
Numerical and spatial patterning of yeast meiotic DNA breaks by Tel1
- Howard Hughes Medical Institute, Molecular Biology Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA
- Corresponding author: s-keeney{at}ski.mskcc.org
Abstract
The Spo11-generated double-strand breaks (DSBs) that initiate meiotic recombination are dangerous lesions that can disrupt genome integrity, so meiotic cells regulate their number, timing, and distribution. Mechanisms of this regulation remain poorly understood. Here, we use Spo11-oligonucleotide complexes, a byproduct of DSB formation, to reveal aspects of the contribution of the Saccharomyces cerevisiae DNA damage-responsive kinase Tel1 (ortholog of mammalian ATM). A tel1Δ mutant has globally increased amounts of Spo11-oligonucleotide complexes and altered Spo11-oligonucleotide lengths, consistent with conserved roles for Tel1 in control of DSB number and processing. A kinase-dead tel1 mutation similarly increases Spo11-oligonucleotide levels but mutating known Tel1 phosphotargets on Hop1 and Rec114 does not, implicating Tel1 kinase activity and clarifying roles of Tel1 phosphorylation substrates. Deep sequencing of Spo11 oligonucleotides demonstrates that Tel1 shapes the genome-wide DSB landscape in unexpected ways. Early in meiosis, Tel1 absence causes widespread changes in DSB distributions across large chromosomal domains. Many of these changes are erased as meiosis proceeds, however, illustrating homeostatic behavior of DSB regulatory systems. We further find that effects of Tel1 are distinct but partially overlapping with previously described contributions of the recombination regulator Cst9 (also known as Zip3). Finally, we provide evidence indicating that Tel1-dependent DSB interference influences the population-average DSB landscape but also demonstrate that locally inhibitory effects of an artificial hotspot insertion can be both Tel1-independent and chromosomal context-dependent. Our findings delineate Tel1 roles in regulating number and location of DSBs and illuminate the complex interplay between Tel1 and other pathways for DSB control.
To balance the positive (chromosome segregation promoting) and negative (mutagenic) roles of meiotic DSBs, cells possess overlapping, homeostatic pathways that control the number and repair of DSBs and, in doing so, shape the recombination distribution (Keeney et al. 2014; Cooper et al. 2016). The DSB “landscape,” i.e., the distribution of DSBs across the genome, is defined by complex hierarchical influences operating over different size scales (Pan et al. 2011; Lam and Keeney 2015a; Cooper et al. 2016). Some genomic regions, encompassing tens to hundreds of kb, experience more or less DSB formation than others, and within such DSB-“hot” and DSB-“cold” domains, DSBs are particularly enriched in small (typically <250 bp) regions known as “hotspots.” In Saccharomyces cerevisiae, most hotspots correspond to evolutionarily conserved nucleosome-depleted promoters (Lam and Keeney 2015b). However, the factors shaping the DSB landscape at larger size scales remain poorly understood, particularly those factors related to the intersecting feedback mechanisms that regulate Spo11 activity (Keeney et al. 2014; Cooper et al. 2016).
It has been proposed that one such pathway involves negative regulation of break formation by the DSB-responsive serine/threonine kinase ATM/Tel1 (ataxia telangiectasia mutated in mammals; telomere maintenance in S. cerevisiae). ATM controls cell-cycle checkpoints and promotes DNA repair in somatic cells and is essential for meiosis in mammals (Shiloh and Ziv 2013). Testes from Atm−/− mice display a large increase in DSB numbers, as indicated by a >10-fold rise in the amount of covalent SPO11-oligonucleotide (oligo) complexes, a quantitative by-product of meiotic DSB formation (Lange et al. 2011). In Drosophila melanogaster oocytes, mutation of ATM elevates numbers of DSB-associated γ-H2AV foci (Joyce et al. 2011), and in S. cerevisiae, Tel1 loss increases recombination at the HIS4LEU2 hotspot (Zhang et al. 2011) and genome-wide (Anderson et al. 2015). In a rad50S background, in which unrepaired DSBs accumulate, more DSBs are observed at HIS4LEU2 and on at least one whole chromosome (Carballo et al. 2013; Garcia et al. 2015), although such increases were not apparent on all chromosomes (Argunhan et al. 2013; Blitzblau and Hochwagen 2013; Garcia et al. 2015).
Recently, Tel1 was shown to suppress formation of multiple DSBs nearby on the same DNA molecule (Garcia et al. 2015). In wild-type cells, formation of two DSBs at hotspots on the same chromatid occurs less frequently than expected by chance based on the population-average DSB frequency at each individual hotspot. This “DSB interference” can be detected across 70- to 100-kb distances in wild type but is lost in tel1Δ mutants. Furthermore, over smaller distances (less than ∼10–15 kb), multiple cutting occurs in the absence of Tel1 at substantially elevated frequency compared to expectation from random, indicating that Tel1 normally restrains a tendency for Spo11 to generate clusters of DSBs (Garcia et al. 2015). ATM/Tel1 action may also ensure that DSBs rarely form at the same location on both sister chromatids (Lange et al. 2011; Anderson et al. 2015; Garcia et al. 2015).
Together, these findings have revealed that ATM/Tel1 negatively regulates meiotic DSB numbers in many species. However, the extent to which Tel1 contributes to genome-wide DSB control in yeast has remained controversial and unclear. Furthermore, it has remained an open question if and how Tel1 imposes spatial patterns on DSB formation. To address these issues, we quantified Spo11-oligo complexes and mapped the genome-wide changes in DSB locations upon loss of Tel1. The data provide insight into characteristic genome-wide features of Tel1 loss and relationships among the negative feedback pathways that restrain DSB formation.
Results
Increased Spo11-oligo numbers in tel1Δ
In yeast, global DSB levels are often estimated by Southern blotting of chromosomes separated on pulsed-field gels. Such studies of tel1Δ mutants gave conflicting results (Argunhan et al. 2013; Blitzblau and Hochwagen 2013; Carballo et al. 2013; Garcia et al. 2015). One issue is the difficulty in controlling for DSB lifespan, since in wild-type cells, DSBs disappear as they are repaired by recombination (Hunter and Kleckner 2001). Because Tel1 promotes normal bias in favor of using the homolog rather than the sister chromatid for DSB repair (Carballo et al. 2008; Joshi et al. 2015), absence of Tel1 may alter average DSB lifespan and affect the ability to detect ongoing de novo DSB formation as meiosis proceeds. The use of DSB repair-defective backgrounds was once thought to obviate the lifespan issue, but recent studies have shown that these mutants introduce confounding effects by severing or hyperactivating negative feedback pathways that regulate DSB numbers (Cooper et al. 2014; Keeney et al. 2014; Thacker et al. 2014). Finally, assays based on indirect end-labeling of electrophoretically separated genomic DNA are often unable to distinguish lone DSBs from the clustered DSBs that form in the absence of Tel1, leading to underestimated DSB levels (Garcia et al. 2015).
To circumvent limitations of gel-based quantification of broken chromosomes, we assayed Spo11-oligo levels instead. Spo11 makes a DSB by forming covalent protein-DNA complexes that are then endonucleolytically cleaved to release Spo11 bound to a DNA oligo, with two Spo11-oligo complexes generated from each DSB (Supplemental Fig. S1A; Neale et al. 2005). Because Spo11-oligo complexes are a direct by-product of DSB formation and because their lifespan appears to be tied to progression out of the first meiotic prophase rather than being tied to progression of the recombination reaction, they can be used to quantify relative DSB levels genome-wide in an otherwise wild-type background, mitigating confounding effects of repair-defective mutations (Lange et al. 2011; Thacker et al. 2014).
Whole-cell extracts were prepared from equivalent numbers of wild-type and tel1Δ cells harvested at different times after meiotic induction. Phenotypically normal protein-A-tagged Spo11 was affinity-purified, 3′ ends of Spo11 oligos were radiolabeled using terminal deoxynucleotidyl transferase and [α-32P] dCTP, and Spo11-oligo complexes were separated on SDS-PAGE gels and detected by phosphorimager (Fig. 1A, top). A Western blot for total Spo11 confirms that Spo11 protein expression is induced to a similar extent in wild-type and tel1Δ cultures (Fig. 1A, bottom). Wild-type cells displayed the typical kinetics of appearance and disappearance of Spo11-oligo complexes (Fig. 1A,B), previously shown to coincide with DSB formation (Neale et al. 2005; Thacker et al. 2014). In the tel1Δ mutant, Spo11-oligo complexes first appeared with similar timing and levels as wild type but continued to accumulate, reaching a maximum at 5 h that was 2.2-fold higher on average than in wild type (Fig. 1A,B). Increased numbers of Spo11-oligo complexes were also apparent in the Western blot detecting total Spo11: A slower-migrating band was now apparent that comigrated with the upper radioactive signal. (The species corresponding to the lower radioactive signal was masked on the Western blot by the signal for free Spo11.)
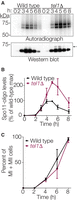
Elevated levels of Spo11-oligo complexes in tel1Δ mutants. (A) Representative time course. Autoradiograph of SDS-PAGE gel (top) shows radiolabeled Spo11-oligo complexes at the indicated times in sporulation medium. Anti-protein A Western blot (bottom) monitors total Spo11. Note that most signal in the Western blot is free Spo11, i.e., not bound to oligos. The arrow indicates the small amount of Spo11 protein that comigrates with slower migrating Spo11-oligo species, visible in the tel1Δ sample because of increased DSB numbers. (B) Quantification of radiolabeled Spo11-oligo complexes in tel1Δ cells, relative to wild-type cultures collected in parallel. Mean ± SD for three pairs of cultures is shown. (C) Meiotic progression (the percentage of cells completing one or both divisions). Mean ± SD for the same cultures as in B; ≥100 cells counted per time point.
DSB formation is inhibited when cells exit from the pachytene stage of prophase I (for review, see Keeney et al. 2014). Thus, DSBs can continue to form under conditions of prophase I arrest, e.g., in response to unrepaired DSBs or in the absence of the transcription factor Ndt80. However, tel1Δ cells divided with similar kinetics as wild-type cells (Fig. 1C), indicating that the increased Spo11-oligo levels are not a consequence of either increased DSBs or altered Spo11-oligo-complex lifespan caused by delayed meiotic progression. Thus, quantification of Spo11-oligo complexes, combined with prior analyses of recombination products (Zhang et al. 2011; Anderson et al. 2015) and some physical analyses of broken genomic DNA (Carballo et al. 2013; Garcia et al. 2015), supports the conclusion that loss of Tel1 increases DSBs globally. Furthermore, the kinetics of accumulation of Spo11-oligo complexes (Fig. 1B) indicate that DSB formation continues over a longer period in tel1Δ than in wild type.
Roles of Tel1 kinase activity and Tel1 phosphorylation targets
An unresolved question is whether negative regulation of DSB numbers by ATM/Tel1 depends on its kinase activity and, if so, what the relevant phosphorylation targets are. To address these issues, we first asked if a known tel1 kinase-dead mutation (“tel1-kd”) (Ma and Greider 2009) increased DSBs. Indeed, Spo11-oligo complexes were elevated in tel1-kd cultures to an extent similar to that in tel1Δ mutants (Fig. 2A). These results suggest that Tel1 can suppress DSB formation via the phosphorylation of one or more target proteins.
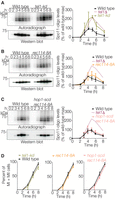
Spo11-oligo complexes in tel1-kd, rec114-8A, and hop1-scd mutants. (A–D) Representative time courses and quantification of (A) two, (B) four, and (C) six independent mutant cultures processed in parallel with (A) three, (B) four, and (C) four independent wild-type cultures are shown as in Figure 1. To aid comparison, tel1Δ data are duplicated from Figure 1. (D) Meiotic division profiles for the same cultures.
A candidate target is Rec114, which is required for DSB formation and bears matches to the S/TQ phosphorylation motif for Tel1 and its related kinase Mec1 (Carballo et al. 2013). Mec1 phosphorylates Rec114 in vitro (Tel1 was not tested); Mec1 and Tel1 are redundantly required for DSB-dependent phosphorylation of Rec114 in vivo; and mutation of eight S/TQ sequences in Rec114 to AQ (“rec114-8A”) yielded indirect evidence of increased and/or earlier DSB formation (Carballo et al. 2013). We therefore asked if rec114-8A phenocopies absence of Tel1. Strikingly, although the rec114-8A mutant showed a slight increase in Spo11-oligo complexes at late time points, this was substantially less than in the tel1Δ or tel1-kd mutants (Fig. 2B). We also combined rec114-8A with mutation of three S/TQ residues of Hop1, another known Tel1 and Mec1 target (known as the hop1-scd allele) (Carballo et al. 2008). However, even the small increase seen at late times with rec114-8A alone was not apparent in the rec114-8A hop1-scd double mutant (Fig. 2C). These mutations had little or no effect on division timing (Fig. 2D). We infer that Rec114 and Hop1 phosphorylation are largely dispensable for Tel1-mediated DSB suppression.
Altered Spo11-oligo sizes in tel1 mutants
Most Spo11-oligo complexes are in two size classes that differ in the lengths of the attached oligos (Fig. 1A; Neale et al. 2005). The ratio between these classes appeared altered relative to wild type in the tel1Δ mutant, with more of the higher molecular weight species (Fig. 1A). To examine this change, Spo11 oligos were separated on a sequencing gel (Fig. 3A). Consistent with the SDS-PAGE, tel1Δ showed an increase in oligos >25 nt long relative to those <20 nt, including increased abundance of very large oligos near the top of the gel (Fig. 3A,B). The distribution of larger oligos was also altered: A modest shoulder at ∼35 nt in wild type became more prominent in tel1Δ, and even longer oligos (from ∼40 to ∼70 nt) displayed a more pronounced banding pattern in the mutant (Fig. 3A,B).
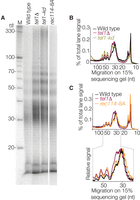
Sizes of Spo11 oligos in Tel1 pathway mutants. (A) Radiolabeled, de-proteinized Spo11 oligos separated on a denaturing 15% polyacrylamide sequencing gel. Lanes are from a single gel with an intervening lane omitted. (M) 10-nt ladder. Note that oligos from lower-molecular-weight Spo11-oligo complexes are underrepresented because most of the shortest oligos (less than ∼12 nt) are lost upon ethanol precipitation. (B,C) Lane profiles from panel A.
The tel1-kd mutant was similar with respect to increased abundance of longer Spo11 oligos (those >25 nt relative to those <20 nt), the more prominent population at ∼35 nt, and the banding at ∼40–70 nt (Fig. 3A,B). Interestingly, these alterations were even more extreme in tel1-kd than in tel1Δ, indicating that presence of a kinase-dead protein does not precisely phenocopy absence of Tel1.
The distribution of oligo sizes was also altered in the rec114-8A mutant, in similar ways but to a lesser degree than in tel1Δ (Fig. 3A,C). This finding is consistent with Rec114 phosphorylation contributing to regulation of the formation and/or processing of DSBs, but the results also reinforce the conclusion that Rec114 is not the sole Tel1 substrate important for DSB control.
The DSB landscape responds differently to Tel1 loss at different stages of meiosis
Having confirmed that Tel1 regulates DSB numbers, we investigated how it shapes the DSB distribution. Analysis of recombination products (Anderson et al. 2015) supported an earlier hypothesis that Tel1 shapes the DSB landscape (Lange et al. 2011), but recombination maps are indirect and low-spatial-resolution indicators of DSB positions. We deep-sequenced Spo11 oligos purified from wild-type and tel1Δ cultures at 4 h in sporulation conditions, when DSBs are maximal in wild type and the tel1Δ mutant has only slightly elevated total Spo11-oligo levels, and at 5 h and 6 h, when the tel1Δ mutant has substantially elevated Spo11 oligos (Fig. 1B; Supplemental Table S1). As expected, the map at 5 h consistently showed patterns intermediate between the 4-h and 6-h time points. Two tel1Δ biological replicates were highly similar (Fig. 4A; Supplemental Fig. S2) and were averaged together. Wild-type samples displayed little systematic variation over this time window, so maps from six samples (two each from 4, 5, and 6 h) were averaged together to generate a consensus wild-type map used for all comparisons to mutant maps.
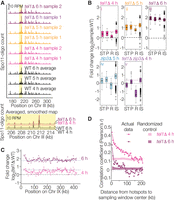
Spo11-oligo landscapes. (A) Reproducibility of Spo11-oligo maps. A representative segment from Chr III is shown at the top and a close-up view of averaged biological replicates in the yellow-highlighted region is shown below. Both Spo11-oligo maps were smoothed with a 201-bp Hann window. (B) Subchromosomal domains. Boxplots show the log-fold change in Spo11-oligo counts in each mutant relative to wild type for hotspots in the indicated genomic regions. (ST) Subtelomeric, 20 kb at each chromosome end (62 hotspots), (P) pericentric, 20 kb centered at each centromere (82 hotspots), (R) rDNA-proximal region (Chr XII: 451,577–467,570; 20 hotpots), (IS) interstitial, i.e., all other genomic regions. Thick horizontal lines are the medians, box edges are the 25th and 75th percentiles, whiskers indicate lowest and highest values within 1.5-fold of the interquartile range, and open circles are outliers. Colored dashed lines show the genome-average fold change for each mutant (1.5 for tel1Δ 4 h, 2.3 for tel1Δ 5 h, 3.5 for tel1Δ 6 h, 1.8 for zip3Δ 5 h, and 1.3 for tel1Δ zip3Δ 4 h). Black dashed lines at zero denote no change. Wild-type data for these and related comparisons are the averaged data for 4-, 5-, and 6-h samples. (C) Regional variation in the response of hotspots to the absence of Tel1. Chr IX is shown as an example. Each point is the log-fold change in Spo11-oligo count relative to wild type. Solid lines are local regression curves; dashed lines indicate genome-wide average fold change or no change. (D) Local domains of correlated behavior. Each point compares the log-fold change in hotspots with the change in their neighbors located in a 5-kb window the indicated distance away. Shaded regions show 95% confidence intervals for hotspots randomized within-chromosome; randomized data at 4 and 5 h show higher correlation because of the residual chromosome size effect (see Fig. 5A and Supplemental Fig. S3D). Subtelomeric, pericentric, and rDNA-proximal regions were excluded.
At short (sub-kb) size scales, Spo11 oligos from tel1Δ cells mapped preferentially to hotspots highly similar in location and width to those from wild type (Fig. 4A; Supplemental Fig. S3A,B). We conclude that the influence of local chromatin structure on hotspot position and shape is largely unaffected by tel1Δ and that the increase in Spo11-oligo levels is not principally from DSBs in regions that were previously inaccessible to Spo11. Interestingly, these findings contrast with those in mouse, where absence of ATM caused more hotspots to become detectable and caused hotspots to become wider (Lange et al. 2016) (see Discussion).
When assessed on larger size scales, however, tel1Δ showed substantial alterations that changed over time in meiosis. At 4 h, Spo11-oligo frequencies had increased less than average in hotspots within 20-kb zones at telomeres and centromeres and had increased even less near the ribosomal DNA (rDNA) (Fig. 4Bi). Accumulation of Spo11 oligos in these regions continued to lag behind the genome average at 5 h (Fig. 4Bii), but by 6 h, hotspots in these domains were similar to or higher than the genome average, i.e., were all about equally elevated (Fig. 4Biii). Thus, these domains, which are suppressed for DSBs in wild type (Pan et al. 2011), are initially somewhat insulated from the elevation in DSB levels caused by Tel1 deficiency but over time continue to accumulate additional DSBs to reach a similar extent as elsewhere in the genome. It is possible that the behavior of subtelomeric regions is influenced by telomere changes that occur during vegetative growth in the tel1Δ mutant (Lustig and Petes 1986).
The remaining interstitial hotspots showed variable responses to the tel1Δ mutation at all time points (Fig. 4Bi,Bii,Biii,C) but with only a modest correlation between the 4-h and 6-h samples (Pearson's r = 0.34) (Supplemental Fig. S3C). The 5-h sample showed intermediate correlation but was more similar to 6 h (Supplemental Fig. S3C). Local regression of the fold change in Spo11-oligo signal along chromosomes displayed peaks and valleys, suggesting that groups of hotspots respond to the tel1Δ mutation in a similar manner (Fig. 4C). To test this conclusion, we measured the correlation in response between hotspots and their neighbors. At 4 h, the change in each hotspot's Spo11-oligo count was positively correlated with the change in its neighbors, with correlation strength decaying with distance but still above randomized controls up to ∼125 kb away (Fig. 4D). In contrast, the 6-h time point had a weaker starting correlation for close hotspots (≤5 kb), and this correlation decayed to background levels by 15 kb (Fig. 4D). The 5-h time point had intermediate behavior: Similar to 6 h, correlation strength decayed to background within a short distance, but background (randomized) correlation levels were higher as in the 4-h time point, reflecting chromosome size effects discussed below (Supplemental Fig. S3D). Thus, while hotspots appear to respond to absence of Tel1 in a “domainal” fashion early in meiosis, this organization is largely erased as meiosis progresses. The convergence of the tel1Δ Spo11-oligo map with the wild-type map is coincident with the period when additional DSBs are forming in the tel1Δ mutant (Fig. 1B). Therefore, we conclude that loss of Tel1 leads to substantial alterations in the DSB landscape, and these alterations change over time as cells continue to make DSBs. The “normalization” of the tel1Δ Spo11-oligo map at later times indicates that the remaining (Tel1-independent) DSB regulatory systems have the net effect of homeostatic maintenance of the DSB distribution despite changes in DSB numbers.
Relationship between Tel1 and the homolog engagement pathway for DSB control
DSB numbers are also elevated in mutants lacking “ZMM” proteins (Thacker et al. 2014), factors needed to complete recombination and synaptonemal complex formation (Lynn et al. 2007). This and findings from other organisms (Wojtasz et al. 2009; Hayashi et al. 2010; Kauppi et al. 2013) have led to the hypothesis that homologous chromosomes that successfully engage one another largely stop making DSBs (Kauppi et al. 2013; Keeney et al. 2014; Thacker et al. 2014). This negative feedback regulation, which contributes to DSB spatial patterning (Thacker et al. 2014), is proposed to be distinct from Tel1-mediated DSB control (Keeney et al. 2014). To test this proposal, we compared how the DSB landscape changes in the absence of Tel1 to how it changes in the absence of the ZMM protein Cst9 (referred to hereafter by its more commonly used alias, Zip3). Spo11-oligo maps from a zip3Δ mutant are currently available only for 5 h in sporulation (Thacker et al. 2014), so the degree to which DSB patterns change over time is unknown. Nonetheless, comparisons with patterns in the tel1Δ mutant proved informative.
In wild type, smaller chromosomes generate more DSBs per kb than larger ones, revealed as an inverse correlation between Spo11-oligo density and chromosome size (Pan et al. 2011). This anti-correlation is absent in zip3Δ at 5 h, indicating a role for ZMM proteins and thus homolog engagement (Fig. 5A; Thacker et al. 2014). The tel1Δ 4-h time point displayed an attenuated version of the zip3Δ 5-h pattern, i.e., a reduced but not absent anti-correlation (Fig. 5A). Similarly, the responses of subtelomeric, pericentric, and rDNA-proximal regions to the absence of Tel1 at 4 h were qualitatively similar to those in the zip3Δ mutant but in a milder form (Fig. 4Bi,Biv), and there was a positive correlation between the tel1Δ 4-h and zip3Δ 5-h samples for changes at interstitial hotspots (r = 0.53) (Supplemental Fig. S3C). The two data sets also displayed similar correlation strengths and length scales for the local correlation in hotspot behavior (Fig. 5B).
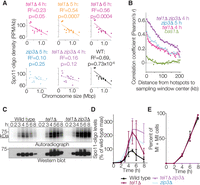
Relationship between Tel1- and Zip3-dependent control pathways. (A) Negative correlation between Spo11-oligo density and chromosome size requires Zip3 but not Tel1. zip3Δ data are from Thacker et al (2014). (B) Local domains of correlated behavior (as in Fig. 4D). Data for tel1Δ 4 h are reproduced from Figure 4D. See Supplemental Figure S4C for randomized controls. Data from a mutant lacking the transcription factor Bas1 serve as a negative control for this analysis, because this transcription factor affects DSB formation within a subset of individual hotspots rather than coordinately influencing local domains containing multiple hotspots (Zhu and Keeney 2015). (C–E) Spo11-oligo time courses as in Figure 1. Mean ± SD for four pairs of cultures is shown. Data from zip3Δ are reproduced from Thacker et al (2014) for comparison (see also Supplemental Fig. S4B).
However, essentially all similarity with the zip3Δ mutant was erased by 6 h in tel1Δ, whether compared in specific subchromosomal domains (Fig. 4B), in local domains of correlated behavior (Fig. 4D), or across all interstitial hotspots (r = −0.01) (Supplemental Fig. S3C). Furthermore, at 6 h, the negative correlation between Spo11-oligo density and chromosome size mirrored the pattern in wild type (Fig. 5A). The 5-h tel1Δ time point was intermediate for all comparisons but generally more similar to the 6-h time point (Figs. 4B,D, 5A; Supplemental Fig. S3C,D). Thus, Tel1 is dispensable for chromosome size-related control of DSB frequency, although it is needed for timely establishment.
To further characterize the apparent similarities between the tel1Δ 4-h and zip3Δ 5-h DSB landscapes, we examined a tel1Δ zip3Δ double mutant. At time points up to ∼5 h, the double mutant yielded levels of Spo11-oligo complexes that were indistinguishable from the tel1Δ single mutant, i.e., with maximum elevated ∼2.4-fold relative to wild type (Fig. 5C,D). For comparison, we previously found that the peak was elevated by 1.8-fold in the zip3Δ mutant (Fig. 5D; Supplemental Fig. S4B; Thacker et al. 2014). These findings indicate that loss of both Tel1 and ZMM function does not cause an additive increase in DSB numbers early in meiosis, suggesting at least some degree of overlap between these mechanisms of DSB control. At later time points, the tel1Δ zip3Δ double mutant maintained Spo11-oligo complexes at high levels (Fig. 5C,D). This is unlike the tel1Δ single mutant in which Spo11-oligo complexes decreased over these time points but is more reminiscent of the pattern in the zip3Δ single mutant (Supplemental Fig. S4B). Absence of Tel1 does not alleviate the arrest caused by the zip3Δ mutation (Fig. 5E); the block to meiotic progression may explain why amounts of Spo11-oligo complexes remain elevated at later times in the tel1Δ zip3Δ double mutant.
We next generated Spo11-oligo maps from the tel1Δ zip3Δ double mutant (Supplemental Fig. S4A), focusing on the 4-h time point when Spo11-oligo complexes are elevated to similar degrees in the double mutant as in each single mutant (Fig. 5D). Analysis of these maps indicated that the epistasis relationships between the tel1Δ and zip3Δ mutations are complex. There was no longer a residual anti-correlation between Spo11-oligo density and chromosome size in the double mutant, indistinguishable from the zip3Δ single mutant at 5 h (Fig. 5A). Similarly, the double mutant more closely mirrored the stronger patterns seen in the zip3Δ single mutant for hotspots in the subtelomeric and rDNA-proximal regions but less so in pericentric regions (Fig. 4Bv). Thus, zip3Δ is epistatic to tel1Δ for these features of the DSB landscape.
For other features, however, Spo11-oligo maps revealed largely nonepistatic relations instead. For interstitial hotspots, the log-fold change of Spo11-oligo counts in the double mutant correlated best with the zip3Δ single mutant map (r = 0.79) (Supplemental Fig. S3C), indicating that most of the change in the double mutant can be accounted for by the zip3Δ mutation. However, there was also a stronger correlation of the tel1Δ 4-h and 5-h data sets with tel1Δ zip3Δ than with the zip3Δ single mutant (r = 0.62 vs. 0.53 for comparisons involving tel1Δ at 4 h; r = 0.51 vs. 0.30 for comparisons involving tel1Δ at 5 h) (Supplemental Fig. S3C), and the tel1Δ 6-h map now displayed a modest correlation with the tel1Δ zip3Δ map that was not seen with the zip3Δ map (r = 0.18 vs. −0.01) (Supplemental Fig. S3C). These observations suggest that the DSB landscape in the double mutant combines changes that are shared in common between the two single mutants with features that are unique to either tel1Δ or zip3Δ. Furthermore, neighboring hotspots displayed more strongly correlated behavior that spread over longer distances (up to ∼200 kb) in the tel1Δ zip3Δ double mutant than in either single mutant (Fig. 5B). This suggests that effects of tel1Δ or zip3Δ mutations are additive (nonepistatic) for this feature of the DSB landscape. Possible explanations for this complex behavior are provided in the Discussion.
Interactions between DSB sites in cis along chromosomes
Inserting a strong artificial hotspot suppresses DSB formation nearby when measured on a population average (Wu and Lichten 1995; Xu and Kleckner 1995). This suppressive effect decays with distance but has been documented to extend ≥60 kb (Wu and Lichten 1995; Jessop et al. 2005; Robine et al. 2007). Tel1-dependent DSB interference operates over a similar size scale (see Introduction) (Garcia et al. 2015), so interference can, in principle, explain the local effect of strong artificial hotspots, assuming that interference includes both sister chromatids. Site-specific endonucleolytic DSBs (i.e., not made by Spo11) also suppress Spo11-generated DSBs nearby (Fukuda et al. 2008), consistent with an inhibitory signal that spreads after DSB formation. Importantly, however, it remains unknown whether the effect of artificial Spo11 hotspot insertion is the same as DSB interference: To meet the formal definition of interference, occurrence of multiple events on the same chromosome must be shown to occur less often than expected from the product of the single-event frequencies, but this has not been tested. An alternative possibility is that artificial hotspot insertion diminishes the intrinsic DSB potential of nearby hotspots, for example, by altering large-scale chromosome structures.
We reasoned that, if artificial hotspot insertion works solely via DSB interference, then the DSB suppressive effect should require Tel1. To test this prediction, we inserted the arg4-URA3 artificial hotspot (Wu and Lichten 1995) at either of two positions in the genomes of otherwise wild-type or tel1Δ cells: at HIS4 on Chr III or POR2 on Chr IX. We then examined the effects of the hotspot insertions in cis by compiling Spo11-oligo maps from 4-h meiotic cultures.
As previously demonstrated (Wu and Lichten 1995), hotspot insertion on Chr III in a TEL1+ background caused a substantial drop in DSB formation nearby: Spo11-oligo density was decreased by fourfold or more in hotspots flanking the insertion site (Fig. 6A,B). Suppression decayed with distance, extending ∼60 kb to the left (nearly to the telomere) and ∼130 kb to the right, extending past the centromere and even spanning, albeit more weakly, the DSB-cold domain from CEN3 to MAT. In tel1Δ, DSBs were suppressed to a similar degree from the insertion site to the left and from the insertion site to the centromere, but the CEN3–MAT region was no longer suppressed (Fig. 6A,B). We conclude that, for this insertion site, Tel1 is dispensable for DSB suppression over distances up to ∼60 kb. We further conclude that the suppressive effect of this hotspot insertion is not simply a consequence of DSB interference, because interference is fully Tel1-dependent when measured in this same region of Chr III (Garcia et al. 2015). We will use the term “hotspot competition” to distinguish the Tel1-independent phenomenon from Tel1-dependent DSB interference (Keeney et al. 2014). Further studies are needed to test whether DSB interference explains the Tel1 dependence of DSB inhibition over longer distances in the CEN3–MAT interval.
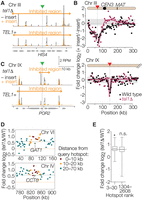
Communication between hotspots in cis along chromosomes. (A–C) Effect of artificial hotspot insertions. A hotspot construct containing arg4 and URA3 on a bacterial plasmid backbone (Wu and Lichten 1995) was inserted within the HIS4 gene on Chr III or the POR2 gene on Chr IX (green triangles and vertical dotted lines). (A,C) Spo11-oligo maps (smoothed with a 201-bp sliding Hann window) are shown for wild-type and tel1Δ strains with and without the insertion. (B) Each point shows, for each 5-kb segment moving outward from the insertion points, the log-fold change in the Spo11-oligo count caused by the presence of the artificial hotspot insertion (position of insertions denoted by red triangles). Curves were drawn manually to highlight the trends in the data. Gray shading encloses ± 1 SD for all 5-kb bins genome-wide (excepting Chr III and IX) to illustrate the range of sampling noise in this analysis. (D,E) Signature consistent with Tel1-dependent DSB interference around the strongest natural hotspots. In panel D, each point displays the log-fold change in Spo11-oligo count (4-h tel1Δ vs. wild type) for hotspots located the indicated distance away from either the GAT1 (top) or CCT6 (bottom) promoter hotspots. Horizontal dashed lines indicate genome-average change or no change. In panel E, boxplots summarize the log-fold change for hotspots located within 10 kb of the next 28 hottest hotspots (rank numbers 3–30) or within 10 kb of the middle third of hotspots (rank numbers 1304–2608). (*) P = 0.0011 for a one-sided Wilcoxon rank-sum comparison to all hotspots genome-wide, (n.s.) not significant (P = 0.61). Horizontal dotted line indicates genome-average change. Boxplots are as defined in Figure 4B.
Chr IX gave a strikingly different result. In the TEL1+ background, this hotspot insertion caused a substantial drop in Spo11-oligo counts for hotspots immediately flanking the insertion site, yielding a similar magnitude of suppression as on Chr III (Fig. 6B,C). However, the suppressive effect covered a much smaller zone, only ∼20 kb to the left and ∼15 kb to the right. The more limited distance of suppression may be, in part, a consequence of the artificial hotspot being ∼4.5-fold weaker at the Chr IX location, based on normalized Spo11-oligo counts in TEL1+ (Supplemental Fig. S5; Supplemental Table S4). Context dependence for DSB frequency within this insert was documented previously (Borde et al. 1999). In the tel1Δ background, the hotspots closest to the insertion (<2 kb away) were still suppressed, but the next hotspots further away (≥5 kb) were unaffected by the insertion (Fig. 6B,C). Thus, the zone of DSB suppression is much smaller at this location, and Tel1 is essential beyond the most immediate vicinity of the new hotspot. More broadly, these results show that the local effects of a hotspot insertion can be context-dependent in terms of both the distance over which Tel1-independent hotspot competition operates and the degree to which Tel1-dependent processes (presumably DSB interference) contribute.
A natural hotspot in the HIS4 promoter can exert a suppressive effect on an artificial one inserted nearby (Fan et al. 1997), and adjacent natural hotspots can be suppressed when DSBs are artificially targeted to novel sites by fusing Spo11 to the DNA binding domain of Gal4, illustrating functional interactions between natural yeast DNA sequences (Robine et al. 2007). However, other studies found no evidence of such interactions between two natural hotspots near HIS2 (Bullard et al. 1996) or between Bas1- or Ino4-regulated hotspots and their neighboring hotspots (Zhu and Keeney 2015). It has thus been unclear to what extent suppressive interactions occurring between natural hotspots can contribute to population-average DSB frequencies. To address this question, we asked if absence of Tel1 causes a greater than average increase in DSBs for hotspots located near strong natural hotspots, because weak hotspots should be more highly suppressed by Tel1 signals emanating from their stronger neighbors.
To test this prediction, we examined GAT1, the strongest natural hotspot in the SK1 strain background (Pan et al. 2011). At 4 h in the tel1Δ mutant, this hotspot displayed a Spo11-oligo count that was slightly below the genome-average increase (Fig. 6D; Supplemental Fig. S6A). In contrast, nearly all of its neighbors within ∼10–20 kb on either side experienced above-average increases in Spo11-oligo levels; hotspots further away were not consistently increased above the genome average (Fig. 6D). Similarly, most hotspots in the vicinity of another exceptionally strong natural hotspot, CCT6, also increased more than average (Fig. 6D; Supplemental Fig. S6B), as did hotspots located up to ∼10 kb away from the next 28 hottest hotspots considered as a group (Fig. 6E). This pattern meets the prediction for Tel1-dependent inhibitory interactions between strong hotspots and their weaker neighbors and is thus consistent with being a consequence of DSB interference. Notably, however, we detected no reliable hotspot-suppression signature for hotspots that are less active than this elite group (Fig. 6E).
Taken together, these findings support the hypothesis that Tel1-dependent DSB interference can shape the population-average DSB landscape but that it provides a quantitatively modest contribution that is readily detected only at relatively strong hotspots and only over short distances. Our findings also demonstrate a Tel1-independent component to interactions between artificial hotspots and their surroundings. This hotspot competition is context-dependent, as insertions on Chr IX and Chr III behave differently.
Discussion
Quantification of Spo11-oligo complexes reinforces the conclusion that more DSBs form when Tel1 is missing in S. cerevisiae, which was indicated by many studies (Zhang et al. 2011; Carballo et al. 2013; Anderson et al. 2015; Garcia et al. 2015) but not all (Argunhan et al. 2013; Blitzblau and Hochwagen 2013). However, although it is now clear that Tel1/ATM has an evolutionarily conserved function in regulating DSB numbers, the mechanism remains poorly understood. Tel1 kinase activity appears to be critical, but the relevant phosphotarget(s) is unknown. Prior studies implicated Rec114 phosphorylation (Carballo et al. 2013), but we found that mutating the presumed S/TQ phosphoacceptor sites mimicked absence of Tel1 only weakly if at all. It is therefore likely that DSB suppression proceeds via another phosphotarget(s) and does so either exclusively of or redundantly with the tested S/TQ motifs of Rec114 and Hop1. We favor the redundancy interpretation because it provides a straightforward explanation for the modest increase in DSB levels and partial alteration of Spo11-oligo sizes in the rec114-8A mutant (which together indicate that Rec114 is indeed a functional target of Tel1) and because of the fact that phosphomimetic mutations at Rec114 S/TQ sites substantially reduce DSBs (Carballo et al. 2013) (which may indicate that Rec114 phosphorylation is sufficient but not necessary for DSB inhibition). Identifying the other phosphotarget(s) will be an important next step.
Tel1 and DSB processing
We find that Tel1 also has a conserved role in modulating the lengths of Spo11 oligos. Atm−/− spermatocytes have alterations in SPO11-oligo sizes that are strikingly similar to those observed in tel1Δ and tel1-kd, namely, fewer short oligos relative to long ones, a more pronounced banding pattern for oligos in the ∼40- to 70-nt range, and an increase in abundance of very long oligos (Lange et al. 2011). One model to explain these findings is that Tel1 acts early in meiotic DSB processing. Indeed, Tel1 phosphorylates proteins necessary for Spo11 removal from DNA ends, such as Mre11 and Sae2 (Cartagena-Lirola et al. 2006; Terasawa et al. 2008), and Tel1 is required for both the efficient initiation of DSB resection and the generation of normal-length exonucleolytic resection tracts (Mimitou et al. 2017). Thus, altered oligo sizes may reflect changes in the positions of Mre11-dependent endonucleolytic cleavage (Cannavo and Cejka 2014) and/or decreased 3′→5′ exonuclease activity of Mre11 (Garcia et al. 2011).
An alternative, nonexclusive model is that some of the Spo11 oligos with unusual sizes arise from adjacent unresected DSBs on the same DNA molecule, i.e., with 3′ ends of these Spo11 oligos derived from nearby Spo11 cuts rather than from Mre11-dependent endonucleolytic cleavage (Supplemental Fig. S1B; Garcia et al. 2015). In this model, the size changes are a consequence of aberrant control of DSB numbers along with a change in nucleolytic DSB processing. This model is consistent with detection of a substantial subpopulation of unresected DSBs in Tel1-deficient cells (Mimitou et al. 2017). This model also readily accounts for the apparent contribution of Rec114 phosphorylation and for the observation that reducing DSB numbers (by Spo11+/− heterozygosity) in an Atm−/− background partially suppresses the changes in SPO11-oligo sizes (Lange et al. 2011).
In either model, it is unclear why presence of catalytically inactive Tel1 should exacerbate the changes. In mice, an ATM kinase-dead mutation causes embryonic lethality, whereas deletion mutants are viable, providing precedent for critical functional differences between the two types of allele (Daniel et al. 2012; Yamamoto et al. 2012).
Tel1 shapes the DSB landscape
Because Tel1/ATM is activated in the vicinity of DSBs, it was argued that it would contribute to spatial patterning of DSBs across the genome (Lange et al. 2011). Spo11-oligo mapping affirmed this prediction but also unexpectedly demonstrated that the effect of Tel1 absence differs over the course of meiosis. At 4 h, the DSB landscape in tel1Δ resembled that in zip3Δ at 5 h, but by 6 h that similarity was largely gone and the tel1Δ mutant instead displayed a unique but modest set of differences from wild type. Maps from a later zip3Δ culture are not available, so it is unknown whether the DSB landscape becomes more like wild type as a zip3Δ meiosis continues or if it instead stays profoundly altered. However, our data clearly demonstrate that the zip3Δ mutant has a much stronger defect than tel1Δ at 5 h and that zip3Δ is epistatic to tel1Δ at 4 h for many of the more severe defects.
We consider two straightforward ways to rationalize the similarity between the early tel1Δ map and the zip3Δ 5-h map. First, subchromosomal domains that are more or less sensitive to Tel1-mediated DSB suppression might also be correspondingly more or less sensitive to ZMM-dependent suppression. For example, domains enriched for Tel1 substrates might be more sensitive to Tel1 regulation; of note, enrichment for Hop1, Red1, and Rec114 is correlated with stronger ZMM-dependent DSB suppression (Thacker et al. 2014). A second, nonexclusive possibility follows from noting that Tel1 promotes inter-homolog bias early in meiosis (Joshi et al. 2015). Meiotic recombination favors the use of the homolog rather than the sister chromatid as the repair partner for DSBs (Hunter and Kleckner 2001), and this inter-homolog bias depends on Tel1 or its paralog Mec1 (related to mammalian ATR) (Carballo et al. 2008). Tel1 is principally responsible for this function early in meiosis, when DSB levels are relatively low, but it is rendered redundant with Mec1 later, when DSB levels are high (Joshi et al. 2015). We therefore reason that tel1Δ mutants, because of reduced inter-homolog bias, may experience a delay in homolog engagement and may as a consequence initially display many of the alterations in the DSB landscape that are diagnostic of the ZMM mutation zip3Δ. In this scenario, substitution of Mec1 for Tel1 activity eventually overcomes the transient homolog engagement defect. One implication is that the homeostatic compensation that occurs dynamically over the course of meiosis is generally invisible to methods that rely on examining only the final outcome of recombination, e.g., tetrad analysis.
Both models provide an explanation for the complex epistasis relationship between the tel1Δ and zip3Δ mutations, in which some measurements, such as amount of Spo11-oligo complexes, indicate a nonadditive (i.e., at least partially epistatic) relationship, whereas other measures, including genome-wide DSB distributions, show additive (nonepistatic) behaviors. Taken together, the results are consistent with the hypothesis that Tel1 and ZMM proteins regulate DSB formation via distinct mechanisms but also indicate that these pathways for DSB control overlap with one another because of the chromosome-associated proteins that implement the control and/or because both pathways are needed to support proper engagement between homologous chromosomes.
What then are the unique contributions of Tel1 to shaping the DSB landscape? Perhaps the most prominent effect is via Tel1 control of DSB interference, which discourages cutting of the same chromatid more than once within a small distance (Garcia et al. 2015). DSB interference presumably works via spreading in cis along chromosomes of an inhibitory signal comprising Tel1-dependent phosphorylation of Rec114 and/or other targets. However, DSB interference manifests itself principally at the level of individual cells and is thus predicted to leave a relatively small quantitative footprint in the population-average DSB map (Zhang et al. 2011; Garcia et al. 2015). Our finding that clear signatures of Tel1-dependent interactions between hotspots are readily detected only at the hottest hotspots is consistent with this prediction. Also consistent with this view, the 6-h tel1Δ Spo11-oligo map displayed only relatively modest changes relative to wild type, indicating that the net effect of Tel1 absence is that the extra DSBs end up accumulating largely in the same places as they do in wild type. It is possible that Tel1 dependence varies between genomic locations, but we also consider it likely that a hotspot with relatively high DSB frequency is necessary to detect this behavior (Zhang et al. 2011; Zhu and Keeney 2015; Cooper et al. 2016). This, in turn, may explain why studies of medium-strength natural hotspots failed to display similar interactions between neighbors (Bullard et al. 1996; Zhu and Keeney 2015). Redundancy with Mec1, especially later in meiosis (Joshi et al. 2015), may also contribute to Tel1 having a relatively modest net effect on the DSB landscape. Interestingly, the major Tel1 effects at the population level appeared to be highly local (within several kb), as judged by the short distance over which correlated changes were observed in the tel1Δ mutant at 6 h. It is possible that this pattern reflects a major function for Tel1 in controlling DSBs within single chromatin loops and, to a lesser extent, between adjacent loops, as previously suggested (Garcia et al. 2015).
Mice experience much greater quantitative changes in the DSB landscape in the absence of ATM. Unlike in tel1Δ yeast, Atm−/− mouse spermatocytes display an increase in the number of SPO11-oligo hotspots called at a given threshold over the genome average; weaker hotspots show a disproportionate increase in strength; hotspots across relatively large (up to ∼10–15 Mb) regions display correlated behavior; and the magnitude of increase in hotspot heat is negatively correlated with local SPO11-oligo density in wild type (Lange et al. 2016). These findings all point to ATM-dependent DSB inhibition working over relatively large (Mb-scale) domains, with this suppression having a substantial effect on the overall DSB landscape. At a local level, hotspot width also increases. These differences from yeast may reflect different functions for ATM vs. Tel1 or may simply reflect that ATM plays a quantitatively larger role in DSB control in mouse: SPO11-oligo complexes increase more than 10-fold in Atm−/− male mice (Lange et al. 2011) compared with only approximately twofold in tel1Δ yeast.
Nonetheless, the negative feedback regulation of DSB numbers via ATM/Tel1 and the fact that this regulation contributes to shaping the DSB landscape are common threads linking these evolutionarily distant species. It has been argued that the elements that shape DSB distributions can be divided into “intrinsic” factors (the DSB machinery and its chromatin substrate) and more “extrinsic” (or “reactive”) factors that provide homeostatic feedback regulation (Keeney et al. 2014; Cooper et al. 2016). Our results place Tel1 firmly on the list of such extrinsic factors and illustrate the complex interplay between Tel1-dependent and ZMM-dependent pathways for controlling both the number and distribution of DSBs.
Communication in cis along chromosomes: more than just DSB interference via Tel1
Our findings provide insight into the phenomenon by which creation of a new DSB hotspot influences behavior of other hotspots nearby. Some of the earliest studies of this phenomenon were on Chr III (Wu and Lichten 1995; Xu and Kleckner 1995; Fan et al. 1997). When we began this project, we envisioned that DSB suppression would be a consequence of the same interference phenomenon recently documented by Neale and colleagues (Garcia et al. 2015). We were therefore surprised to find that DSB suppression did not require Tel1 even though Tel1-dependent DSB interference is readily detectable across the same regions on Chr III (Garcia et al. 2015).
We were also surprised by the different behavior of the hotspot insertion on Chr IX. The Tel1-dependent component of suppression of adjacent DSBs can be explained as a reflection of DSB interference. Whether the Tel1-independent component is the same as on Chr III remains unknown, but regardless of whether it is the same mechanism or not, our results indicate that hotspot competition is either highly context-dependent, proportional to the strength of the hotspot insertion (which is itself context-dependent), or both.
How might hotspot competition work? One possibility is that it is also an interference phenomenon (i.e., it results from double cutting occurring less often than expected from single-cutting frequencies). If so, it involves the spread of an inhibitory signal from a DSB via a mechanism that does not require Tel1 signaling. For example, propagation along chromosome axes of relief of mechanical stress has been proposed as a mechanism to impose spatial patterning on DSBs (Kleckner et al. 2004; Keeney 2008). Another possibility is that different genomic sites compete with one another by titrating locally limited factors. A third possibility is that insertion of heterologous DNA sequences lowers the intrinsic DSB-forming potential in flanking regions by disrupting normal formation of higher-order chromosome structures that support Spo11 activity, for example, the loop-axis organization of meiotic chromosomes and/or binding of DSB-promoting chromosome structure proteins such as Red1, Hop1, or cohesins. All three of these models are compatible with locus-to-locus variation depending on local chromosomal features such as loop attachment sites, chromatin structure, replication timing, and intrinsic affinity for binding of chromosome structure proteins.
Methods
Yeast strains
Yeast strains are of the SK1 background (Supplemental Table S2). Tagged Spo11 constructs were as previously described (Thacker et al. 2014). The hop1-scd allele hop1-S298A, S311A, T318A (Carballo et al. 2008) was generously provided by R. Cha. The tel1-kd allele of Ma and Greider (2009) (tel1-D2612A,N2617A,D2631A) was generated via QuickChange mutagenesis on a plasmid-borne fragment of TEL1 and integrated to replace the endogenous TEL1 sequence. We recreated the rec114-8A allele described by Carballo et al. (2013) (S148A, T175A, T179A, S187A, S229A, T238A, S256A, S307A) using synthetic DNA fragments and integrated it to replace the endogenous REC114 gene. The artificial hotspot insertions were created as follows: Fragments of the yeast genome containing coding regions from HIS4 (+726 to +2062 with respect to ATG) or POR2 (−380 to +1001 with respect to ATG) were cloned into the EcoRI sites of pMJ113 (Wu and Lichten 1995), cut with either XbaI (HIS4 hotspot) or BstXI (POR2 hotspot), and transformed into the appropriate SK1 strains. All integrations were confirmed by Southern blotting and/or sequencing of DNA amplified from genomic DNA.
Spo11-oligo complexes
Cells were cultured and harvested from meiotic time courses as previously described (Thacker et al. 2014). Denaturing extracts were prepared from yeast cells using 10% trichloroacetic acid and agitation in a bead beater, then Spo11-ProtA was affinity-purified on rabbit IgG-coupled magnetic beads. Spo11-oligo complexes were end-labeled with [α-32P] dCTP and terminal deoxynucleotidyl transferase (TdT), resolved on SDS-PAGE gels, then transferred onto PVDF membranes and visualized by phosphorimager. Western blotting to monitor total Spo11 was done using an anti-Protein A antibody (Sigma). To evaluate lengths of Spo11 oligos, Spo11-oligo complexes were affinity-purified and radiolabeled, and then digested with proteinase K. The de-proteinized Spo11 oligos were ethanol-precipitated, resuspended in formamide dye, and run on a 15% denaturing polyacrylamide sequencing gel with radiolabeled 10-nt DNA ladder as a marker (Life Technologies).
Spo11-oligo sequencing
Culture conditions and volumes were identical to those described previously (Thacker et al. 2014) except that 200 mL of SPM culture was removed for each time point. Affinity-purification of Spo11-ProtA with Fast Flow IgG Sepharose beads (GE) and sequencing library preparation were as previously described (Thacker et al. 2014). Briefly, ∼200 fmol of Spo11 oligos were tailed with GTP at their 3′ ends using TdT, then ligated to a duplex DNA adapter with a 3′ CCCC overhang using T4 RNA ligase 2 (gift of S. Shuman, MSKCC). Complementary strands of Spo11 oligos were synthesized with Klenow fragment of DNA polymerase I, gel-purified on denaturing polyacrylamide gels, then tailed with GTP and ligated to a second duplex adapter. The resulting ligation products were amplified by low cycle numbers of PCR to add Illumina sequencing adapters, and PCR products were gel-purified and sequenced using Illumina HiSeq in the MSKCC Integrated Genomics Operation.
R version 3.1.1 (R Core Team 2016) and GraphPad Prism 6.0e were used for analyses. The Illumina reads were mapped to the yeast genome using a pipeline that was described previously (Supplemental Table S1; Pan et al. 2011; Thacker et al. 2014). Briefly, adapter sequences were removed from the 5′ and 3′ ends of the reads, and the remaining sequence was mapped to the S. cerevisiae genome (SGD version June 2008, that is, sacCer2) using gmapper-ls (2_1_1b) from the SHRiMP mapping package (Rumble et al. 2009). Reads that mapped uniquely to the genome (i.e., the vast majority of all reads) were used in subsequent analyses. Each map was normalized to the total number of reads mapping uniquely to a chromosome (discarding reads mapping to the 2-µ plasmid and mitochondrial DNA), and biological replicate maps were averaged.
A new list of hotspots was compiled to take advantage of current availability of a large number of deeply sequenced wild-type Spo11-oligo preparations (Thacker et al. 2014; Zhu and Keeney 2015; this study; X Zhu, M van Overbeek, S Keeney, unpubl.). These maps were generated using untagged, HA-tagged, FLAG-tagged, or Protein A-tagged Spo11, the latter two having been shown to be phenotypically indistinguishable from untagged with respect to DSB levels (Thacker et al. 2014). Hotspots were called on an averaged wild-type Spo11-oligo map using a previously described algorithm (Pan et al. 2011). Briefly, candidate hotspots were first identified as regions where the Spo11-oligo map smoothed with a 201-bp Hann window was >0.2 RPM per bp (i.e., 2.41 times the genome average Spo11-oligo density). Hotspot boundaries within these candidate locations were defined as the leftmost and rightmost coordinates to which at least one Spo11 oligo had mapped. Hotspots separated by <200 bp were merged. In this manner, we compiled 3908 hotspots. This serves as a definitive list for wild-type SK1 under these conditions (Supplemental Table S3).
In analyses presented in Figures 4B–D, 5B, 6D, and Supplemental Figure S4C, where we evaluated the log-fold change in the heat of hotspots, the heat in the wild type was a trimmed mean of all wild-type maps at all time points (two biological replicates at 4, 5, and 6 h) such that the lowest and highest value was omitted from the calculation of the average. We assumed a global increase in Spo11-oligo number of 1.5-fold for the tel1Δ 4-h time point, 2.3-fold for tel1Δ 5-h, 3.5-fold for tel1Δ 6-h, and 1.3-fold for tel1Δ zip3Δ 4-h, based on the difference in Spo11-oligo levels relative to wild-type at the indicated time points (Figs. 1B, 5D). For ratios, we added a small constant (+20 RPM) to numerator and denominator to prevent dividing by zero and to minimize sampling variability for ratios involving small denominators. To examine the spatial correlations between change in hotspot heats as shown in Figures 4D, 5B, and Supplemental Figure S4C, we calculated the Pearson's correlation coefficient between the log-fold change at each hotspot and the log-fold change for neighboring hotspots within a 5-kb window located from 5 to 200 kb away in steps of 5 kb. Randomized controls for this analysis were generated by randomly reassigning the log-fold change among hotspots within the same chromosome.
Data access
Sequence reads and compiled Spo11-oligo maps from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE84696. Source code for Spo11-oligo mapping is available in the Supplemental Material (Supplemental File 1) and at http://cbio.mskcc.org/public/Thacker_ZMM_feedback.
Acknowledgments
We thank A. Viale (MSKCC Integrated Genomics Operation) for sequencing and N. Socci (MSKCC Bioinformatics Core) for mapping the sequencing reads. We thank S. Shuman (MSKCC) for gifts of T4 RNA ligase 2 and M. Lichten (NCI) and R. Cha (Bangor Univ.) for strains and plasmids. This work was supported by the Howard Hughes Medical Institute and by National Institutes of Health (NIH) grants R01 GM058673 and R35 GM118092. Core facilities are supported by the NIH/NCI Cancer Center Support Grant P30 CA008748.
Author contributions: N.M. performed the experiments. N.M. and S.K. designed the project, analyzed and interpreted the data, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.213587.116.
- Received July 28, 2016.
- Accepted November 30, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








