained an initial reference data set for Mus musculus (GRCm38/mm10) from GtRNAdb (http://gtrnadb.ucsc.edu/genomes/eukaryota/Mmusc10/). The high confidence set of this data set contained 400 standard decoding tRNAs and one selenocysteine tRNA, and the set of notable atypical predictions included five tRNAs with anticodon/isotype mismatch(es). We first merged all entirely identical tRNA isodecoders to reduce redundancy. For mitochondrial tRNAs, we obtained an initial mitochondrial reference genome for M. musculus from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) (accession number NC005089). Mitochondrial tRNA genes were identified based on annotations and using the tRNAscan-SE search server (version 2.0, http://lowelab.ucsc.edu/tRNAscan-SE/) (Lowe and Chan 2016).
The merged cytosolic and mitochondrial reference tRNA data sets contained 216 isodecoders (21 amino acid isotypes and 49 isoacceptors including the selenocysteine tRNA) and 22 isoacceptors (20 amino acid isotypes), respectively. Thereafter, we mapped randomly selected raw sequencing data to these two data sets using Geneious Prime (version 2019.2.3) (https://www.geneious.com) to eliminate the single nucleotide polymorphisms (SNPs) in tRNAs between the mice used in the current study and the two data sets. Briefly, apart from the wobble anticodon positions of several cytosolic tRNAs (e.g., tRNAAla-AGC and tRNAArg-ACG), the bases showing a mutation rate >0.98 to only one of the other three bases in the mapping alignments were regarded as SNPs and modified. Most tRNA methylation sites incorporate different bases rather than just the cognate nucleotide type in the reference genome during reverse transcription. For a specific methylation site, the incorporated bases should not have a strong bias to any one of the other three different bases in general. Therefore, this threshold can effectively distinguish the SNPs from the methylation sites in tRNAs.
Mapping of tRNA-seq data
Prior to mapping, all raw sequencing data were filtered to remove de-multiplexing, adapters, primers, and low-quality reads using Trimmomatic (version 0.32) as clean data. Because different bases will be incorporated where tRNA methylations are located by TGIRT during cDNA synthesis, we first determined the types and positions of known methylations within mammalian tRNAs (Suzuki and Suzuki 2014; Clark et al. 2016; Suzuki et al. 2020). Considering the average numbers of potential mutation sites within target tRNAs, we set the maximal mismatch percentages for subsequent mapping at 9% and 5% for the modified cytosolic and mitochondrial reference tRNA data sets, respectively. As the most common methylation type closest to the 3′ end (16 bp in general) within cytosolic tRNAs, m1A58 hinders cDNA synthesis by causing RT stops from the 14th base to the 3′ end. Therefore, the minimal overlap between the mapped reads and the tRNA databases was set as 14 bp for evaluating the obstacles caused by m1A58. Briefly, all clean reads from the filtered sequencing data of ≥ 14 bp were mapped to targeted tRNAs under the allowed mismatch settings. We simultaneously mapped all clean reads of each sample to the modified cytosolic and mitochondrial reference tRNA data sets according to the above-mentioned parameters without iteration using Geneious Prime.
Expression-related analyses of tRNA-seq data
In the current study, we focused mainly on the transcriptome-wide expression and modification differences in host tRNA profiles between the GF and SPF mice and among the four examined tissue types.
The length of most cDNAs was shorter than that of the Illumina reads, so generally only one end read of paired-end reads contained effective information, whereas the other end read was lost during quality control or mapping. Therefore, prior to the calculation of tRNA expression, we first reduced the influence of paired-end sequencing on the expression measurements. Briefly, for each mapping alignment of the targeted tRNA gene in the databases, we counted the mapped reads and the mapped paired-end reads. The number of mapped tRNA molecules (including full-length tRNA molecules and partial tRNA fragments) could be computed for each sample (Supplemental Table S1). Thereafter, the mapped tRNA molecules were normalized to calculate tRNA expression according to FPKM values using the following formula: FPKM = mapped cytosolic or mitochondrial tRNA molecules/(mapped total cytosolic or mitochondrial tRNA molecules [millions] × tRNA length [kbp]). The FPKM values at higher tRNA levels (isoacceptors and amino acid isotypes) were calculated according to mammalian genetic codes.
Prior to the performance of expression-related analyses, we first evaluated the accuracy of the calculated FPKM values via PCA of the cytosolic (216 tRNA isodecoders) and mitochondrial tRNAs (22 tRNA isoacceptors) of the +DM and −DM libraries using the R (version 4.0.1) (R Core Team 2020) packages FactoMineR (version 2.4) (Lê et al. 2008) and factoextra (version 1.0.7) (https://CRAN.R-project.org/package=factoextra), respectively. The PCAs indicated that the accuracy, stability, and replicability of tRNA expression in the −DM libraries were better than those in the +DM libraries. The most likely cause might be the nonuniform or nondirectional tRNA loss of the +DM libraries during additional experimental treatments. Therefore, we performed subsequent expression-related analyses based mainly on the data from the −DM libraries.
To further verify the reliability of the calculated FPKM values, we first performed a correlation analysis of tRNA expression in the three intestinal biological replicates of the GF mice based on the FPKM values of the 238 tRNAs. Volcano plots based on the FPKM values of the 238 tRNAs were generated with the R package ggplot2 (version 3.3.2) (Wickham 2016) to evaluate tissue-specifically up- and down-regulated genes between the GF and SPF mice. P < 0.05 and log2 (fold change) > 1 were set as the thresholds of significance. Using the FPKM values of the 21 cytosolic and 20 mitochondrial tRNA amino acid isotypes as the input data, tRNA expression in the four tissues at the amino acid isotype level was visualized. Significant differences between the GF and SPF mice were determined with the unpaired Student's t-test. We further calculated the percentage of each tRNA isoacceptor among total cytosolic or mitochondrial tRNA isoacceptors for each sample and visualized the results using a heat map. In addition, we performed a tRNA coexpression correlation analysis at the amino acid isotype level based on the data from the four tissues with the R package corrplot (version 0.84) (https://github.com/taiyun/corrplot) to investigate the tRNA coexpression correlations among the tissues and between the GF and SPF mice.
Modification-related analyses of tRNA-seq data
According to our previous studies related to DM-tRNA-seq (Zheng et al. 2015; Clark et al. 2016), in −DM libraries, the specific tRNA sites of the main Watson-Crick face methylations that were sensitive to demethylase treatment could be easily identified because they generally show high mutation fractions. The levels of these tRNA methylations could be reflected by using the mutation and stop fractions as semiquantitative indicators for both the −DM and +DM libraries, respectively. In this case, the differences in these tRNA methylations between the −DM and +DM libraries, among the tissues, or between the GF and SPF mice in the same library type could be compared to some extent. For the calculation of mutation and stop fractions, we used previously described formulas and protocols (Clark et al. 2016). The mutation fraction of each modification site in each sample was calculated based on the sequencing reads that covered the modification site while excluding shorter reads that stopped before the site. As a common case, deletions were also considered and counted as a mutation signature. To efficiently count and calculate the mutation fractions at the tRNA isodecoder level, we first merged the clean data from biological replicates and performed mapping again with the same parameters. Thereafter, the mutation fractions at the tRNA isoacceptor level were calculated according to mammalian genetic codes. The mutation fractions of the main methylations at the Watson-Crick face, including m1A, m1G, m1I, and m3C, were displayed using heat maps generated with the R package pheatmap (version 1.0.12) (https://CRAN.R-project.org/package=pheatmap).
To compare the differences between the +DM and −DM sequencing data, we randomly selected two cytosolic amino acid isotypes (tRNAHis and tRNAPhe) and two mitochondrial isoacceptors (tRNALeu-UAG and tRNAPhe). All tRNA isodecoders of each selected cytosolic tRNA isotype were merged as a single tRNA gene using degenerate bases. Thereafter, we performed mapping again for the −DM and +DM libraries from the brains (randomly selected tissue type) with the same parameters and further compared the mapping coverage and mutation fraction variations between them. A representative cytosolic isoacceptor (tRNALeu-CAG) was selected to show the mutation and stop fractions at every position of the tRNA to explore tissue-specific and microbiome-dependent differences in tRNA modifications for the −DM libraries. The stop fractions of and after m1A58 were not accurately quantified by tRNA-seq and are not shown.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE150355.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (K01 DK111764 to X.W. and R01 GM113194 to T.P.), National Natural Science Foundation of China (32070615 to X.W.), and Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (recipient in 2019 to X.W.).
Author contributions: X.W. and T.P. conceived and initiated the study. X.W. and W.C. designed and performed the experiments. J.H. conducted the analyses. J.H., X.W., and T.P. wrote the manuscript. F.Z., Z.P., and L.W. contributed to the manuscript preparation. All authors read, proofread, and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.272153.120.
-
Freely available online through the Genome Research Open Access option.
- Received September 29, 2020.
- Accepted April 7, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








