
Time course regulatory analysis based on paired expression and chromatin accessibility data
- 1Department of Statistics, Stanford University, Stanford, California 94305, USA;
- 2CEMS, NCMIS, MDIS, Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing 100080, China;
- 3Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, 650223, China;
- 4Department of Biomedical Data Science, Bio-X Program, Center for Personal Dynamic Regulomes, Stanford University, Stanford, California 94305, USA
-
↵5 These authors contributed equally to this work.
Abstract
A time course experiment is a widely used design in the study of cellular processes such as differentiation or response to stimuli. In this paper, we propose time course regulatory analysis (TimeReg) as a method for the analysis of gene regulatory networks based on paired gene expression and chromatin accessibility data from a time course. TimeReg can be used to prioritize regulatory elements, to extract core regulatory modules at each time point, to identify key regulators driving changes of the cellular state, and to causally connect the modules across different time points. We applied the method to analyze paired chromatin accessibility and gene expression data from a retinoic acid (RA)–induced mouse embryonic stem cells (mESCs) differentiation experiment. The analysis identified 57,048 novel regulatory elements regulating cerebellar development, synapse assembly, and hindbrain morphogenesis, which substantially extended our knowledge of cis-regulatory elements during differentiation. Using single-cell RNA-seq data, we showed that the core regulatory modules can reflect the properties of different subpopulations of cells. Finally, the driver regulators are shown to be important in clarifying the relations between modules across adjacent time points. As a second example, our method on Ascl1-induced direct reprogramming from fibroblast to neuron time course data identified Id1/2 as driver regulators of early stage of reprogramming.
In time course expression analysis, gene expression is measured at multiple time points during a natural biological process such as spontaneous differentiation of progenitor cells, or during an induced biological process such as cellular response to a stimulus or treatment (Storey et al. 2005). In the last two decades, many methods were developed to infer gene regulatory networks (GRNs) from time course gene expression data, for example, information theory–based methods (Margolin et al. 2006; Hempel et al. 2011; Kinney and Atwal 2014), Bayesian network–based methods (Perrin et al. 2003; Zou and Conzen 2005), ordinary differential equation–based methods (Bansal et al. 2006; Wang et al. 2006), and permutation-based methods (Hempel et al. 2011). Such analysis is popular because the expression data, which is inexpensive to measure, can provide a rich description of the changes in the cellular states during the time course. Conversely, because the regulation of gene expression involves the interaction of transcription factors with DNA on regions with open chromatin structure, the measurement of gene expression alone is not sufficient for the study of the regulation (Duren et al. 2017; Miraldi et al. 2019). Much deeper understanding can be revealed by time course regulatory analysis, in which both gene expression and chromatin accessibility are measured at each time point in a time course experiment. With the advent of cost-effective technologies (i.e., Assay for Transposase-Accessible Chromatin using sequencing [ATAC-seq]) for measuring chromatin accessibility (Buenrostro et al. 2013), paired expression and accessibility data are now becoming available in many time course experiments, such as FOS-induced neuronal activities (Su et al. 2017), epidermal development (Li et al. 2019), myeloid cell differentiation (Ramirez et al. 2017), early cardiomyocyte differentiation (Liu et al. 2017), iPSC reprogramming (Wapinski et al. 2017; Cao et al. 2018), and induced neuron reprogramming (Wapinski et al. 2017; Cao et al. 2018). Here, we present a methodology for the analysis of data from studies with such experimental designs.
Figure 1 presents an outline of our methodology (for detail, see Methods). First, we infer context-specific GRN from matched ATAC-seq and RNA-seq data at each time point to output reliable regulatory relations. Using the inferred GRN, we define two types of scores for regulatory relations. The regulatory strength of a transcription factor (TF) on a target gene (TG) is quantified by the trans-regulation score (TRS), which is calculated by integrating information from multiple regulatory elements (REs) that may mediate the activity of the TF to regulate the TG. Here, a prior TF-TG correlation across external public data (Supplemental Table S1) is included in the TRS definition to distinguish the TFs sharing the same binding motif (i.e., TFs from the same family). The regulatory strength of an RE on a target gene is quantified by the cis-regulation score (CRS), which is calculated by integrating the TRS of TFs with binding potential on the RE. Based on these scores, we use non-negative matrix factorization to extract the core regulatory modules that characterize different biological processes and/or subpopulations of cells. Finally, we identify driver TFs (i.e., TFs driving expression changes between adjacent time points) as TFs with large difference TRS scores on up-regulated genes versus other genes. This allows us to causally connect ancestor–descendant regulatory modules along with time points (Methods). Our methodology for time course regulatory analysis is implemented in the software Time Course Regulatory Analysis (TimeReg), which is freely available (https://github.com/SUwonglab/TimeReg).
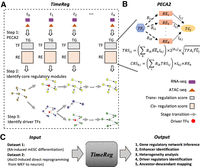
Schematic overview of Time Course Regulatory Analysis (TimeReg) based on paired gene expression and chromatin accessibility data. (A) TimeReg proposes a three-step framework to infer a high-quality gene regulatory network. Step 1: PECA2 infers context-specific GRN from matched ATAC-seq and RNA-seq data at a single time point to output TF-TG and RE-TG regulatory matrix. Step 2: NMF decomposes the regulatory matrix and extracts the core regulatory modules at each time point. Step 3: Driver regulators are identified (Methods). (B) Overview of PECA2 method. (C) Schematic overview of the major results on two data sets.
In this paper, we will validate the utility of TRS and CRS by comparison with TF-TG relations and RE-TG relations defined by data from independent gene knockdown, ChIP-seq, and HiChIP experiments. After validating these key concepts, we will apply our method to study a time course of retinoic acid (RA)–induced differentiation of mouse embryonic stem cells (mESCs), in which gene expression and chromatin accessibility are measured at baseline (day 0) and at days 2, 4, 10, and 20 after RA treatment. We will extract core regulatory modules on each time point, map the modules across time points to trace their development trajectory, and validate the core regulatory modules by single-cell RNA-seq data. As a second illustration, we will apply our method on Ascl1-induced direct reprogramming from fibroblast to neuron time course data to detect heterogeneity, explore the regulatory dynamics, and identify driver regulators in the reprogramming process. By applying our method on different biological problems, we will illustrate that our methodology is capable of providing reliable regulatory relations in time course experiments and dissecting the dynamics at network level.
Results
Chromatin accessibility and expression dynamics of retinoic acid–induced mESC differentiation
Mouse ESC was induced to differentiate by treatment with retinoic acid (RA). We harvested cells at days 0, 2, 4, 10, and 20 (mESC, D2, D4, D10, and D20), and performed ATAC sequencing (ATAC-seq) and RNA sequencing (RNA-seq) to measure the paired chromatin accessibility and gene expression time course data (Fig. 2A). Regulatory elements (REs) at each time point are identified and quantified by its openness score in the same way as in Duren et al. (2017). In total, 174,059 REs are obtained across all time points. From gene expression data, we selected 7975 genes that have at least twofold expression change and the maximum expression level is >10. These sets of RE and gene are used for all subsequent analyses. From the results of principal component analysis (PCA) and Pearson's correlation coefficient (PCC) on ATAC-seq data of twofold dynamic REs (Fig. 2B,C), our genome-wide gene expression and chromatin accessibility profiling show high-quality reproducibility among biological replicates. We also find a sharp change in the chromatin accessibility landscape during the time course, whereas the changes in gene expression are more moderate (Fig. 2D,E). The change in accessibility between day 0 and day 2 is particularly large. This suggests that the immediate responses to RA treatment are large changes in chromatin accessibility, which then induces subsequent gene expression changes in the time course.
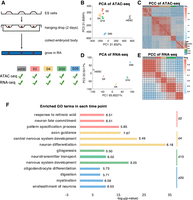
Genome-wide profiling of gene expression and chromatin accessibility during RA induction reveals landscape for RA-driven lineage transition. (A) Schematic outline of study design. (B,C) PCA and heat map of the Pearson's correlation matrix on ATAC-seq data. (D,E) PCA and heatmap of the Pearson's correlation matrix on RNA-seq data. (F) Enriched GO terms in the top 200 specific genes at each time point. The horizontal axis is −log10(P-value) and the number behind the bar represents fold enrichment.
We performed Gene Ontology (GO) term enrichment analysis on the 200 most specifically expressed genes (Methods) at each time point. Because the differentiation is induced by RA, as a positive control we check whether the GO term “response to retinoic acid” is enriched in day 2. Indeed, this term is highly enriched in day 2 (P = 3.67 × 10−10, fold change = 6.51). This gives us confidence that the enrichment analysis on specifically expressed genes is capable of detecting biologically relevant signals. Figure 2F presents the most significantly enriched GO terms at each time point. It suggests that RA-induced differentiation into multiple cell types, with neuronal cells arising early in the time course and glial lineages emerging later. The enrichment of “digestion” on day 20 suggests that the differentiation also gave rise to other cell types beyond neurons and glia (Fig. 2F).
Context-specific inference of gene regulatory relations by the PECA2 method
We developed PECA2 as a method to infer gene regulatory relations (TF-TG relations and RE-TG relations) in a cellular context based on gene expression and chromatin accessibility data in that context (Methods). First, the trans-regulation score (TRS) for a given TF-TG pair is defined by integrating information from multiple REs that may mediate the activity of the TF to regulate the TG (Fig. 1B; Methods). Before applying it to analyze our time course data, we first validate the usefulness of TRS using gene perturbation experiments. On mESCs, we performed shRNA knockdown (separately) of transcription factors Pou5f1, Sox2, Nanog, Esrrb, and Stat3 and measured gene expression changes following the knockdown. Regarding the most differentially expressed genes (FDR<0.01), see Guan et al. (2019) following the knockdown as true target genes of the TF, we calculated the area under the ROC curve (AUC) of target prediction based on ranking by TRS. As a comparison, we collected ChIP-seq data for these TFs on mESC and used the potential target gene score from BETA (Wang et al. 2013) to predict the target genes. Figure 3A shows that TRS-based prediction has substantially higher AUCs than predictions based on ChIP-seq data. We also compared our methods with different baseline methods (different combination of features from expression and accessibility data) (Methods) on TF-TG prediction. TRS-based predictions get much higher accuracy then these baseline methods (Supplemental Fig. S1). These results validated the usefulness of TRS in predicting TF-TG relations.
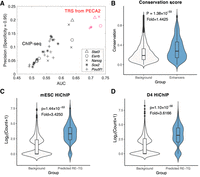
PECA2 infers accurate gene regulation supported by ChIP-seq, shRNA knockdown, and HiChIP experiments. (A) Comparison of PECA2 TRS with ChIP-seq experiment on five important regulators in mESC by taking the knockdown data as ground truth. Shapes represent different transcription factors and colors represent different methods. Red represents results from PECA2 and black represents results from ChIP-seq experiment. (B) Conservation score distribution comparison between REs predicted to regulate at least one gene and randomly selected REs. (C,D) Validation of PECA2 predicted RE-TG pairs by the HiChIP experiment on mESC and RA D4. Background RE-TG pairs are randomly selected to have the same distance distribution as the predicted RE-TG pairs. “Fold” represents fold change of average read count of predicted RE-TG pairs versus background RE-TG pairs.
After we have the trans-regulation score, we define the cis-regulation score (CRS) for each RE-TG pairs by integrating the TRS of TFs with binding potential on the RE (Fig. 1B; Methods). To validate the usefulness of CRS, we downloaded the publicly available H3K27ac HiChIP data on mESC (Mumbach et al. 2017) and also performed H3K27ac HiChIP experiments on RA D4 (Zeng et al. 2019). The RE-TG pairs from CRS-based prediction (Methods) have 17.72 and 15.30 HiChIP reads on average on mESC and RA D4, respectively. As a control, we selected random RE-TG pairs (under the constraint that they have the same distance distribution as our predicted RE-TG pairs) from all candidate RE-TG pairs. The RE-TG pairs from control groups have 5.17 and 4.23 read counts in HiChIP data on average on mESC and RA D4, respectively (Fig. 3C,D). HiChIP counts are much higher in CRS-predicted RE-TG pairs (one-tailed Wilcoxon rank-sum test, ESC: P-value = 1.4412 × 10−53, Fold = 3.4250; RA D4: P-value = 1.1025 × 10−56, Fold = 3.616). We also constructed two more control RE-TG sets and compared the read counts with our predicted RE-TG pairs. Those controls are selected from RE-TG pairs have the same TG expression distribution and have the same distribution of number of ends covered by H3K27ac ChIP-seq peaks, respectively. Our predicted RE-TG pairs have much higher read counts than both the control groups have (Supplemental Fig. S2). Finally, when we examined the sequences of the RE in the RE-TG pairs, the CRS-predicted REs have significantly higher sequence conservation than those from the control group (one-tailed Wilcoxon rank-sum test, P-value = 1.3801 × 10−33 Fold change = 1.4425) (Fig. 3B). These results validated the usefulness of CRS in predicting RE-TG relations. Taken together, PECA2 provides high-quality, genome-wide, and context-specific inference of gene regulatory relations for each single time point with paired gene expression and chromatin accessibility data. PECA2 is available at https://github.com/SUwonglab/PECA.
Identification and annotation of novel regulatory elements by CRS
To identify important REs, we analyzed REs and their target genes. First, we annotated the REs into three groups: (1) promoters, (2) known enhancers (from the mouse ENCODE Project, including developmental stage enhancers), and (3) novel enhancers (Supplemental Table S2). We found that 7% of REs are promoters, 60% are known enhancers, and the remaining 33% are novel enhancers (Fig. 4A). About 69% of the genes are regulated by both known and novel enhancers. To understand the functions of the known and novel enhancers, we divided the genes into two groups (targets of known enhancers, and targets of novel enhancers) depending on whether the associated RE with the maximum CRS score is a known enhancer or a novel enhancer. On average over different time points, known enhancers have 4604.80 target genes and novel enhancers have 3370.20 target genes (Fig. 4B). We found the novel enhancers with maximum CRS scores are highly conserved with a mean conservation score of 0.2034. The conservation score of those enhancers is about twofold higher than random regions, which is higher than the mean conservation of known enhancers (conservation score = 0.1753) and comparable to open promoter regions (conservation score = 0.2368) (Fig. 4C). On each time point, we chose the top 500 specifically expressed genes (based on gene specificity score) (Methods) of known enhancers and novel enhancers, respectively. Figure 4D shows the GO enrichment score (defined as the geometric mean of fold change and −log10[P-value]) of known versus novel enhancers’ targets on RA D10. GO terms such as cerebellar granular layer development, synapse assembly, regulation of AMPA receptor activity, and hindbrain morphogenesis are only enriched in novel enhancers’ targets, but not enriched in known enhancers’ targets. These results suggest that enhancer annotation from the ENCODE Project on those GO terms associated genes is still incomplete. The reason is that the mouse ENCODE Project contains brain tissue but does not contain specific neuron cellular context. By combining experimental and computational analysis, we identified new enhancers whose inclusion will greatly enhance the interpretation of data from the time course.
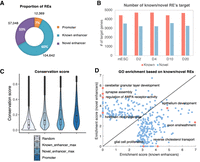
Identification and annotation of novel enhancers. (A) Pie charts of the promoter, known enhancers, and novel enhancers in REs. (B) Bar plot of numbers of known enhancers and novel enhancers at different time points. (C) Conservation score distribution of different sets of REs. (D) Comparison of GO enrichment on known enhancers’ targets and novel enhancers’ targets on D10. Top 500 specifically expressed genes in each group are chosen to perform GO analysis. The x-axis represents enrichment score on known enhancers’ targets, and the y-axis represents that on novel enhancers’ targets. Each dot represents one GO term. The enrichment score is defined as the geometric mean of fold change and −log10 P-value.
Core regulatory modules of TFs and TGs reveal subpopulation characteristics
To better understand the regulatory network inferred by PECA2, we divided the TF-TG networks, which is represented by a TRS matrix (rows represent TF and columns represent TG), into several modules (dense subnetworks) by non-negative matrix factorization (NMF) (Brunet et al. 2004; Wang et al. 2008) based module detection method (Methods; Fig. 5A). We used RA D4 data to illustrate and validate this approach.
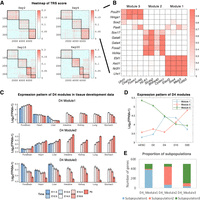
Core regulatory modules extracted from gene regulatory networks are supported by subpopulation from single-cell RNA-seq data. (A) Heatmap of reordered normalized TRS scores at D2 to D20. The black line represents the detected modules from NMF. (B) Heatmap of D4 normalized TRS on selected specific genes and TFs. (C) Mean expression pattern of genes from three different modules of RA D4 on the developmental stage of seven tissues. (D) Mean expression pattern of genes from three different modules of RA D4 on RA time course. (E) Distribution of RA D4 top 500 module-specific genes’ maximum expressed subpopulations from single-cell RNA-seq data.
NMF analysis of the TRS matrix at D4 yielded three modules, in which the TRS scores between TFs and TGs within the same module are much higher than those between different modules. It means we have divided the TF-TG networks into three groups of nodes with dense connections internally and sparser connections between groups. Figure 5B shows this pattern for selected TFs and TGs. We found genes from different modules have different expression patterns on mouse tissue development data (Gorkin et al. 2017) as well as the RA induction data (Fig. 5C,D). Module 1 contains TFs such as Ascl1, Ebf1, Nr2f1, and Lhx1. They were previously reported to be relevant to neuronal development. Using data from Gorkin et al. (2017), we also found that Module 1 associated TGs have a high and increasing expression pattern in embryonic brain development (Fig. 5C, top). Module 2 contains mesodermal and endodermal related regulators such as Sox17, Gata4, Gata6, Foxa2, and Hnf4a. Consistently, target genes in Module 2 are highly expressed in the liver, lung, heart, kidney, intestine, and stomach, but lowly expressed in the brain (Fig. 5C, middle). Module 3 associated TFs such as Pou2f1, Hmga1 (Nishino et al. 2008), Sox2 (Graham et al. 2003), and Pax6 (Sansom et al. 2009) are known to be involved in the maintenance of neural stem and progenitor cells. Consistently, we found that the expression of Module 3 associated TGs has decreasing patterns during embryonic development in all tissues (Fig. 5C, bottom). These pieces of evidence show that the biological function of module-specific regulators is well-matched to the cellular context suggested by the expression pattern of the corresponding target genes.
Because the cell population after 4 d of differentiation is expected to be a mixture of subpopulations of cells representing different developmental lineages, it is likely that the modules identified above may reveal the regulatory characteristics of these subpopulations. We used single-cell RNA-seq data to test this hypothesis. In a previous study (Duren et al. 2018), we performed scRNA-seq experiments on day 4 of the RA-induced time course and found that there are three distinct subpopulations of cells (Duren et al. 2018). We selected the top 500 module-specific genes (Methods) in each module and assigned each gene to one of the three subpopulations (the one with the maximum expression). We found that 76.20% of Module 1–specific genes are matched to the subpopulation 1, 75.00% of Module 2–specific genes are matched to the subpopulation 2, and 79.40% of Module 3–specific genes are matched to the subpopulation 3 (Fig. 5E). This result shows that the modules are well-matched to the subpopulations and therefore can help us to elucidate the regulatory relations within the subpopulations. In other words, our analysis of bulk expression and accessibility data provided useful information on subpopulation-specific gene regulatory networks.
Finally, we applied the same analysis for each time point and found that there are three modules in D2, three modules in D4, four modules in D10, and four modules in D20 (Fig. 5A; Supplemental Figs. S3–S5).
Associations of regulatory modules across time points
With these functionally meaningful regulatory modules indicating subpopulation at each time point, we next investigated the relationships among them across time points. Figure 6A shows the similarities between modules in adjacent time points. It is seen that the three modules in D2 are well-matched to the three modules in D4 (Jaccard similarities are 0.77, 0.82, and 0.85), the four modules in day 10 are well-matched to the four modules in D20 (Jaccard similarities are 0.69, 0.74, 0.88, and 0.66). This suggests that the analysis of a set of matched modules across time points may reveal useful biological insight. There are four modules in D10 but only three in D4. Modules 2 and 3 in D4 are similar to Modules 2 and 3 in D10 (Jaccard similarities are 0.74 and 0.88, respectively). However, it is not obvious how Modules 1 and 4 in D10 are related to the D4 modules.
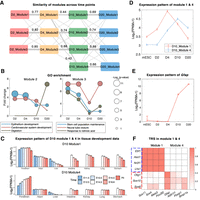
Core regulatory modules decompose the mixture populations consistently across time points. (A) Jaccard similarity of modules between neighboring time points. Line width represents the Jaccard similarity, and the similarity value is labeled on the line if it is >0.1. (B) GO analysis on modules at each time points. Top 100 specifically expressed genes of each module at each time point are selected for GO enrichment analysis. The x-axis represents time points, and the y-axis represents fold enrichment. The size of circles represents the enrichment P-value. (C) Mean expression pattern of genes from D10_Module1 and D10_Module4 on developmental stage of seven tissues. (D) Mean expression pattern of genes from D10_Module1 and D10_Module4 on RA time course. (E) Expression pattern of the glial marker gene Gfap on RA time course. (F) Heatmap of D10 normalized TRS on selected specific genes and TFs.
To explore the function of the modules, we extracted the top 100 most specifically expressed genes from each module (Methods) in each of the time points and performed GO enrichment analysis (Fig. 6B). Epithelium development and cardiovascular system development are enriched in Module 2 of earlier time points. In D20 of Module 2, digestion is enriched, but the enrichment levels of epithelium development and the cardiovascular system development are decreased. Those enriched GO terms suggest Module 2 involves endoderm and mesoderm development, which is consistent with the function of subpopulation 2 from the scRNA-seq data (Duren et al. 2018). Stem cell population maintenance is enriched in Module 3 and has a downward trend along with time, which is consistent with the expression pattern of Module 3–specific genes in RA induction and tissue development data (Fig. 5C,D), indicating that the stem cell population is becoming smaller. Module 3 also enriches neural tube closure, which suggests Module 3 contains neural stem cells.
To see the difference between Module 1 and Module 4 in D10, we checked the expression pattern of those genes on mouse developmental stage data and RA time course data (Fig. 6C,D). In the developmental stage, genes from Module 1 are always highly expressed in forebrain, but genes from Module 4 have an upward trend. These results suggest that both Module 1 and Module 4 are brain-related cell populations, but Module 1 is much earlier than Module 4 in development. In RA induction time course data, we also see a similar pattern (Fig. 6D). From the results of GO enrichment analysis, we find they are enriched in different functions. Module 4 in D10 and D20 are enriched in glial cell differentiation which is not enriched in any of the modules in previous time points. Module 1 in all the time points are enriched in axon guidance (Supplemental Fig. S6). GO enrichment analysis show Module 1 is neuron related and Module 4 is glial related.
Next, we checked the specific trans-regulators of these two modules (Fig. 6F). We find genes from Module 1 are regulated by Ebf1, Ascl1, Nr2f1, and Lhx1, whereas genes from Module 4 are regulated by Olig1, Sox10, and Sox8. From the similarity of regulators and enriched GO terms on target genes, Module 1 in D10 is very similar to Module 1 in D4. Therefore, Module 1 would correspond to neuron subpopulation. The GO enrichment analysis and module-specific regulators indicate that the newly generated module in D10 (Module 4) is the glial population. If it were true, the glial marker genes should be highly expressed in D10 but low expressed in D4. We examined the expression pattern of the astrocyte marker gene Gfap, which is specific to Module 4. Indeed, Gfap is very highly expressed in D10 but almost not expressed in D4 (Fig. 6E). From these results, we conclude that Module 4 corresponds to the glial population and has emerged between D4 and D10. To clarify the origin of Module 4 further, we need to identify the regulators that drive the expression and accessibility changes. We will return to this question subsequently.
Driver regulators shed light on cell lineage transition
Under RA treatment, mESCs differentiated (or transitioned) into three different subpopulations after 2 d. To explore the regulatory mechanism behind the transition, we identified driver regulators in each module. Driver regulators are defined as TFs that (1) are up-regulated during the transition from day 0 to day 2 by at least 1.5-fold, and (2) with TRS score on up-regulated genes being significantly higher than that on non-up-regulated genes (one-tailed rank-sum test, FDR < 0.05). Figure 7A gives the driver regulators of the three modules in RA day 2. In Module 2, driver regulators include Gata4, Gata6, Rxra, Sox17, Foxa2, and Hnf4a. Sox17, Foxa2, and Hnf4a are involved in endoderm development. Gata4 and Gata6 are known to be involved in mesoderm development. We find Rxra, an important cofactor of retinoic acid receptors, is involved in endoderm and mesoderm development. The expression level of retinoic acid receptor cofactor Rxra is not very high on day 2 (FPKM 14.47, ranked 3897 in 7975 dynamic expressed genes, top 48.87%), but it is identified as a driver regulator. We find that the expression of Rxra is correlated with up-regulated genes (on ENCODE data, rank-sum test, P = 6.44 × 10−104) (Fig. 7B). Furthermore, we find the motif of RXRA is enriched in REs associated with up-regulated genes (rank-sum test, P = 1.98 × 10−26) (Fig. 7C). Previous literature indicated that knocking out of Rxra leads to an abnormal phenotype in the cardiovascular system (Sucov et al. 1994; Chen et al. 1998; Mascrez et al. 2009). Even though the expression level is not very high, Rxra is likely to be an important driver of the transition from stem cell state to mesoderm and endoderm state. In Module 1, the driver regulators are neuron-related factors like Ascl1, Nr2f1, Pou3f2-4, Hox family, Meis family, Pbx family, Rarb, and other factors. In Module 3, driver regulators are Pax6, Dbx1, Gli1, Gli3, and other factors. Pax6 is known to be important in neural stem cell development. We also find that developing brain homeobox factor Dbx1 is one of the important drivers.
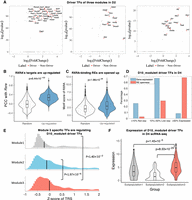
Identification of driver regulators reveals ancestor–descendant fates for regulatory modules. (A) Driver regulators of D2_Module1, D2_Module2, and D2_Module3. The x-axis is log2 fold change; the y-axis is −log10(P-value). (B) Distribution comparison of Rxra’s PCC with up-regulated genes and randomly selected genes. (C) Distribution comparison of RXRA’s motif enrichment on REs of up-regulated genes and randomly selected genes. (D) Distribution of D10_Module4 driver TF expression (blue bar) and their RE openness (red bar) on D4. Expression or openness greater than the median are labeled as “Expressed,” less than decile are labeled as “Not exp,” and the remaining are labeled as “Low exp.” (E) Distribution of Z-score of TRS between top 50 module-specific TFs and driver TFs of D10_Module4. (F) Expression distribution of the Module4's driver regulators on D4 scRNA-seq data. Columns represent subpopulations identified from scRNA-seq data.
To determine the origin (ancestor) of the new population in D10, we checked the expression of driver regulators of D10_ Module4 (Sox8, Nfix, Olig1, Nfib, Hoxc8, Nkx2-2, Foxo6, Cpeb1, Lbx1, Hoxa6, Pou6f1, and Rfx4) in D4. We find 11 of 12 driver regulators have not been expressed (greater than the median expression level of all genes, noted as ≥50%) (Fig. 7D) in D4 yet. However, 118 of 174 (67.82%) REs of those driver TFs are already open in D4 (Fig. 7D). So although driver TFs are not expressed yet, it is still possible to determine the ancestor of Module 4 by comparing the TRS score of module-specific TFs targeting on those driver TFs. Figure 7E shows the distribution of normalized TRS score (Z-score) of the top 50 module-specific TFs of each module targeting on the Module 4 driver TFs. The results show that TFs from Module 3 in D4 have significantly higher TRS scores than those from Module 1 and Module 2 (one-tailed rank-sum test, P-values are 0.0014 and 1.97 × 10−6, respectively), which indicates that the new module in D10 is likely to have arisen from Module 3 in D4. To test this hypothesis, we compared the expression of these driver TFs in scRNA-seq from D4. It is seen that the driver regulators are more highly expressed in subpopulation 3 than subpopulations 1 and 2 (Fig. 7F), which is consistent with our hypothesis, because Module 3 has been matched to subpopulation 3 in our previous analysis (Fig. 5E). As further validation, we compared the expression of neural stem cell markers (Sox2 and Nes) in the three subpopulations in D4 (Supplemental Fig. S7B,C). In most of the subpopulation 3 cells, Sox2 and Nes are expressed, which indicates that subpopulation 3 is enriched for neural stem cells from which the glial cells in D10 emerged.
Based on driver regulators, it is possible to construct the developmental path of the subpopulations. We define a transition score between the modules in adjacent time points to construct the ancestor–descendant map for the subpopulations in the time course (Methods). The ancestor–descendant mapping results are topologically similar to the mapping based on module similarities (Fig. 6A) except for the TFs driving the transition Module 4 in D10 (Fig. 8). After RA induction, three subpopulations—neuron, mesoderm and endoderm, and neural stem cell—are generated between mESC to D2. Glia subpopulation is generated from neural stem cells between D4 and D10.
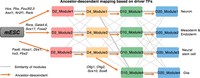
TimeReg suggests the developmental trajectory of the subpopulations for RA-driven lineage transition. Schematic overview of the subpopulations at each stage. The gray line represents the similarity of neighboring regulatory modules. The orange line represents the ancestor–descendant mapping among regulatory modules. TFs on the orange lines represent the important driver regulators causally connecting the modules.
TimeReg identifies novel driver regulators of direct reprogramming from fibroblasts to neurons
In addition to mESC differentiation, we applied TimeReg to study the regulatory network for direct lineage reprogramming, which can convert mouse embryonic fibroblasts (MEFs) to induced neuronal (iN) cells, and its underlying mechanism to overcome epigenetic barriers is fundamental for differentiation and development. A previous study (Wapinski et al. 2013) found that Ascl1 is sufficient to induce neuron from fibroblast and generated time course gene expression data for MEF to iN reprogramming. Follow-up study generated time course chromatin accessibility data (Wapinski et al. 2017), observed rapid chromatin changes in response to Ascl1 at day 5, and identified Zbtb18, Sox8, and Dlx3 as key TFs downstream from Ascl1 for major transition at day 5. We collected gene expression data and chromatin accessibility data of Ascl1-induced reprogramming from these two previous studies. Two levels of time course data can be paired on day 0, day 2, and day 5. This allows us to run TimeReg integrative analysis on this iN reprogramming data. We asked how the cell triggers the follow-up signals, remodeling the chromatin structure in a genome-wide way, and turns on and off lineage-specific gene expression within 48 h at early stage.
Core regulatory modules are consistent with single-cell data
We find two well-separated modules in the day 2 network (Fig. 9A). Module 1 contains TFs like Ascl1, Id2, Sox4, Sox8, Sox11, Dlx3, and Zbtb18, and contains TG like Cplx2, Dner, Nkain1, and Eda2r. Module 2 contains TFs like Klf4, Tead3, AP1 complex factors, Egr1, Prrx1, and contains TG like Myof, Emp1, Actn1, and Bst2. In reprogramming time course data, Module 1–specific genes have an increasing pattern, but Module 2–specific genes have a decreasing pattern (Supplemental Fig. S8A). Furthermore in public ENCODE data, Module 1–specific genes have a high and increasing expression pattern in embryonic brain development and low and decreasing patterns on heart, kidney, liver, and lung tissue development, whereas Module 2–specific genes have the opposite trend (Supplemental Fig. S8B). GO enrichment analysis shows that Module 1–specific genes are enriched in neuron-related functions, and Module 2–specific genes are enriched in muscle and epithelial related functions, which are consistent with the expression pattern in developmental stages (Supplemental Fig. S8C). Collectively, TimeReg reveals two functional modules, which are consistent with our expectation that the neuron subpopulation is induced to become larger, and other populations are repressed to become smaller. This finding is supported by the single-cell RNA-seq data (Treutlein et al. 2016) of this Ascl1 reprogramming, which shows two well-separated subpopulations (Fig. 9B). We find that 90.40% of Module 1–specific genes are matched to the subpopulation 1 (Fig. 9C), and 88.80% of Module 2–specific genes are matched to the subpopulation 2 (mapped to the subpopulation with the maximum expression). In single-cell RNA-seq data, the neuron-specific gene Ascl1 is specifically expressed in subpopulation 1 and the muscle-specific gene Myof is specifically expressed in subpopulation 2 (Fig. 9D,E). These results show that the modules identified by TimeReg indeed capture subpopulation characteristics and therefore can help us to elucidate the regulatory relations within the subpopulations.
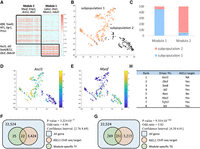
TimeReg analysis on direct reprogramming from fibroblast to neuron. (A) Heatmap of reordered normalized TRS scores at day 2. The black line represents the detected modules from NMF. (B) t-SNE plot of single-cell RNA-seq data. Color represents clustering label. (C) Distribution of day 2 top 500 module-specific genes’ maximum-expressed subpopulations from single-cell RNA-seq data. (D,E) Expression of module-specific genes on t-SNE plot. (F,G) Module 1–specific genes are enriched in ASCL1 ChIP-seq target genes. (H) List of driver regulators of Module 1 in day 2.
Neuron-related module-specific genes are enriched in Ascl1 ChIP-seq targets
As the reprogramming is induced by pioneer factor Ascl1, Module 1–specific regulators and target genes should contain Ascl1 target genes. To validate the module-specific regulators and module-specific genes, we compared them with the Ascl1 target genes from ChIP-seq data at day 2. The results show both Module 1–specific TFs and Module 1–specific target genes are significantly overlapped with the Ascl1 target genes (Fisher's exact test, P-values 3.22 × 10−7 and 9.35 × 10−62, odd ratios 4.90 and 5.02, respectively) (Fig. 9F,G).
Discovery of novel driver regulators
The TimeReg analysis identified eight driver regulators for the neuron-related Module 1 at day 2 (Fig. 9H). Ascl1 ranked the first in the list, which means our method successfully detected this super-regulator. Among the other seven driver regulators, six of seven regulators contain Ascl1 ChIP-seq peaks in their regulatory region. Dlx3 and Sox8 ranked second and third, respectively. This is consistent with the fact that they were also previously reported to be important regulators of reprogramming at day 5 (Wapinski et al. 2017). Except for these known regulators, our method detected some novel driver regulators. The forth-ranking regulator is Id2, which is not reported in the two separate studies from gene expression level and chromatin accessibility level, respectively. Id1, one important paralog of Id2, is also identified as a driver regulator. Previous research shows that dimerization of ID1/2 with bHLH family factor MYOD would prevent MYOD from binding to DNA and inhibits muscle differentiation (Sun et al. 1991; Jen et al. 1992). Therefore, activation of Id1/2 by Ascl1 will inhibit muscle differentiation and drive the cell into the neural state. Id1 and Id2 would be important driver regulators of the reprogramming process. Another interesting driver regulator is Tcf12, which is known to be involved in the initiation of neuronal differentiation (Rebhan et al. 1997). Our computational model TimeReg successfully identified these driver regulators by integrating gene expression and chromatin accessibility data, which may play an important role in reprogramming within 48 h and gain new insights for early regulation.
Discussion
In this paper, we proposed a time course regulatory analysis tool TimeReg from paired gene expression and chromatin accessibility data. To reduce dimensionality, TimeReg integrates expression and chromatin accessibility levels, aggregates the REs and TFs to quantitatively infer TF-TG and RE-TG regulatory strength, and narrows down to regulatory module level by highlighting the important role of driver TFs. The GRN is validated by experimental data, and the core regulatory modules characterize different subpopulations of cells. Based on the driver regulators’ sequential expression and binding on chromatin accessible regions, we causally connect the modules (subpopulations) in adjacent time points and reveal the developmental trajectory for RA induced differentiation.
Inferring ancestor–descendant fates from developmental time courses is a challenging problem, especially in the presence of large gaps (≥48 h) between time points (Schiebinger et al. 2019). In this study, we found a newly generated population at D10 that was absent in the previous time point D4. It is challenging to infer ancestor–descendant in this 6-d gap development stage even if one has single-cell data on each time point. From scRNA-seq data on D4, we see ∼72% of the D10 Module 4–specific genes are highly expressed in subpopulation 1 (only 15% are highly expressed in subpopulation 3) (Supplemental Fig. S7A), but driver regulators are more highly expressed in subpopulation 3 (Fig. 7F). There are two reasons why Module 4 specific genes are more highly expressed in subpopulation 1 than subpopulation 3: (1) Many expressed genes are shared in neuron and glial, and (2) glial-specific genes have not been expressed in D4 yet. Because of these reasons, the expression-based analysis will not be able to map the ancestor of Module 4 correctly. By exploiting paired expression and accessibility data in the time course, our method is capable of inferring such mappings reliably.
We note that because knockdown of a TF may result in secondary changes in addition to those caused by the knocked down TF, TF ChIP-seq data would not be expected to predict all the transcriptional changes. Conversely, ATAC-seq data before and after the knockdown could reveal changes related to the knocked down TF as well as secondary effects caused by changes in other TFs. Therefore, time course experiments to collect paired RNA-seq and ATAC-seq data before and after knockdown of key TFs will be a powerful approach to gene regulatory analysis.
Finally, we discuss the limitation of our method. The incorporation of prior knowledge from external data into our model has allowed us to greatly reduce the complexity of the model. The caveat is that the cellular context used in the prior calculation is currently incomplete and this may cause modeling bias. The validation results reported above show that in spite of this, our method is already useful for many types of inferences and predictions. We expect that the bias associated with the use of external data will be further minimized as these data become more complete in the future. One possible extension of this paper is to identify the union of all modules across multiple time points simultaneously, which would describe the module dynamics more clearly.
Methods
Gene regulatory network inference from a single time point by PECA2
Our previous method PECA takes paired expression and chromatin accessibility data across diverse cellular contexts as input,
models how trans- and cis-regulatory elements work together to affect gene expression in a context-specific manner, and outputs the transcriptional
regulatory network with TF-RE-TG as the building block (Duren et al. 2017). PECA2 aims to infer the regulatory network in a new cellular context different from those used in training the model by
selecting active REs, specifically expressed TFs, and expressed TGs in this context. Unlike PECA, which requires diverse cellular
context data as input, PECA2 only requires one paired data (i.e., one sample) as input and infers the gene regulatory network.
PECA2 first improves the TF-TG accuracy by taking the combinatorial regulation among cis-regulatory elements into account and aggregating the REs used by the same TF to regulate a given TG. An upstream TF regulating
a TG should satisfy three conditions: (1) The TF should be expressed, (2) motifs of the TF should be enriched in the REs of
this TG, and (3) the TF should be coexpressed with this TG across diverse cellular contexts. By combining this information,
we define the trans-regulation score (TRS) between given ith TF and jth TG as follows: where Bik is motif binding strength of ith TF on kth RE, which is defined as the sum of binding strength (motif position weight matrix-based log-odds probabilities; see HOMER
software for detail) of all of the binding sites on this RE; and
where Bik is motif binding strength of ith TF on kth RE, which is defined as the sum of binding strength (motif position weight matrix-based log-odds probabilities; see HOMER
software for detail) of all of the binding sites on this RE; and 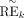 represents the normalized accessibility [REk × REk/median(REk)] of kth RE. The first term REk represents the actual accessibility of the RE, and the second term represents relative accessibility compared to the median
accessibility level of this RE on external data. If one RE is accessible in the given cellular context, and the accessibility
is also much higher than the accessibility level on other contexts, then this RE is specifically accessible in the given cellular
context;
represents the normalized accessibility [REk × REk/median(REk)] of kth RE. The first term REk represents the actual accessibility of the RE, and the second term represents relative accessibility compared to the median
accessibility level of this RE on external data. If one RE is accessible in the given cellular context, and the accessibility
is also much higher than the accessibility level on other contexts, then this RE is specifically accessible in the given cellular
context; 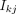 represents the interaction strength between the kth RE and jth TG, which is learned from the PECA model on diverse cellular contexts.
represents the interaction strength between the kth RE and jth TG, which is learned from the PECA model on diverse cellular contexts. 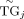 represents the normalized expression level of the jth TG [TGj × TGj/median(TGj)]. TFAi represents the activity of the ith TF (geometric mean of normalized expression and motif enrichment score on open region); Rij is the expression correlation of the ith TF and jth TG across diverse cellular contexts. Higher regulation score TRSij implies the jth TG is more likely to be regulated by the ith TF. In deciding which TRSij are statistically significant, we randomly select some {TFi − TGj} pairs and take these pairs as negative controls. We choose the threshold of the regulation score by controlling the false
discovery rate (FDR) at 0.001.
represents the normalized expression level of the jth TG [TGj × TGj/median(TGj)]. TFAi represents the activity of the ith TF (geometric mean of normalized expression and motif enrichment score on open region); Rij is the expression correlation of the ith TF and jth TG across diverse cellular contexts. Higher regulation score TRSij implies the jth TG is more likely to be regulated by the ith TF. In deciding which TRSij are statistically significant, we randomly select some {TFi − TGj} pairs and take these pairs as negative controls. We choose the threshold of the regulation score by controlling the false
discovery rate (FDR) at 0.001.
PECA2 then improves the RE-TG accuracy by taking the TF combinatorial regulation into account and aggregating the TFs using
a given RE. We define cis-regulation score (CRS) for RE-TG pairs based on the binding TFs’ TRS as
We approximate the distribution of non-zero log2(1+CRS) by a normal distribution, and predict RE-TG associations by selecting the pairs that have P-value < 0.05.
Regulatory module detection by matrix factorization
To detect key TF-TG subnetworks (core modules) from the TRS, we use non-negative matrix factorization (NMF). Before matrix
factorization, we perform the following transformations for TRS matrix: (1) log2 transformation  ; (2) normalize across rows and columns,
; (2) normalize across rows and columns,  , where function Z(x) represents Z transformation for each row of matrix x; and (3) set the negative values to zero. We assign the TFs and TGs into the clusters based on the maximum factor loading.
To determine the number of modules, we run NMF 50 times and calculate consistency based on the cophenetic correlation coefficient
(Brunet et al. 2004). We choose K from the range of 2–7 and choose the K which gives the most consistent results.
, where function Z(x) represents Z transformation for each row of matrix x; and (3) set the negative values to zero. We assign the TFs and TGs into the clusters based on the maximum factor loading.
To determine the number of modules, we run NMF 50 times and calculate consistency based on the cophenetic correlation coefficient
(Brunet et al. 2004). We choose K from the range of 2–7 and choose the K which gives the most consistent results.
Module-specific genes
Given one gene and its corresponding module, we define a module specificity score for this gene by comparing the normalized TRS of in-module TFs and out-module TFs (one-tailed rank-sum test). If an in-module TF's normalized TRS score is significantly higher than that of the out-module TF's, then this gene is specific to this module. Module specificity score is defined as the product of −log10(P-value) and the mean TRS score with the TFs in the same module. We take the 500 genes that have the highest module specificity score as module-specific genes. Similarly, we define module-specific TFs by comparing the normalized TRS of in-module genes and out-module genes.
Ancestor–descendant mapping via driver TF
To map the modules in adjacent time points, we define a transition score TSijt for the ith module from the tth time point, and the jth module from the (t + 1)th time point by mean normalized TRS score of source TF set Si,t to target TF set Tj,t+1 as where normalized TRS
where normalized TRS 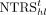 is the Z-score of the TRS score, is defined as TRS minus mean TRS score of hth TF and divided by the standard deviation of hth TF. Source TF set Si,t is the top 50 module-specific TFs from the ith module of tth time point. Target TF set Tj,t+1 is defined as the top 20 driver TFs from the jth module of the (t + 1)th time point. If the number of driver TFs is <20, we add some TFs from top-ranking module-specific TFs to get a target
TF set Tj,t+1 containing 20 TFs. The ancestor–descendant relation should have a higher transition score. So we map the jth module from the (t + 1)th time point to the module at the tth time point, which has a maximum transition score.
is the Z-score of the TRS score, is defined as TRS minus mean TRS score of hth TF and divided by the standard deviation of hth TF. Source TF set Si,t is the top 50 module-specific TFs from the ith module of tth time point. Target TF set Tj,t+1 is defined as the top 20 driver TFs from the jth module of the (t + 1)th time point. If the number of driver TFs is <20, we add some TFs from top-ranking module-specific TFs to get a target
TF set Tj,t+1 containing 20 TFs. The ancestor–descendant relation should have a higher transition score. So we map the jth module from the (t + 1)th time point to the module at the tth time point, which has a maximum transition score.
Baseline methods for TRS score
We have five baseline methods for TRS: Pearson's correlation coefficient (PCC), summation of TF binding strength in nearby
open regions (BO), summation of TF binding strength in interacting regions (BI), summation of TF binding strength in interacting
open regions (BOI), and combination of PCC with BOI. PCC-based TRS is defined as PCC between TF expression and TG expression
from diverse cellular contexts. Baseline TRS are defined as follows:
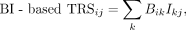

 where dkj represents the distance between the kth RE and the jth TG; d0 is a constant; and we choose 40 kb as a default value. We only consider the REs within 200 kb distance from the TGs.
where dkj represents the distance between the kth RE and the jth TG; d0 is a constant; and we choose 40 kb as a default value. We only consider the REs within 200 kb distance from the TGs.
Identification and quantification of REs
We define regulatory elements (REs) by the union of open peaks from ATAC-seq (called by MACS2) data on each time point. In
the whole induction process, we get 174,059 REs in total. To quantify the accessibility of the peaks, we calculate the openness
score for each sample in each region. Given a certain region of length L, we treat this region as foreground and denote by X the count of reads in the region. To remove the sequencing depth effect, we choose a background region with a length L0 and denote by Y the count of reads in this background window. The openness score is defined as the fold change of read counts per base pair
as where δ is a pseudocount (the default value of δ is 5 in our implementation).
where δ is a pseudocount (the default value of δ is 5 in our implementation).
Gene specificity and GO analysis
To select specific genes in each time point, we define gene specificity score based on expression in the current condition
and its median expression level on publicly available diverse context data and RA time course data. Gene specificity of the
ith gene on the tth time point is defined as follows: where the median(FPKMi, public) represents the median expression level of the ith gene in public data, median(FPKMi, RA) represents the median expression level of the ith gene in RA time course data, FPKMi,t represents the expression level of the ith gene on the tth time point. We select the top specific 200 genes and GO enrichment analysis based on PANTHER Version 14.1 (Thomas et al. 2003).
where the median(FPKMi, public) represents the median expression level of the ith gene in public data, median(FPKMi, RA) represents the median expression level of the ith gene in RA time course data, FPKMi,t represents the expression level of the ith gene on the tth time point. We select the top specific 200 genes and GO enrichment analysis based on PANTHER Version 14.1 (Thomas et al. 2003).
Reprogramming data analysis
To do TimeReg analysis on reprogramming data, we set K as [1, 2, 2] on the three time points, respectively. ASCL1 ChIP-seq target genes are genes that have ASCL1 ChIP-seq peak on promoters or enhancers. Driver TFs are ranked by −log10(P-value) × fold, where fold is the mean TRS score of up-regulated genes divided by mean TRS score of non-up-regulated genes.
Experimental design of retinoic acid–induced mESC differentiation
Mouse ES cell lines R1 were obtained from the American Type Culture Collection (ATCC Cat. no. SCRC-1036). The mESCs were first expanded on an MEF feeder layer previously irradiated. Then, subculturing was carried out on 0.1% bovine gelatin-coated tissue culture plates. Cells were propagated in mESC medium consisting of Knockout DMEM supplemented with 15% knockout serum replacement, 100 µM nonessential amino acids, 0.5 mM beta-mercaptoethanol, 2 mM GlutaMAX, and 100 units/mL Penicillin-Streptomycin with the addition of 1000 units/mL of LIF (ESGRO, Millipore).
mESCs were differentiated using the hanging drop method (Wang and Yang 2008). Trypsinized cells were suspended in a differentiation medium (mESC medium without LIF) to a concentration of 50,000 cells/mL. Then, 20 µL drops (∼1000 cells) were placed on the lid of a bacterial plate, and the lid was upside down. After 48 h incubation, embryoid bodies (EBs) formed at the bottom of the drops were collected and placed in the well of a six-well ultralow attachment plate with fresh differentiation medium containing 0.5 µM retinoic acid for up to 20 d, with the medium being changed daily.
We followed the ATAC-seq protocol published by Buenrostro et al. (2013) with the following modifications. The EBs were first treated with 0.25% Trypsin + EDTA for 10–15 min at 37°C with pipetting. The pellet was then resuspended in the transposase reaction mix (25 µL 2× TD buffer, 2.5 µL transposase, and 22.5 µL nuclease-free water) and incubated for 30 min at 37°C. After purification, DNA fragments were amplified using 1:30 dilution of 25 µM Nextera Universal PCR primer and Index primer under the following conditions: for 5 min at 72°C, for 30 sec at 98°C, and a total 10 cycles of 10 sec at 98°C, 30 sec at 63°C, and 1 min at 72°C. The library of mESC, D2, D4, and D20 was sequenced on NextSeq with 50-bp paired-end reads; the library of D10 was sequenced on HiSeq with 100-bp paired-end reads.
Total RNA was extracted using the Qiagen RNeasy mini kit. Libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs) with the following modifications. mRNA was first isolated from 1 µg of total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module. Then it was fragmented for 12 min at 94°C before the first strand and the second strand cDNA synthesis. The double-stranded cDNA was then end repaired and ligated with NEBNext adaptor, followed by AMPure XP beads purification (Beckman Coulter). Each library was amplified using NEBNext Universal PCR primer and Index primer (for detail see NEBNext multiplex oligo for Illumina [E7335, New England Biolabs]) under the following conditions: for 30 sec at 98°C, then a total of six cycles of 10 sec 98°C, 30 sec at 65°C, and 30 sec at 72°C, with a final extension for 5 min at 72°C. Additional PCR (four to six cycles) were necessary to obtain enough DNA for sequencing. Finally, an equal amount of DNA from each library was pooled together, and a 400-bp fragment was selected by 2% E-Gel SizeSelect Gels (Thermo Fisher Scientific) and purified with AMPure XP beads. The library was sequenced on Illumina HiSeq with 100-bp paired-end reads.
Experimental design of shRNA knockdown in mESC
For plasmids and shRNA, the pSuper.puro vector containing the human H1 RNA promoter for ectopic expression of small hairpin RNA (shRNA) was obtained from Oligoengine. The shRNA constructs for each target were designed using the Clontech RNAi Target Sequence Selector program. DNA oligonucleotides were first synthesized by Elim Biopharmaceutical, Inc. To anneal oligos, DNA was incubated in the following conditions (for 30 sec at 95°C; for 2 min at 72°C; for 2 min at 37°C; and for 2 min at 25°C). Then, the annealed oligo inserts were ligated with pSuper.puro vector linearized with BglII and either HindIII or XhoI restriction enzymes. Following transformation, the positive clones were selected by digesting with EcoRI and XhoI restriction enzymes and run in 1% agarose gel. In addition, the presence of the correct insert within pSuper.puro vector was confirmed by sequencing before transfection in mammalian cells (sequencing primer: 5′-AAGATGGCTGTGAGGGACAG). The list of shRNA constructs used in this study is shown in Supplemental Figure S9A. The primers used to measure target gene knockdown efficiency in qRT-PCR are shown in Supplemental Figure S9B.
RNA interference (RNAi) experiments were performed with Nucleofector technology. Briefly, 12 µg of plasmid DNA was transfected into 3.5 × 106 mouse ES cells using the Mouse ES cell Nucleofector kit (Lonza). After nucleofection, the cells were incubated in 500 µL warm ES medium for 15 min. Then, the cells were split into four gelatin-coated 60-mm tissue culture plates containing 5 mL of warm ES medium. Puromycin selection was introduced 18 h later at 1 µg/mL, and the medium was changed daily. Then at 30 h, 48 h, and 72 h after puromycin selection, the cells were collected for RNA isolation.
To minimize genomic DNA contamination, RNA was extracted with RNeasy mini kit (Qiagen), and on-column DNA digestion was performed using RNase-free DNase kit (Qiagen). Microarray hybridizations were performed on the MouseRef-8 v2.0 expression beadchip arrays (Illumina). To prepare the sample, 200 ng of total RNA was reverse transcribed, followed by a T7 RNA polymerase-based linear amplification using the Illumina TotalPrep RNA Amplification kit (Applied Biosystems). After amplification, 750 ng of biotin-labeled cRNA was hybridized to gene-specific probes attached to the beads, and the expression levels of transcripts were measured simultaneously.
Software availability
Software and processed data of RA time course are available at GitHub (https://github.com/SUwonglab/TimeReg, https://github.com/SUwonglab/PECA), and as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE136312.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Lingjie Li, Shining Ma, and Zhanying Feng for helpful discussions. We also thank Miranda Lin Li for her help in the experiment. This work was supported by grants R01HG010359 and P50HG007735 from the National Institutes of Health (NIH). Y.W. was supported by The National Natural Science Foundation of China (NSFC) under grants nos. 11871463, 61671444 and the Strategic Priority Research Program of the Chinese Academy of Sciences (no. XDB13000000).
Author contributions: W.H.W. conceived the project. Z.D. designed the analytical approach, performed data analysis with the help of J.X., and wrote the software. X.C. performed all biological experiments. Y.W. and W.H.W. supervised the research. All authors wrote, revised, and contributed to the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.257063.119.
-
Freely available online through the Genome Research Open Access option.
- Received September 23, 2019.
- Accepted March 9, 2020.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








