
Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions
- Kyuha Choi1,2,
- Xiaohui Zhao1,
- Andrew J. Tock1,
- Christophe Lambing1,
- Charles J. Underwood1,3,4,
- Thomas J. Hardcastle1,
- Heïdi Serra1,
- Juhyun Kim2,
- Hyun Seob Cho2,
- Jaeil Kim2,
- Piotr A. Ziolkowski1,
- Nataliya E. Yelina1,
- Ildoo Hwang2,
- Robert A. Martienssen3 and
- Ian R. Henderson1
- 1Department of Plant Sciences, University of Cambridge, Cambridge, CB2 3EA, United Kingdom;
- 2Department of Life Sciences, Pohang University of Science and Technology, Pohang, Gyeongbuk, 37673, Republic of Korea;
- 3Howard Hughes Medical Institute–Gordon and Betty Moore Foundation, Watson School of Biological Sciences, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA
Abstract
Meiotic recombination initiates from DNA double-strand breaks (DSBs) generated by SPO11 topoisomerase-like complexes. Meiotic DSB frequency varies extensively along eukaryotic chromosomes, with hotspots controlled by chromatin and DNA sequence. To map meiotic DSBs throughout a plant genome, we purified and sequenced Arabidopsis thaliana SPO11-1-oligonucleotides. SPO11-1-oligos are elevated in gene promoters, terminators, and introns, which is driven by AT-sequence richness that excludes nucleosomes and allows SPO11-1 access. A positive relationship was observed between SPO11-1-oligos and crossovers genome-wide, although fine-scale correlations were weaker. This may reflect the influence of interhomolog polymorphism on crossover formation, downstream from DSB formation. Although H3K4me3 is enriched in proximity to SPO11-1-oligo hotspots at gene 5′ ends, H3K4me3 levels do not correlate with DSBs. Repetitive transposons are thought to be recombination silenced during meiosis, to prevent nonallelic interactions and genome instability. Unexpectedly, we found high SPO11-1-oligo levels in nucleosome-depleted Helitron/Pogo/Tc1/Mariner DNA transposons, whereas retrotransposons were coldspots. High SPO11-1-oligo transposons are enriched within gene regulatory regions and in proximity to immunity genes, suggesting a role as recombination enhancers. As transposon mobility in plant genomes is restricted by DNA methylation, we used the met1 DNA methyltransferase mutant to investigate the role of heterochromatin in SPO11-1-oligo distributions. Epigenetic activation of meiotic DSBs in proximity to centromeres and transposons occurred in met1 mutants, coincident with reduced nucleosome occupancy, gain of transcription, and H3K4me3. Together, our work reveals a complex relationship between chromatin and meiotic DSBs within A. thaliana genes and transposons, with significance for the diversity and evolution of plant genomes.
During meiosis, a single round of DNA replication is coupled to two rounds of chromosome segregation, producing haploid gametes (Villeneuve and Hillers 2001; Hunter 2015). Homologous chromosomes pair and recombine during meiotic prophase I, which can result in reciprocal crossover (Villeneuve and Hillers 2001; Hunter 2015). Meiotic recombination initiates from DNA double-strand breaks (DSBs), which can be repaired using a homologous chromosome to produce crossovers or noncrossovers (Szostak et al. 1983; Villeneuve and Hillers 2001; Kauppi et al. 2004; Baudat et al. 2013; Hunter 2015). Meiotic DSBs are generated by SPO11 topoisomerase-like transesterases, which act as dimers to cleave opposing phosphodiester backbones using catalytic tyrosine residues (Keeney and Kleckner 1995; Keeney et al. 1997; Neale et al. 2005; Pan et al. 2011). In plants, SPO11-1 and SPO11-2 interact with MEIOTIC TOPOISOMERASE VIB (MTOPVIB), which forms a conserved catalytic core complex (Grelon et al. 2001; Hartung et al. 2007; Robert et al. 2016; Vrielynck et al. 2016). Following phosphodiester cleavage, the SPO11 catalytic tyrosine remains covalently bound to the target site 5′ end (Keeney and Kleckner 1995; Neale et al. 2005; Pan et al. 2011). In budding yeast, endonucleases including Sae2 and Mre11 then generate additional DNA backbone cuts 3′ to the DSB site, which together with exonucleases cause release of SPO11-oligonucleotide complexes (Neale et al. 2005; Garcia et al. 2011; Lam and Keeney 2014). Purification and sequencing of SPO11-oligonucleotides, which are typically about 20–40 bases in length, have provided a high-resolution method to profile meiotic DSB patterns genome-wide in budding yeast, fission yeast, and mouse (Pan et al. 2011; Fowler et al. 2014; Lange et al. 2016).
Meiotic DSB and crossover frequency vary extensively along eukaryotic chromosomes and often concentrate in ∼1–2 kb hotspots (Kauppi et al. 2004; Baudat et al. 2013; Choi and Henderson 2015). Genetic and epigenetic information make varying contributions to the control of hotspot location and activity in different eukaryotic lineages (Kauppi et al. 2004; Baudat et al. 2013; Choi and Henderson 2015). For example, budding yeast DSB hotspots form predominantly in nucleosome-depleted regions in gene promoters and rarely in exons and gene terminators (Wu and Lichten 1994; Fan and Petes 1996; Pan et al. 2011; Lam and Keeney 2015). Local base composition, higher-order chromosome structure, transcription factor binding, and ATM/ATR kinase signaling have further been shown to modify DSB frequency in budding yeast (de Massy 2013; Lam and Keeney 2014; Székvölgyi et al. 2015; Cooper et al. 2016). In contrast, mouse SPO11-oligonucleotides form at specific DNA sequence motifs that are bound by the meiotic KRAB zinc finger protein PRDM9, which additionally possesses a SET domain that catalyzes histone H3K4me3 and H3K36me3 (Mihola et al. 2009; Baudat et al. 2010; Myers et al. 2010; Parvanov et al. 2010; Grey et al. 2011, 2017; Brick et al. 2012; Lange et al. 2016; Powers et al. 2016). As a consequence, PRDM9-dependent SPO11 hotspots occur in nucleosome-depleted regions with well-positioned flanking nucleosomes that acquire H3K4me3 and H3K36me3 during meiosis (Brick et al. 2012; Baker et al. 2014; Lange et al. 2016; Powers et al. 2016; Grey et al. 2017). In budding yeast, H3K4me3 is bound by the Spp1 COMPASS complex subunit, which simultaneously interacts with meiotic chromosome axis protein Mer2 and thereby tethers chromatin to axial repair sites (Borde et al. 2009; Acquaviva et al. 2013; Sommermeyer et al. 2013). Because the mouse COMPASS subunit CXXC1 interacts with both PRDM9 and the IHO1 axis protein (Imai et al. 2017), this suggests a conserved mechanism for chromatin loop-tethering during meiotic DSB repair.
Plant crossovers are enriched in euchromatin at the chromosome scale and are in proximity to gene promoters and terminators at the fine scale (Fu et al. 2002; Horton et al. 2012; Choi et al. 2013; Drouaud et al. 2013; Hellsten et al. 2013; Wijnker et al. 2013; Choulet et al. 2014; Shilo et al. 2015). Crossovers in plant genomes also show positive associations with H3K4me3, histone variant H2A.Z (Liu et al. 2009; Choi et al. 2013; Drouaud et al. 2013; Wijnker et al. 2013; Shilo et al. 2015), and A-rich and CTT/CNN-repeat DNA sequence motifs (Choi et al. 2013; Wijnker et al. 2013; Shilo et al. 2015). Furthermore, plant crossover hotspots can be directly suppressed by acquisition of heterochromatic modifications, such as DNA methylation and H3K9me2 (Yelina et al. 2015). Transposons are typically heterochromatic and show RNA polymerase II suppression caused by epigenetic modifications (Slotkin and Martienssen 2007). Repetitive sequences are also frequently crossover-suppressed during meiosis to limit nonallelic homologous recombination and genome instability (Sasaki et al. 2010). Despite this, evidence exists for specific transposon families promoting meiotic recombination in plants, fungi, and animals (Myers et al. 2005; Yandeau-Nelson et al. 2005; Shi et al. 2010; Horton et al. 2012; Sasaki et al. 2013; He et al. 2017; Yamada et al. 2017). However, genome-wide meiotic DSB patterns and their relationship to chromatin, DNA sequence, and crossover frequency have yet to be reported in a plant genome. To further explore the relationship between meiotic recombination and epigenetic information, in plant genes versus repeats, we mapped SPO11-1-oligonucleotides, crossovers, and chromatin throughout the ∼135 Mb Arabidopsis thaliana genome, which contains diverse DNA and RNA transposons.
Results
Purification and sequencing of A. thaliana SPO11-1-oligonucleotides
To map meiotic DSBs throughout the A. thaliana genome, we sought to purify and sequence SPO11-1-oligonucleotides (Grelon et al. 2001; Pan et al. 2011). We generated a 6×Myc translational fusion at the C terminus of A. thaliana SPO11-1, driven by the endogenous promoter, which fully complements spo11-1 infertility and crossover frequency, as measured using fluorescent recombination reporter lines (Fig. 1A,B; Supplemental Table S1; Grelon et al. 2001; Berchowitz and Copenhaver 2008). To analyze SPO11-1-Myc expression during the stages of meiosis, we performed immunocytology on pollen mother cells using α-Myc antibodies and coimmunostained for the ASYNAPTIC1 (ASY1) HORMA domain protein and stained chromatin with DAPI (Armstrong et al. 2002). Axis-associated SPO11-1-Myc foci showed a comparable number (mean = 204.6 foci, n = 10) and duration (leptotene until pachytene) to those reported for its partner protein MTOPVIB (Fig. 1C; Supplemental Fig. S1; Vrielynck et al. 2016). No α-Myc signal was detected above background in wild-type meiotic cells or in SPO11-1-Myc somatic cells (Fig. 1C). Therefore, SPO11-1-Myc is functional and accumulates on meiotic chromosomes, coincident with endogenous DSB formation (Sanchez-Moran et al. 2007; Vrielynck et al. 2016).
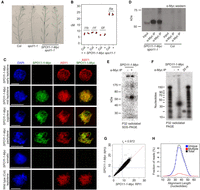
Purification and sequencing of A. thaliana SPO11-1-oligonucleotides. (A) Inflorescences from wild type (Col), spo11-1, and SPO11-1-Myc spo11-1. Greater silique length indicates higher fertility. (B) Crossover frequency (cM) measured using fluorescent crossover reporter lines (FTLs I1b, I1f, I2f, and I5a) in wild type (Col) or SPO11-1-Myc spo11-1 (+). Mean values are shown in red. (C) Nuclei from SPO11-1-Myc or wild type (Col) immunostained using α-Myc (green) or α-ASY1 (red) antibodies and stained for DAPI (blue). Scale bars, 10 μM. (D) α-Myc Western blot from SPO11-1-Myc or Col extracts, before and after α-Myc immunoprecipitation (α-Myc-IP). Biological replicate samples are shown. (E) Detection of end-radiolabeled SPO11-1-Myc complexes following immunoprecipitation and SDS polyacrylamide gel electrophoresis (SDS-PAGE). (F) Detection of end-radiolabeled, purified SPO11-1-oligos following proteinase K digestion of immunoprecipitates and polyacrylamide gel electrophoresis (PAGE). A labeled 20-base oligonucleotide (20 nt) was run alongside as a size control. (G) Scatter plot showing correlation of library size normalized SPO11-1-oligo values calculated in adjacent 10-kb windows between wild-type libraries RPI1 and RPI3. Blue dotted lines indicate genome average values. Spearman's rank correlation coefficient (rs) is printed above the plot. (H) Histogram showing lengths of uniquely aligning (blue), multiply aligning (red), and total (black) SPO11-1 reads from library RPI48.
Following protein extraction from meiotic-stage floral buds, SPO11-1-Myc was detectable as a ∼54 kDa band using Western blotting (Fig. 1D). Oligonucleotides covalently attached to SPO11-1-Myc were radioactively 3′-end labeled using terminal transferase (Neale and Keeney 2009), which revealed 60–70 kDa complexes (Fig. 1E). No signal was observed when the protocol was repeated without antibody (Fig. 1E). Following proteinase K digestion of SPO11-1-Myc immunoprecipitates and polyacrylamide gel electrophoresis (PAGE) separation, we detected radiolabeled SPO11-1-oligonucleotides approximately 30–40 bases in length (Fig. 1F). SPO11-1-oligos in this size range were gel purified and used to generate sequencing libraries, using a protocol adapted from budding yeast (Supplemental Fig. S2; Pan et al. 2011). As an additional control, we repeated SPO11-1-oligo purification and end labeling from epitope-untagged wild type (Col), SPO11-1-Myc spo11-1, and SPO11-1-Myc prd2. The prd2 mutant is disrupted in the A. thaliana MEI4 ortholog, which phenocopies spo11-1 in showing sterility and univalent chromosomes at metaphase I (De Muyt et al. 2009; Kumar et al. 2015). Following PAGE separation, we observed that SPO11-1-Myc showed ∼18-fold greater radiolabeled signal, compared with wild type or SPO11-1-Myc prd2, in the 30–40 nt size range (Supplemental Fig. S3). This demonstrates that our assay has a low level of background and that PRD2 is required for SPO11-1-oligo formation in A. thaliana.
Three biological replicate SPO11-1-Myc spo11-1 libraries were sequenced to high depth (11–28 million mapped SPO11-1-oligo reads) (Supplemental Tables S2–S4). Replicate libraries showed high correlation at multiple scales (Fig. 1G; Supplemental Fig. S4; Supplemental Table S5). For example, Spearman's rank correlation coefficients between replicate libraries using 10-kb adjacent windows were between 0.96 and 0.97 (Supplemental Table S5). We also generated sequencing libraries following α-Myc IP from untagged wild-type Col alongside SPO11-1-Myc positive controls. We observed that ∼98%–99% of the reads from the Col libraries corresponded to primer or adapter sequences and only 0.01%–0.02% of reads mapped to the A. thaliana genome, further demonstrating the low level of background in our assay (Supplemental Table S6). To serve the function of an input control, we used single-end reads from a Columbia genomic DNA (gDNA) library, trimmed to lengths of 50 bp, which were aligned and processed as for the SPO11-1-oligos. Following library size normalization, we calculated log2(SPO11-1-oligos/gDNA) values for each position in the genome. Finally, Z-score standardization was applied, such that scores represent the signed number of standard deviations from the mean.
The majority (92.2%–93.4%) of SPO11-1-oligo reads aligned uniquely (Fig. 1H; Supplemental Table S2). Multiple mapped reads showed similar alignment lengths to uniquely mapped reads; for example, mean wild-type alignment lengths were 39.1 bp for uniquely aligned reads and 39.0 bp for multiply aligned reads (Fig. 1H; Supplemental Table S3). We tested three methods for assignment of multiply mapping reads. First, “best match” mapping selected the alignment with highest MAPQ score, or if alignments had equal scores, one location was randomly chosen. Second, “fractional” mapping allocated an equal fractional value to each valid alignment. Third, CSEM uses the local density of uniquely mapping reads to assign a weighted value to each multiple mapping position (Chung et al. 2011; Lange et al. 2016). We assessed these strategies by analyzing SPO11-1-oligo frequency along chromosomes and within transposable element families and did not observe substantial differences between these methods (Supplemental Fig. S5). Therefore, for all subsequent analysis best match mapping was used.
The genomic landscape of SPO11-1-oligonucleotides, crossovers, and chromatin
To visualize SPO11-1-oligo levels throughout the genome, we analyzed adjacent 10-kb windows and plotted values along the A. thaliana chromosomes (Fig. 2A). At the chromosome scale, SPO11-1-oligos are highest in the gene-rich chromosome arms and depleted in the heterochromatic pericentromeres, which show high levels of DNA cytosine methylation (Fig. 2A; Yelina et al. 2015). To assess chromatin structure, we analyzed micrococcal nuclease sequencing data (MNase-seq) (Supplemental Table S7; Choi et al. 2016), which was normalized using paired-end reads from a Columbia genomic DNA (gDNA) library (Z-score standardized log2[MNase-seq/gDNA]). Nucleosome occupancy showed a strong negative correlation with SPO11-1-oligos in both the chromosome arms (rs = −0.70, P < 2.2 × 10−16) and pericentromeres (rs = −0.95, P < 2.2 × 10−16) (Fig. 2A,B; Supplemental Table S7; Choi et al. 2016). This is consistent with DNA accessibility, governed by nucleosome occupancy, being a major driver of A. thaliana SPO11-1-oligo formation at the chromosome scale. To compare with a euchromatic mark, we performed ChIP-seq for H3K4me3 (Z-score standardized log2[ChIP-seq/input]) (Supplemental Table S8), which correlated positively with gene density (chromosome scale rs = 0.84, P < 2.2 × 10−16) and negatively with DNA methylation (CG rs = −0.76, P < 2.2 × 10−16) and transposable element (TE) density (rs = −0.93, P < 2.2 × 10−16), as expected from previous work (Zhang et al. 2006, 2009). Interestingly, although H3K4me3 was positively correlated with SPO11-1-oligos in the pericentromeres (rs = 0.85 P < 2.2 × 10−16), it showed a negative correlation in the chromosome arms (rs = −0.82, P < 2.2 × 10−16) (Fig. 2A,B).
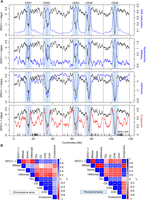
Genomic landscape of SPO11-1-oligonucleotides, crossovers, euchromatin, and heterochromatin. (A) SPO11-1-oligos (black, Z-score standardized log2[SPO11-1-oligos/gDNA]), nucleosome occupancy (blue, Z-score standardized log2[MNase/gDNA]) (Choi et al. 2016), and H3K4me3 (blue, Z-score standardized log2[ChIP/input]) levels were calculated in adjacent 10-kb windows and plotted along the A. thaliana chromosomes, using a rolling average. DNA methylation (blue, proportion of methylation in all sequence contexts) (Stroud et al. 2013) and crossovers (red) (Choi et al. 2016; Serra et al. 2018) were analyzed in the same way using 200-kb and 10-kb windows, respectively. The centromeric assembly gaps are indicated by vertical dashed lines, and telomere positions are indicated by vertical solid lines. The pericentromeres are shaded light blue and are defined as regions surrounding the centromere with greater than average DNA methylation. x-Axis ticks indicate the positions of NBS-LRR gene homologs (Choi et al. 2016). (B) Matrices showing Spearman's rank correlation coefficient between the listed parameters, with 10-kb windows used for correlations and shading proportional to the value. Matrices were calculated separately for the chromosome arms and pericentromeres.
To compare SPO11-1-oligos with crossover frequency, we used 3320 crossover events mapped in 437 Col×Ler F2 plants by genotyping-by-sequencing (Fig. 2A; Supplemental Table S9; Rowan et al. 2015; Choi et al. 2016; Serra et al. 2018). Crossovers were identified between Col/Ler single-nucleotide polymorphism (SNP) markers and mapped to a mean resolution
of 970 bp. At the chromosome scale, there was a positive correlation between SPO11-1-oligos and crossover frequency, which
was stronger in the pericentromeres (rs = 0.86, P < 2.2 × 10−16) compared with the arms (rs = 0.45, P < 2.2 × 10−16) (Fig. 2A,B). However, there was also significant variation in the ratio of SPO11-1-oligos to crossovers along the chromosome arms
(Fig. 2A). To further explore associations with crossovers, we used a generalized linear model. We considered Col/Ler SNP windows
used to detect crossovers (479,888 SNPs, mean intermarker distance = 248 bp) and calculated SPO11-1-oligo, nucleosome, H3K4me3,
and DNA methylation levels within these windows. Data were modeled using the binomial family with a logistic link function.
The formula for the final model was:

This model revealed a significant positive effect on crossovers for SPO11-1-oligos (0.176, P = 3.23 × 10−11) and negative effects for nucleosomes (−0.334, P = 1.32 × 10−10) and DNA methylation (−4.108, P < 2.0 × 10−16) (Fig. 3A; Supplemental Table S10). Therefore, crossover incidence is significantly associated with higher SPO11-1-oligos and the euchromatic signatures of lower nucleosome occupancy and reduced DNA methylation.
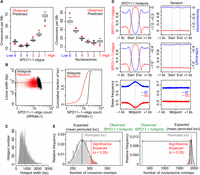
A. thaliana SPO11-1-oligonucleotide hotspots and crossovers. (A) Plots showing observed (red) overlap of SNP intervals with crossovers per megabase. SNP intervals (n = 479,888) were divided into six groups (hexiles) following ranking by mean SPO11-1-oligo levels (log2[SPO11-1-oligos/gDNA]; group 1 = highest, group 6 = lowest). Data were modeled with the glm2 function in R, using the binomial family with logistic link function and the formula: CO ∼ SPO11-1-oligos + nucleosomes + H3K4me3 + DNA methylation + width + interactions. Using this logistic model, we then obtained the probability of windows within each hexile overlapping a crossover (black box plots). (B) SPO11-1-oligo counts per hotspot (RPKM + 1) plotted against hotspot widths (bp; black) compared with equivalent analysis of random loci of the same number and widths (red). Also shown is a cumulative distribution curve plotting hotspots (black) and random loci (red), according to ranked SPO11-1-oligo counts (RPKM + 1). (C) Histogram of ranger-defined SPO11-1-oligo hotspot widths (bp), with the mean indicated by the red vertical dashed line. (D) Average profiles of SPO11-1-oligos (red, Z-score standardized log2[SPO11-1-oligos/gDNA]), nucleosomes (blue, Z-score standardized log2[MNase/gDNA]), and H3K4me3 (blue, Z-score standardized log2[ChIP/Input]) within SPO11-1-oligo hotspots, scaled to a common width, and in 1-kb flanking windows (left). Equivalent randomly positioned loci were analyzed in the same way (right). Also shown are plots analyzing the relative frequency of AT (blue) and GC (red) bases around SPO11-1-oligo hotspots or random loci. (E) Histograms show the relative frequency distribution of 10,000 sets of permuted (randomly positioned) loci, which are of the same number and widths as the SPO11-1-oligo hotspots, with regard to the number of permuted loci that overlap one or more crossovers (left) or highly positioned nucleosomes (right; gray bars). The number of SPO11-1-oligo hotspots observed (green vertical line) or expected (black vertical line; mean overlaps for permuted loci) to overlap one or more loci, together with the significance threshold (red vertical line; α = 0.05) for a difference between observed and expected overlaps, are indicated.
Previous work defined crossover hotspot regions in the A. thaliana genome (Yelina et al. 2012, 2015; Choi et al. 2013, 2016; Drouaud et al. 2013). We observed that pollen-typing hotspots 3a, 3b, RAC1, I4a, and 130x, for which we have highest resolution crossover data, show higher than average SPO11-1-oligo levels. Furthermore, a permutation test using 10,000 sets of five randomly positioned loci of equivalent widths found observed mean SPO11-1-oligo levels to be higher than expected, although not significantly, at these crossover hotspots (observed mean coverage = 0.142, expected [permuted] mean coverage = 0.001, P = 0.1441). However, variation in SPO11-1-oligo and cM/Mb values within the hotspots were not significantly correlated, except at 3a and RAC1, where weak negative correlations were observed (3a rs = −0.54, P = 9.93 × 10−3 and RAC1 rs = −0.23, P = 5.37 × 10−5) (Supplemental Figs. S7–S18). Therefore, despite a positive relationship genome-wide, there is substantial variation in the ratio of SPO11-1-oligos:crossovers within known crossover hotspots. This indicates that factors, including interhomolog sequence divergence and other epigenetic features, influence patterns of crossover designation at the fine scale, in addition to DSB levels.
SPO11-1-oligo hotspots in nucleosome-depleted gene regulatory regions
To identify SPO11-1-oligo hotspots, we used the ranger tool within the PeakRanger suite, which identified regions of enrichment and summits in each wild-type replicate library, with the gDNA reads provided as a background control (Feng et al. 2011). Peaks were ranked by their −log10-transformed false discovery rate (FDR), and this ranking was compared between wild-type replicates RPI1 and RPI8 to give an irreproducible discovery rate (IDR). SPO11-1-oligo peaks with IDR <0.05 were retained as high-quality hotspots, and any overlapping peaks were merged. This approach identified 5914 hotspots genome-wide with a mean width of 823 bp (Fig. 3B–D). We did not observe a relationship between hotspot width and activity (RPKM + 1) (Fig. 3B). Consistent with analysis of SPO11 hotspots in mouse and budding yeast, rank ordering of A. thaliana hotspots by activity produced a smooth continuum (Fig. 3B; Pan et al. 2011; Lange et al. 2016). Our hotspots show strong SPO11-1-oligo enrichment that was significantly correlated with depletion of both nucleosomes (rs = −0.95, P < 2.2 × 10−16) and H3K4me3 (rs = −0.91, P < 2.2 × 10−16) (Fig. 3D). SPO11-1-oligo hotspots also show enrichment of AT over GC base frequency, consistent with AT-richness excluding nucleosomes (Fig. 3D; Segal and Widom 2009). We also tested overlap of SPO11-1-oligo hotspots with the GBS crossovers (n = 3320) by performing permutation tests using the regioneR package (Supplemental Table S11; Gel et al. 2016). Here 10,000 sets of 5914 randomly positioned loci with the same widths as the SPO11-1-oligo hotspots were evaluated for overlap with crossovers and other genome features. This identified significantly greater than expected overlap between SPO11-1-oligo hotspots and both crossovers (P < 0.0001) and gene promoters (P < 0.0001) (Fig. 3E; Supplemental Table S11). In contrast, significantly fewer than expected SPO11-1-oligo hotspots overlap highly positioned nucleosomes (P < 0.0001) and gene open reading frames (TSS–TTS) (P < 0.0001) (Fig. 3E; Supplemental Table S11).
Given the significant overlap of SPO11-1-oligo hotspots with gene promoters, we analyzed SPO11-1-oligo density in relation to chromatin around gene transcriptional start sites (TSSs) and termination sites (TTSs) (Fig. 4A,B). Similar to budding yeast, SPO11-1-oligos were highly elevated in A. thaliana nucleosome-free regions in gene promoters (Fig. 4A–C). Interestingly, we also observed strong SPO11-1-oligo enrichment in nucleosome-depleted transcriptional terminators, where plant crossovers are elevated (Fig. 4A–C; Choi et al. 2013; Wijnker et al. 2013). A further difference is that A. thaliana genes possess on average 6.7 exons (Cheng et al. 2017), whereas budding yeast genes lack introns (Pan et al. 2011). We observe that A. thaliana introns have higher SPO11-1-oligos and lower nucleosomes compared with exons (Supplemental Fig. S19). However, SPO11-1-oligos are overall suppressed within relatively nucleosome-dense gene bodies, compared with flanking nucleosome-depleted promoter and terminator regions (Fig. 4A–C; Supplemental Table S11). H3K4me3 shows prominent enrichment at the +1-nucleosome position, immediately downstream from TSSs, in proximity to the region of gene promoter SPO11-1-oligo enrichment (Fig. 4B; Zhang et al. 2009).
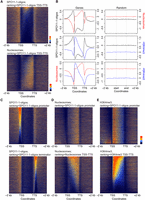
SPO11-1-oligonucleotide enrichment in gene promoter and terminator nucleosome-free regions. (A) Heat maps of SPO11-1-oligos (upper) and nucleosome occupancy (lower) within gene transcriptional units (between transcriptional start [TSS] and termination [TTS] sites) and 2-kb flanking regions. The rows represent individual genes, which have been ordered by descending SPO11-1-oligo values between TSS and TTS. For shading, SPO11-1-oligo and nucleosome values equal to defined quantiles were mapped linearly to a vector of six colors: dark blue (lowest), blue, light blue, yellow, orange, red (highest). (B) Density of SPO11-1-oligos (black; log2[SPO11-1-oligos/gDNA]), nucleosome occupancy (red, nucleosomes; log2[MNase/gDNA]), and H3K4me3 (blue; log2[ChIP/input]) in wild type, across gene transcriptional units (TSS to TTS), and in flanking 2-kb windows. The right-hand column shows results from analysis of the same number of randomly chosen positions. (C) Heat maps as for A, but showing SPO11-1-oligos around genes ordered by descending SPO11-1-oligo levels in gene promoters (upper, 500 bp upstream of TSS) or gene terminators (lower, 500 bp downstream from TTS). (D) Heat maps as for A, but showing nucleosomes around genes ordered by descending SPO11-1-oligo levels in gene promoters (upper), or by descending nucleosome levels within TSS–TTS (lower). (E) Heat maps as for A, but showing H3K4me3 around genes ordered by descending SPO11-1-oligo levels in gene promoters (upper) or by descending H3K4me3 levels within TSS–TTS (lower).
To investigate variation in DSB levels, we ranked genes according to SPO11-1-oligos in 500-bp windows upstream of gene TSSs (promoters) or downstream from TTSs (terminators) (Fig. 4C). Promoter SPO11-1-oligos did not strongly associate with terminator levels, showing that meiotic recombination initiation varies independently at opposite ends of A. thaliana genes (Fig. 4C). We used the SPO11-1-oligo promoter ranking to look at associated variation in nucleosome occupancy and H3K4me3 levels. High SPO11-1-oligo levels strongly associate with lower nucleosome occupancy at gene promoters, further consistent with DNA accessibility being a major determinant of A. thaliana SPO11-1-oligo levels (Fig. 4D). In contrast, H3K4me3 levels within genes did not show a strong association with promoter SPO11-1-oligo levels (Fig. 4E), consistent with analysis of mouse and budding yeast SPO11-oligonucleotides (Tischfield and Keeney 2012; Lange et al. 2016).
SPO11-1-oligo hotspots and coldspots in transposable elements
To explore SPO11-1-oligo levels within repetitive sequences, we selected 29,150 transposable elements from 10 DNA and RNA families for analysis (Supplemental Table S12; Buisine et al. 2008). Extensive SPO11-1-oligo variation was observed between transposon families, with high levels in Helitrons, which transpose via rolling-circle replication, and Pogo/Tc1/Mariner and MuDR “cut-and-paste” DNA transposons (Fig. 5A; Supplemental Table S12; Kapitonov and Jurka 2001; Slotkin and Martienssen 2007). In contrast, retrotransposons that replicate via RNA intermediates, including LTR and non-LTR families, were SPO11-1-oligo coldspots (Fig. 5A; Supplemental Table S12; Beauregard et al. 2008). As observed for genes (Fig. 4), variation in transposon family SPO11-1-oligos negatively correlated with nucleosome occupancy (r = −0.972, P = 2.57 × 10−6) (Fig. 5B; Supplemental Table S12). We divided transposons into six groups (hexiles) after ranking by within-element SPO11-1-oligos (hexile 1 = highest, hexile 6 = lowest) (Fig. 5C). This grouping showed strong correlations between higher SPO11-1-oligos and reduced transposon lengths (r = −0.935, P = 0.006) and lower nucleosome occupancy (r = −0.98, P = 6.0 × 10−4) (Fig. 5C; Supplemental Table S13). Further consistent with this, we observed that the SPO11-1-oligo hotspots defined earlier significantly overlapped DNA transposons, including Helitrons, Pogo/Tc1/Mariner, and MuDR (all P < 0.0001), whereas RNA transposons, including Gypsy and Copia LTR and LINE-L1 retroelements, overlapped significantly less than expected by chance (all P < 0.0001) (Supplemental Fig. S6; Supplemental Table S11).
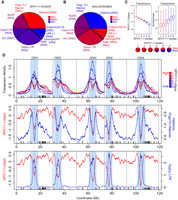
Meiotic recombination and chromatin variation between A. thaliana transposons. (A) Pie chart showing A. thaliana transposon families, with slice size proportional to physical length, and color-coded according to SPO11-1-oligo (Z-score standardized log2[SPO11-1-oligos/gDNA]) levels. Inset colored boxes are shaded equivalent to the genome-wide mean (purple), minimum (blue), and maximum (red) values. (B) As for A, but analyzing nucleosome occupancy (Z-score standardized log2[MNase/gDNA]). (C) Box plots showing SPO11-1-oligo and nucleosome occupancy, according to transposon SPO11-1-oligo hexile groups, with horizontal lines indicating the genome average value. Inset pie charts show the proportion of DNA (red) and RNA (blue) transposons for each SPO11-1-oligo transposon hexile. (D) Density of transposons through the A. thaliana genome according to SPO11-1-oligo hexile: (red) highest; (blue) lowest. x-Axis ticks indicate NBS-LRR gene homolog positions. Plotted beneath are SPO11-1-oligos (red) versus the density of Helitron/Pogo/Tc1/Mariner class DNA transposons (blue), or Gypsy RNA transposons (blue).
At the chromosome scale, high SPO11-1-oligo transposons (e.g., Helitrons and Pogo/Tc1/Mariner) show elevated density in pericentromeric regions and proximal regions of the chromosome arms, whereas low SPO11-1-oligo transposons (e.g., Gypsy LTR) are enriched in proximity to the centromeres (Fig. 5D). Differences in SPO11-1-oligos between transposon families are evident locally; for example, a nucleosome-dense retroelement coldspot ATCOPIA4 shows higher SPO11-1-oligo levels compared with an adjacent cluster of nucleosome-depleted Helitron hotspots (Supplemental Fig. S20). Many SPO11-1-oligo hotspot DNA transposons are short, nonautonomous fragments, although high SPO11-1-oligo levels were also observed within full-length Helitron and Lemi1 Pogo transposons (Supplemental Figs. S21–S25; Feschotte and Mouchès 2000; Kapitonov and Jurka 2001). Hence, despite the expectation that transposons would be suppressed for meiotic DSBs, we observe that nucleosome-depleted repetitive elements contain abundant SPO11-1-oligo hotspots.
Nucleosomes, DNA sequence, and SPO11-1-oligos within genes and transposons
To further investigate spatial relationships between SPO11-1-oligos, chromatin, and DNA sequence, we analyzed 4-kb windows around gene TSSs and TTSs or transposon start and end coordinates, each according to SPO11-1-oligo hexile groups, and compared them to the same number of random positions (Fig. 6A,B; Supplemental Tables S13–S15). In both genes and transposons, a strong negative relationship between SPO11-1-oligos and nucleosome occupancy was observed (Fig. 6A,B; Supplemental Tables S13–S15). We observed quantitative enrichment of AT-rich sequence motifs in high SPO11-1-oligo regions, which have previously been associated with high crossovers (Fig. 6A,B; Supplemental Tables S14, S15; Horton et al. 2012; Choi et al. 2013; Wijnker et al. 2013; Shilo et al. 2015). We propose that these AT-rich motifs exclude nucleosomes, cause higher SPO11-1 accessibility and lead to higher levels of DSB formation and crossover frequency (Fig. 6A,B; Segal and Widom 2009).
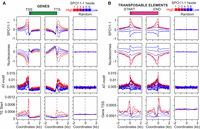
Nucleosomes, AT-sequence motifs, and SPO11-1-oligonucleotides within genes and transposons. (A) Density of SPO11-1-oligos (Z-score standardized log2[SPO11-1-oligos/gDNA]), nucleosome occupancy (Z-score standardized log2[MNase/gDNA]), crossover-enriched AT-motifs (Choi et al. 2013), and TE start coordinates, in 4-kb windows around gene TSSs or TTSs, or the same number of random positions. Genes are grouped according to SPO11-1-oligo promoter or terminator hexile: (red) highest; (blue) lowest. (B) As for A but analyzing transposon SPO11-1-oligo hexiles and showing gene TSS density.
We also note that high SPO11-1-oligo genes and transposons occur in close proximity to one another (Fig. 6A,B). Helitron transposons are known to insert into AT dinucleotides (Kapitonov and Jurka 2001), and Lemi1 Pogo transposons insert into TA dinucleotides (Guermonprez et al. 2008). Therefore, transposon integration site preference likely contributes to DNA element enrichment in AT-rich gene promoters and terminators, where they further contribute to nucleosome exclusion and high meiotic DSB levels (Fig. 6A,B). High recombination rates may also provide an explanation for the tendency of SPO11-1-oligo hotspot transposons to be shorter (Supplemental Tables S12, S13) due to promotion of nonhomologous recombination and sequence rearrangements (Sasaki et al. 2010). Together, these findings reveal intimate connections between DNA sequence, chromatin, and recombination within and around A. thaliana genes and transposons.
SPO11-1-oligo hotspot transposons are enriched in proximity to immunity genes
To investigate genes associated with high DSB levels, we tested for enrichment of Gene Ontology (GO) terms, following ranking by promoter SPO11-1-oligo levels (Fig. 4C). This revealed a strong association with biotic defense GO terms (Supplemental Table S16), which was driven by high-recombination DEFENSIN genes (Supplemental Figs. S26, S27). Defensins are small cysteine-rich peptides with roles in antimicrobial defense and pollen–pistil interactions (Silverstein et al. 2005). Further association of recombination hotspots and immunity genes is evident at the chromosome scale, where high SPO11-1-oligo transposons show elevated density within the nucleotide-binding site–leucine rich repeat (NBS-LRR) immune gene clusters on the right arms of Chromosomes 1 and 5 (Fig. 5D; Choi et al. 2016). NBS-LRR genes encode immune receptors in plants, which are related to the NLR superfamily of proteins that recognize pathogens in animals and fungi (Jones et al. 2016). Plant NBS-LRR genes are among the most diverse gene class observed within and between plant species (Jacob et al. 2013; Jones et al. 2016). In A. thaliana, 73 of 197 NBS-LRR genes are within 500 bp of a SPO11-1-oligo active transposon (Supplemental Table S17). For example, the NBS-LRR crossover hotspots RAC1 and HRG1 are flanked by Helitron and MuDR high-recombination transposons, respectively (Supplemental Figs. S10, S11; Supplemental Table S17; Choi et al. 2016). Further examples include the RPP4 and WRR4 oomycete resistance genes, which contain strong ATREP Helitron SPO11-1-oligo hotspots within their introns (Supplemental Figs. S18, S28; Supplemental Table S17; Van der Biezen et al. 2002; Borhan et al. 2008). Because A. thaliana NBS-LRR genes are sites of natural structural diversity and DNA methylation polymorphism in populations (Kawakatsu et al. 2016; Quadrana et al. 2016; Stuart et al. 2016), we propose that gene-proximal DNA transposons may act as meiotic recombination enhancers, contributing to the high levels of genetic and epigenetic variation observed at these loci.
Epigenetic remodeling of SPO11-1-oligos, chromatin and transcription in met1 DNA methylation mutants
Heterochromatic marks, including DNA methylation, play critical roles in transcriptionally silencing transposable elements and thereby limiting their proliferation within eukaryotic genomes (Slotkin and Martienssen 2007). To directly investigate the role of DNA methylation in recombination, chromatin, and transcription, we compared SPO11-1-oligos, nucleosomes, H3K4me3, and RNA expression genome-wide in wild-type and met1 mutants. MET1 encodes the major CG maintenance DNA methyltransferase in A. thaliana (Kankel et al. 2003; Saze et al. 2003; Stroud et al. 2013). Because met1 mutants show cytological decondensation of heterochromatin and elevated transposon transcription and mobility (Kato et al. 2003; Saze et al. 2003; Mathieu et al. 2007), we sought to test whether related changes in heterochromatic SPO11-1-oligos would occur. For all experiments, we used the null met1-3 allele, which was isolated in a Col background (Saze et al. 2003).
At the chromosome scale, met1 shows a pronounced loss of CG DNA methylation within the pericentromeres (Fig. 7A,B; Stroud et al. 2013). We observe mirrored increases in pericentromeric SPO11-1-oligos in met1 versus wild type, which we analyzed as a differential (ΔSPO11-1-oligos = met1 − wild type) (Fig. 7C,D). The pericentromeres also show reduced nucleosome occupancy in met1, consistent with loss of heterochromatic structure (Fig. 7C,D). The met1 nucleosome differential is highly positively correlated with the CG methylation differential (r = 0.960, P < 2.2 × 10−16) and negatively with the SPO11-1-oligo differential (r = −0.836, P < 2.2 × 10−16) (Fig. 7), all of which show greatest change in the pericentromeres (Fig. 7). H3K4me3 shows a weakly increased pericentromeric signal in met1, which correlates with gain of this modification on a subset of transposons (Fig. 7C,D; Supplemental Figs. S28–S30). The met1 H3K4me3 differential correlated negatively with the CG methylation (r = −0.57, P < 2.2 × 10−16) and nucleosome (r = −0.55, P < 2.2 × 10−16) differentials, and positively with the SPO11-1-oligo differential (r = 0.55, P < 2.2 × 10−16) (Fig. 7). We note that these approaches are not able to quantify total levels of SPO11-1-oligos or chromatin between genotypes. However, as loss of CG methylation in met1 pericentromeres is highly correlated with decreased nucleosome occupancy and gain of SPO11-1-oligos, we propose that this reflects the primary change in the met1 chromatin and recombination landscapes at the chromosome scale (Fig. 7A–D).
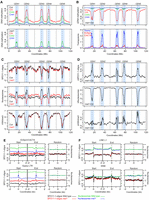
Coordinate epigenetic remodeling of chromatin and SPO11-1-oligonucleotides in met1 DNA methylation mutants. (A) Percent DNA methylation in CG (red), CHG (green), and CHH (blue) sequence contexts plotted from wild type (Col) and met1-3 (Stroud et al. 2013). Values were calculated in adjacent 10-kb windows and plotted along the A. thaliana chromosomes, using a rolling average. Centromeric assembly gaps are indicated by vertical dashed lines, and telomere positions are indicated by vertical solid lines. The pericentromeres are shaded light blue, which are defined as regions with greater than average DNA methylation. (B) The wild type (Col) versus met1 DNA methylation differential (Δ, met1 − wild type), plotted as in A (upper). The density of Gypsy (blue) and EnSpm/CACTA (red) transposons are plotted as in A (lower). (C) SPO11-1-oligos (Z-score standardized log2[SPO11-1-oligos/gDNA]), nucleosome occupancy (Z-score standardized log2[MNase/gDNA], blue), and H3K4me3 (Z-score standardized log2[ChIP/Input], blue) plotted as for A, in wild type (Col, black) and met1-3 (red). (D) The wild type (Col) versus met1 SPO11-1-oligo, nucleosome occupancy and H3K4me3 differentials (Δ, met1 − wild type), plotted as in A (upper). (E) Plots analyzing SPO11-1-oligos in wild type (black) and met1 (red), and nucleosomes in wild type (green) and met1 (blue) for EnSpm/CACTA and Gypsy transposons in 4-kb windows around their start and end coordinates, or the same number of randomly chosen positions. (F) As for E, but analyzing LINE L1 and SINE transposons.
The met1 SPO11-1-oligo differential is strongly positively correlated with the density of pericentromere-enriched Gypsy (r = 0.839, P < 2.2 × 10−16) and EnSpm/CACTA transposons (r = −0.793, P < 2.2 × 10−16) (Fig. 7B–E), which are SPO11-1-oligo coldspots in wild type (Fig. 5A). Indeed, analysis of SPO11-1-oligos and nucleosomes over these transposable elements showed higher accessibility and recombination initiation in met1 (Fig. 7E). These trends are also evident at individual transposable elements (Supplemental Figs. S29–S31). For example, ATENSPM9 and ATENSPM10 EnSpm/CACTA and ATGP3 Gypsy transposons show activation of transcription and SPO11-1-oligos in met1, in addition to showing reduced nucleosome occupancy and gain of H3K4me3 (Supplemental Figs. S29–S31). In contrast, LINE and SINE retroelements showed unchanged nucleosome occupancy and levels of SPO11-1-oligos in met1 (Fig. 7F). Together, this demonstrates coordinate epigenetic remodeling of chromatin, histone modifications, transcription, and meiotic recombination initiation, caused by loss of CG DNA methylation in met1 pericentromeres and specific transposable elements.
Discussion
Despite deep conservation of core meiotic factors, such as SPO11, many aspects of genome architecture, chromatin, and recombination vary between eukaryotes. A. thaliana hotspots are most similar to those observed at budding yeast promoters, which are driven by nucleosome occupancy (Wu and Lichten 1994; Fan and Petes 1996; Pan et al. 2011; Lam and Keeney 2015). We observed a strong influence of AT-sequence richness on A. thaliana SPO11-1-oligo levels. Because AT-richness excludes nucleosomes (Segal and Widom 2009), we propose that these motifs allow increased SPO11-1 access to DNA, and this underlies their association with elevated crossover frequency (Choi et al. 2013; Wijnker et al. 2013; Shilo et al. 2015). In contrast to budding yeast, A. thaliana also shows elevated SPO11-1-oligos within nucleosome-depleted gene terminators and introns, indicating that varying gene architecture can influence meiotic DSB patterns between species. Interestingly, avian crossover hotspots are also observed at both gene promoters and terminators (Singhal et al. 2015), meaning that meiotic recombination hotspots at gene 3′ ends may be widely conserved. Consistent with analysis of budding yeast and mouse SPO11-oligos, we did not observe a positive relationship between H3K4me3 and A. thaliana SPO11-1-oligo levels at either the chromosome or fine scale (Tischfield and Keeney 2012; Lange et al. 2016; Yamada et al. 2017). However, H3K4me3 may promote crossovers downstream from DSB formation, via tethering repair sites to the chromosome axis, as in budding yeast and mammals (Borde et al. 2009; Acquaviva et al. 2013; Sommermeyer et al. 2013; Imai et al. 2017). It is also important to note that in comparison to budding yeast and mouse, A. thaliana SPO11-1 appears to be more promiscuous, which may reflect a difference in SPO11-1 activity or the chromatin substrate.
Although a positive genome-wide association is observed between SPO11-1-oligos and crossovers, weaker correlations exist at the fine scale within defined crossover hotspots (Choi et al. 2013, 2016; Drouaud et al. 2013; Yelina et al. 2015). One contributor to this may be interhomolog polymorphism, which can modify crossover maturation downstream from DSB formation. For example, at the mouse A3 and human NID1 hotspots, local suppression of crossovers occurs in proximity to indel and minisatellite structural polymorphisms, despite the presence of gene conversions (and therefore DSBs) (Jeffreys and Neumann 2005; Cole et al. 2010). Furthermore, data in budding yeast indicate that interhomolog joint molecules may be mobile (Allers and Lichten 2001), and repeated rounds of strand invasion and dissolution may occur during repair (Oh et al. 2008; De Muyt et al. 2012), which could cause differences in the locations of the initiating DSB and final crossover resolution. Finally, crossover:noncrossover ratios are known to vary widely between mammalian hotspots, and in maize evidence for initiation of meiotic recombination exists along the chromosomes despite crossovers being restricted to distal subtelomeric regions (Rodgers-Melnick et al. 2016; He et al. 2017). Therefore, although meiotic DSB frequency is related to crossover frequency in plants, other genetic and epigenetic features may govern crossover designation or direct alternative modes of repair.
Although budding yeast and A. thaliana gene promoter DSB hotspots are similar, their genomes differ in other respects. For example, A. thaliana possesses complex megabase-size centromeres, extensive pericentromeric heterochromatin, and a larger and more diverse transposon complement (Copenhaver et al. 1999; Zhang et al. 2006; Buisine et al. 2008; Lister et al. 2008; Quadrana et al. 2016; Stuart et al. 2016). In contrast, budding yeast strains S288C and SK1 contain 50 and 30 Ty elements, respectively, and possesses point centromeres consisting of a single nucleosome (Sasaki et al. 2013; Vincenten et al. 2015). Using the A. thaliana met1 mutant, we showed that loss of CG DNA methylation causes coordinated loss of nucleosome occupancy, gain of euchromatic marks (H3K4me3), transcription, and SPO11-1-oligos within A. thaliana pericentromeres and in specific EnSpm/CACTA and Gypsy transposons. This is reminiscent of increased SPO11-DSBs detected in retrotransposons in mouse dnmt3l DNA methylation mutants (Zamudio et al. 2015), indicating that epigenetic repression of meiotic recombination in transposons is conserved between plants and mammals. Interestingly, although met1 shows increased pericentromeric SPO11-1-dependent DSBs, CG methylation mutants show decreased pericentromeric crossovers (Colomé-Tatché et al. 2012; Melamed-Bessudo and Levy 2012; Mirouze et al. 2012; Yelina et al. 2012, 2015). In contrast, mutants in the non-CG DNA methylation/H3K9me2 pathway show both increased pericentromeric SPO11-1-oligos and crossovers (Underwood et al. 2018). This indicates that although both CG and non-CG DNA methylation inhibit pericentromeric meiotic DSBs, only non-CG methylation and/or H3K9me2 inhibit crossovers (Underwood et al. 2018).
Despite the expectation that transposons would be recombination suppressed in order to avoid genome instability (Sasaki et al. 2010), we observed that a subset of A. thaliana DNA transposons (Helitron, Pogo/Tc1/Mariner) contain strong SPO11-1-oligo hotspots. These DNA transposons are AT-rich and nucleosome-depleted in wild type and frequently occur in proximity to genes. Because Helitron and Pogo/Tc1/Mariner transposons display TA and AT dinucleotide insertion site preferences (Kapitonov and Jurka 2001; Guermonprez et al. 2008), this likely contributes to their enrichment in AT-rich gene regulatory regions, where they may further contribute to nucleosome exclusion and enhanced SPO11-1-oligo levels. Higher meiotic recombination initiation may also contribute to hotspot transposons tending to occur as shorter, nonautonomous fragments. For example, insertions, deletions, and rearrangements can result from nonallelic recombination between repeated loci (Sasaki et al. 2010). Our data revealed unexpected diversity in the chromatin and recombination landscapes between A. thaliana transposable element families. Because RNA and DNA transposons vary in abundance and chromosomal distribution in plant genomes (Kapitonov and Jurka 2001; Buisine et al. 2008; Guermonprez et al. 2008; Liu et al. 2009; Choulet et al. 2014; Quadrana et al. 2016; Stuart et al. 2016), repetitive elements may directly contribute to the variation in meiotic recombination observed between species (Henderson 2012; Higgins et al. 2014).
A subset of budding yeast Ty retrotransposons contain SPO11-oligo hotspots, which in some cases correspond to nucleosome-free regions (Sasaki et al. 2013). Repetitive sequences overall are suppressed for mouse SPO11-oligos, but specific families of DNA transposons (MULE-MuDR, TcMar, hAT, PiggyBac) contain functional PRDM9 binding sites and DSB hotspots (Yamada et al. 2017). Indeed, it is notable that PRDM9 belongs to a large and rapidly evolving KRAB zinc-finger gene family, members of which play major roles in transcriptional silencing of transposable elements and gene regulatory networks (Imbeault et al. 2017; Yamada et al. 2017). This implies an evolutionary connection between control of repetitive elements and recombination hotspots in mammals. Our SPO11-1-oligo data are consistent with previous observations of elevated meiotic recombination in specific families of plant transposable elements (Yandeau-Nelson et al. 2005; Shi et al. 2010; Horton et al. 2012; He et al. 2017). Therefore, findings from across eukaryotes show that transposable elements can shape patterns of meiotic recombination, with the potential to influence genetic diversity and adaptation in their host genomes.
Methods
Generation of A. thaliana SPO11-1-Myc spo11-1 lines
6×c-Myc epitopes were translationally fused to a genomic clone of SPO11-1 in the pPZP211 binary vector (Supplemental Methods), which was transformed into wild-type A. thaliana (Col-0) using Agrobacterium tumefaciens strain GV3101, via floral dipping. SPO11-1-Myc transformants were crossed with spo11-1-3 (SALK_146172) heterozygotes to perform complementation tests of fertility (Hartung et al. 2007).
Crossover measurements using fluorescent seed and pollen
Crossover measurements using fluorescent seed or pollen were carried out as described (Yelina et al. 2013; Ziolkowski et al. 2015).
Immunocytological analysis
Chromosome spreads of A. thaliana pollen mother cells and immunostaining of ASY1 and SPO11-1-Myc were performed using fresh buds, as described (Armstrong et al. 2002). The following antibodies were used: α-ASY1 (rabbit, 1/500 dilution) (Armstrong et al. 2002) and α-Myc (mouse, 1/50 dilution) (9E10, Santa Cruz Biotechnology). Microscopy was conducted using a DeltaVision Personal DV microscope (Applied Precision/GE Healthcare) equipped with a CDD CoolSNAP HQ2 camera (Photometrics). Image capture and analysis were performed using softWoRx software version 5.5 (Applied precision/GE Healthcare).
Purification and sequencing of SPO11-1-oligonucleotides
Unopened flower buds were collected and used to extract SPO11-1 and perform immunoprecipitation (Supplemental Methods). SPO11-1-Myc immunoprecipitates were used for radioactive end-labeling and SDS-PAGE analysis, or proteinase digestion, followed by purification and sequencing of the oligonucleotides (Supplemental Methods; Supplemental Table S18).
Micrococcal nuclease (MNase) digestion and chromatin immunoprecipitation
ChIP experiments were performed following standard protocols (Supplemental Methods). H3K4me3 antibodies (AbCam ab8580) were used to perform ChIP on chromatin extracts from unopened flower buds. MNase experiments were performed as described (Choi et al. 2016). A detailed description of all bioinformatics analysis is provided in Supplemental Methods. Statistical analyses were performed using the R language (R Core Team 2016).
Data access
All genomic data from this study have been submitted to the ArrayExpress repository (https://www.ebi.ac.uk/arrayexpress/experiments/), hosted by the European Bioinformatics Institute (EBI), European Molecular Biology Laboratory, under accession numbers E-MTAB-5041 (SPO11-1-oligonucleotides), E-MTAB-5042 (MNase-seq), E-MTAB-5048 (H3K4me3 ChIP-seq), E-MTAB-5417 (RNA-seq), E-MTAB-4657, E-MTAB-5476, and E-MTAB-6577 (GBS), and E-MTAB-6257 (control libraries).
Acknowledgments
We thank Gregory Copenhaver (University of North Carolina, Chapel Hill) for FTL lines and Scott Keeney (Memorial Sloan Kettering Cancer Center) for helpful discussions. Research was supported by a Royal Society University Research Fellowship, the Gatsby Charitable Foundation grant GAT2962, BBSRC grant BB/N007557/1, BBSRC grant BB/L006847/1, BBSRC-Meiogenix IPA grant BB/N007557/1, ERA-CAPS/BBSRC “DeCOP” grant BB/M004937/1, ERC “SynthHotSpot” Consolidator Grant, National Natural Science Foundation of China grant 61403318, RDA Next-Generation BioGreen 21 Program PJ01137901, NRF Basic Science Research Program NRF-2017R1D1AB03028374, POSTECH Basic Research Institute and an EMBO long-term postdoctoral fellowship ALT 807-2009.
Author contributions: K.C., C.L., C.J.U., H.S., P.A.Z., N.E.Y., R.A.M., and I.R.H. contributed to design of the study. K.C., C.L., C.J.U., Ju.K., H.S.C., Ja.K., and H.S. performed experiments. K.C., X.Z., A.J.T., C.L., C.J.U., T.J.H., H.S., I.H., R.A.M., and I.R.H. analyzed the data. K.C., X.Z., A.J.T., C.L. C.J.U., T.J.H., H.S., P.A.Z., N.E.Y., R.A.M., and I.R.H. wrote the manuscript.
Footnotes
-
↵4Present address: Vegetable Crop Research, KeyGene, 6708 PW Wageningen, The Netherlands
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.225599.117.
- Received May 26, 2017.
- Accepted February 8, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








