
Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA
- Suresh Ramakrishna1,
- Abu-Bonsrah Kwaku Dad1,
- Jagadish Beloor2,
- Ramu Gopalappa1,
- Sang-Kyung Lee2 and
- Hyongbum Kim1,3
- 1Graduate School of Biomedical Science and Engineering/College of Medicine, Hanyang University, Seoul 133-791, Republic of Korea;
- 2Department of Bioengineering and Institute for Bioengineering and Biopharmaceutical Research, Hanyang University, Seoul 133-791, Republic of Korea
Abstract
RNA-guided endonucleases (RGENs) derived from the CRISPR/Cas system represent an efficient tool for genome editing. RGENs consist of two components: Cas9 protein and guide RNA. Plasmid-mediated delivery of these components into cells can result in uncontrolled integration of the plasmid sequence into the host genome, and unwanted immune responses and potential safety problems that can be caused by the bacterial sequences. Furthermore, this delivery method requires transfection tools. Here we show that simple treatment with cell-penetrating peptide (CPP)–conjugated recombinant Cas9 protein and CPP-complexed guide RNAs leads to endogenous gene disruptions in human cell lines. The Cas9 protein was conjugated to CPP via a thioether bond, whereas the guide RNA was complexed with CPP, forming condensed, positively charged nanoparticles. Simultaneous and sequential treatment of human cells, including embryonic stem cells, dermal fibroblasts, HEK293T cells, HeLa cells, and embryonic carcinoma cells, with the modified Cas9 and guide RNA, leads to efficient gene disruptions with reduced off-target mutations relative to plasmid transfections, resulting in the generation of clones containing RGEN-induced mutations. Our CPP-mediated RGEN delivery process provides a plasmid-free and additional transfection reagent–free method to use this tool with reduced off-target effects. We envision that our method will facilitate RGEN-directed genome editing.
The clustered, regularly interspaced, short palindromic repeat (CRISPR)-associated (Cas) system is an RNA-guided DNA cleavage system found in bacteria and archaea that serves as an adaptive immune system (Horvath and Barrangou 2010; Wiedenheft et al. 2012) and has been utilized as an attractive tool for targeted genome editing in various systems, including bacteria (Jiang et al. 2013), model organisms (Cho et al. 2013b; Dickinson et al. 2013; Friedland et al. 2013; Gratz et al. 2013; Hwang et al. 2013; Li et al. 2013a,c; Wang et al. 2013; Yang et al. 2013), plants (Li et al. 2013b; Nekrasov et al. 2013; Shan et al. 2013), and human cells (Cho et al. 2013a; Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013). Such RNA-guided endonucleases (RGENs) consist of the Cas9 protein and guide RNAs. The natural guide RNAs, which comprise CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), direct Cas9 to a specific genomic locus harboring the target sequence and can be replaced with a chimeric single-guide RNA (sgRNA) (Jinek et al. 2012). Although the short, truncated sgRNAs that were initially used experimentally were less efficient than the native crRNA–tracrRNA duplex (Cong et al. 2013), untruncated sgRNAs harboring the full-length tracrRNA tail are more efficient than the native duplex (Hsu et al. 2013) and are now predominantly used as guide RNAs. The Cas9 nuclease (derived from Streptococcus pyogenes) is targeted to genomic sequences that are complementary to the 20-nucleotide (nt) sgRNA guide sequence and are followed by a 5′-NGG protospacer adjacent motif. After sequence recognition, the Cas9 nuclease makes a double-strand break (DSB) at that specific genomic locus, enabling targeted genome editing.
Efficient genome editing using RGENs requires the successful delivery of guide RNA and Cas9 into cells. Cas9 and sgRNA are typically expressed in cells from plasmids or microinjected into cells as mRNA and RNA, respectively. Such methods require delivery tools such as transfection reagents or instruments for microinjection or electroporation, and the use of such plasmids in new cell types requires the selection of optimized promoters and/or codon optimization. Furthermore, plasmid-based delivery can be complicated with several potential problems: uncontrolled integration of the plasmid DNA into the host genome, unwanted immune responses (Hemmi et al. 2000; Wagner 2001) caused by bacterial sequences, and safety problems if the antibiotic-resistance gene is taken up by pathogenic bacteria. Thus, the development of methods for delivering Cas9 protein and sgRNA directly into cells, without additional tools, would greatly facilitate the use of RGENs. Among the programmable nucleases now used for gene disruption, zinc finger nucleases (ZFNs) (Gaj et al. 2012) and cell-penetrating peptide (CPP)–conjugated transcription activator-like effector nucleases (TALENs) (Liu et al. 2014) have been delivered directly to cells, but RGENs have not. Although recombinant Cas9 protein complexed with guide RNA has been microinjected into gonads and embryos to induce site-specific mutations in Caenorhabditis elegans, zebrafish, and mice (Cho et al. 2013b; Sung et al. 2014), direct delivery into cultured mammalian cells has not been achieved. Furthermore, RGEN delivery may be more complicated than ZFN delivery because both protein and RNA are involved.
Here we modified Cas9 protein and sgRNA by conjugating and complexing them, respectively, to CPP; treatment of cells with these reagents led to efficient gene disruptions in several human cell types with lower off-target mutation frequencies than plasmid transfection. Thus, we expect that our plasmid-free, CPP-mediated direct delivery system will facilitate RGEN-induced genome editing.
Results
Conjugation of CPP to a recombinant Cas9 protein
We postulated that introduction of a CPP (Regberg et al. 2013) into the Cas9 protein might enable its direct delivery into cells. However, when we genetically fused Cas9 to a CPP that consists of four Gly, nine Arg, and four Leu (Matsui et al. 2003; Ding et al. 2010), it was difficult to obtain purified protein in suitable quantities, which is in line with results with ZFNs (Gaj et al. 2012) and TALENs (Liu et al. 2014) fused with several CPPs. Thus, we sought to minimize genetic modification of Cas9 by adding a Cys residue at the C terminus (Fig. 1) to enable CPP conjugation after Cas9 expression. For Cas9 purification and detection, we also added histidine and hemagglutinin (HA) tags at the N and C terminus, respectively. To optimize protein isolation, we cultured Escherichia coli cells transformed with the Cas9-expressing plasmid at two different temperatures. A significant amount of IPTG-induced Cas9 protein was in the soluble fraction in the 30°C overnight culture group, whereas the majority of Cas9 protein was in the insoluble pellet in the 37°C 4-h culture group (Supplemental Fig. 1). From the soluble fraction obtained from the culture at 30°C, we purified Cas9 protein through nickel NTA column-mediated isolation followed by dialysis with 50-kDa cutoff membranes (Supplemental Fig. 2). We next determined whether this isolated protein is functional by using an in vitro cleavage assay, which revealed that the purified Cas9 protein efficiently cleaves a target sequence only in the presence of the sgRNA (Supplemental Fig. 3). To conjugate this Cas9 protein with a maleimide-linked CPP that consists of four Gly, nine Arg, and four Leu (4-maleimidobutyryl-4G9R4L; for brevity, hereafter m9R), we incubated the isolated protein with m9R; the free SH residue in the C-terminal cysteine of Cas9 reacts with the primary amine (−NH2) residue in m9R, forming a thioether bond. Excess, unconjugated m9R was removed using dialysis. Mass spectrometry showed that a suitable fraction of Cas9 protein was conjugated with m9R (Supplemental Fig. 4). Although the unmodified Cas9 also contains two cysteine residues (80C and 574C), this Cas9 failed to be conjugated to m9R when incubated with m9R (Supplemental Fig. 4), suggesting that the m9R was conjugated to the C-terminal cysteine residue that we introduced.
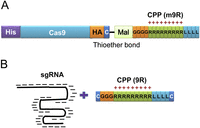
Schematic representation of (A) Cas9 protein conjugated to a cell-penetrating peptide (CPP) and (B) sgRNA complexed with a CPP. The single-letter codes for amino acids are used ([C] cysteine; [G] glycine; [R] arginine; [L] leucine). (His) Histidine tag; (HA) hemagglutinin tag; (Mal) maleimide; (sgRNA) single-guide RNA.
Complexing sgRNA with CPP
To facilitate delivery of sgRNA, the other RGEN component, into cells, we complexed sgRNA (negatively charged) with a similar CPP consisting of one Cys, three Gly, nine Arg, four Leu, and one Cys (for brevity, hereafter 9R; positively charged) at different ratios (Fig. 1). An electrophoretic mobility shift assay revealed that 9R completely retarded the sgRNA at weight ratios of 5 and higher (Fig. 2A), indicating that 9R can condense sgRNA. An ethidium bromide (EtBr) exclusion assay also showed that 9R can decrease the fluorescence intensity of an EtBr:sgRNA complex in a dose-dependent manner with 77% and 85% reduction at weight ratios of 5 and 10, respectively (Fig. 2B), corroborating that 9R condenses sgRNA. Dynamic light scattering (DLS) showed that the average particle size decreased as the 9R:sgRNA weight ratio increased, reaching 408 and 344 nm at ratios of 5 and 10, 1.9- and 2.3-fold smaller than that at a ratio of 2.5, suggestive of tighter complex formation (Fig. 2C). Because the overall positive charge on the surface of the nanoparticle facilitates cellular uptake via interaction with the negatively charged cell membranes, zeta potential was also measured using DLS; a positive charge of +13 mV was observed at a weight ratio of 2.5 and increased to +29 mV as the ratio of 9R:sgRNA increased (Fig. 2D). Scanning electron microscopy revealed that 9R condensed sgRNA into nanoparticles with an average size of ∼500 nm (Fig. 2E). Taken together, these results show that 9R and sgRNA form condensed, positively charged nanoparticles, possibly facilitating the delivery of sgRNA into cells.
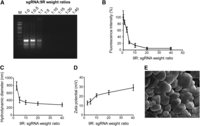
Physiochemical characterization of the sgRNA:9R complex. (A–D) A constant amount of sgRNA was complexed with various amounts of 9R. (A) Electrophoretic mobility shift assay. sgRNA was complexed with 9R at various weight ratios and subjected to electrophoresis. (B) Ethidium bromide (EtBr) exclusion assay. The reduction in EtBr fluorescence of sgRNA by complex formation with 9R at various weight ratios was measured. The relative fluorescence intensity is expressed as the percentage of the fluorescence of the uncomplexed sgRNA–EtBr sample. Data represent the average ± SD (n = 3). (C,D) Particle size distribution (C) and surface charge distribution (D) of the complex at different 9R:sgRNA weight ratios were determined using dynamic light scattering (DLS). Data represent the average ± SD (n = 3). (E) Representative scanning electron microscopic image of sgRNA:9R nanoparticles. Scale bar, 500 nm.
Human cell–based episomal reporter assay to monitor Cas9-m9R and sgRNA:9R function
To investigate whether Cas9-m9R and sgRNA:9R can cleave a target sequence within human cells, we used a reporter assay that involves an RFP-GFP reporter plasmid containing the RGEN target sequence between the RFP and GFP genes (Fig. 3A). Because the GFP gene is fused to the RFP gene out-of-frame, only RFP, but not GFP, is expressed by the reporter. If nuclease-induced DSBs are generated in the target sequence of the reporter, nonhomologous end joining–mediated DSB repair can elicit the formation of small insertions and deletions (indels), which can lead to functional GFP expression through frame-shift in the GFP gene. HEK293T cells were transfected with the reporter plasmid and treated three times sequentially with Cas9-m9R and sgRNA:9R, targeting the CCR5 gene. Flow cytometry showed that the average frequency of GFP-expressing cells in this population was 6.1%, which is comparable to the frequency when cells were transfected with plasmids encoding Cas9 and sgRNA (average frequency, 7.2%) (Fig. 3B). As expected, GFP expression was not observed in negative controls, which included cells only transfected with reporters and those only treated with Cas9-m9R. These data suggest that Cas9-m9R and sgRNA:9R can enter human cells and cleave target DNA sequences.
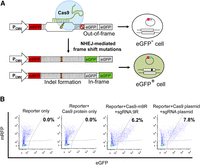
Cas9-m9R and sgRNA:9R-mediated mutagenesis of an episomal target sequence. (A) Schematic representation of the cell-based reporter assay. (B) Representative flow cytometry of reporter-transfected cells sequentially treated with Cas9-m9R and sgRNA:9R three times or those transfected with plasmids encoding Cas9 and sgRNA as the positive control. The cells were analyzed 3 d after the first treatment with Cas9-m9R and sgRNA:9R or transfection with plasmids encoding Cas9 and sgRNA. The percentages of GFP+ cells are indicated.
Endogenous gene disruption in human cells by treatment with Cas9-m9R and sgRNA:9R
To investigate whether Cas9-m9R and sgRNA:9R can edit endogenous genes in human cells without the use of additional delivery tools, we treated HEK293T cells with the two components, in this case with an sgRNA targeting the CCR5 gene, either sequentially or simultaneously. To detect the presence of indels at the target site, we treated amplified DNA fragments representing the target region with mismatch-sensitive T7 endonuclease 1 (T7E1); this assay showed a mutation frequency of 8.7% or 14% in the CCR5 gene after three rounds of sequential or simultaneous treatment, respectively (Fig. 4A). In all following experiments, cells were treated with Cas9-m9R and sgRNA:9R simultaneously unless otherwise specified. Sequencing of the PCR amplicons from the sequential treatment experiment showed that 8.6% of the target sequences were mutated (Fig. 4B), corroborating the T7E1 assay result. In addition, because Cas9 forms a ribonucleoprotein complex with sgRNA (Cho et al. 2013b), we postulated that Cas9 cell entry might promote sgRNA entry. We therefore treated HEK293T cells with Cas9-m9R and sgRNA without 9R. However, neither simultaneous treatment with the sgRNA:Cas9-m9R complex nor sequential treatments with Cas9-m9R and sgRNA achieved targeted genome modification. We speculate that mixing Cas9-m9R with the negatively charged sgRNA might neutralize the positive charge of the CPP, potentially blocking CPP-mediated cell entry of Cas9-m9R. Furthermore, as expected, no mutations were obtained in control cells treated with unmodified Cas9 and sgRNA:9R, either. Our results show that both CPP conjugation to Cas9 and noncovalent complex formation of the CPP with sgRNA are required for genome editing.
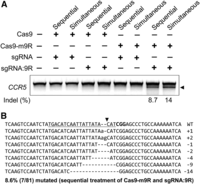
Endogenous gene disruption through CPP-mediated delivery of Cas9 protein and guide RNA. HEK293T cells were either sequentially or simultaneously treated with Cas9-m9R and sgRNA:9R three times. (A) Mutations detected by the T7E1 assay 3 d after the first treatment. The arrow indicates the expected position of DNA bands cleaved by T7E1. Mutations frequencies (indel [%]) were calculated from the band intensities. (B) DNA sequences of the CCR5 wild-type (WT) and mutant clones. The target sequence complementary to sgRNA is underlined. The protospacer adjacent motif is shown in bold. The cleavage site is indicated by an arrowhead. The column on the right indicates the number of inserted or deleted bases.
Factors that affect the genome editing efficiency of Cas9-m9R and sgRNA:9R
We next determined factors that affect the gene disruption efficiency of Cas9-m9R and sgRNA:9R. We first tested the effects of repeated treatments on HEK293T cells and found that this practice enhanced the mutation frequency, as shown by the T7E1 assay (Fig. 5A). The T7E1 assay revealed that repeated treatment of cells with Cas9-m9R and sgRNA:9R with known off-target activities increased off-target as well as on-target mutations (Supplemental Fig. 5). In all the following experiments, cells were treated daily with Cas9-m9R and sgRNA:9R (one treatment per day) for 3 d, and analysis was performed 3 d after the first treatment unless otherwise specified. Next, in experiments that involved treating cells with Cas9-m9R and sgRNA:9R twice, we determined the effect of the sgRNA:9R weight ratio on mutation frequencies, which we found to be similar in the tested range from 1:3 to 1:40 (Fig. 5B). However, cell viability decreased as the weight ratio of 9R increased, starting at a 1:20 (sgRNA:9R) weight ratio. We next investigated the effect of Cas9-m9R concentration and exposure time. The T7E1 assay showed that increasing concentrations of Cas9-m9R up to 2.0 μM resulted in an increase in gene disruption efficiency (Fig. 5C), whereas extending the incubation time >1 h did not further increase the mutation frequency (Fig. 5D). Incubating the cells at 30°C instead of 37°C after the Cas9-m9R and sgRNA:9R treatments mildly increased the gene disruption efficiency (Fig. 5E). All of these results are in line with those previously obtained with ZFNs (Gaj et al. 2012).

Factors affecting the genome editing efficiency of Cas9-m9R and sgRNA:9R. HEK293T cells were sequentially treated with Cas9-m9R and sgRNA:9R targeting the ABCC11 or CCR5 genes under various conditions, and the resulting mutation frequencies were determined using the T7E1 assay. The arrows indicate the expected position of DNA bands cleaved by T7E1. (A) Multiple treatments with Cas9-m9R and sgRNA:9R increase the mutation frequency. (B) The effect of sgRNA:9R ratios on the mutation frequency. The cells were treated twice with 1 μM Cas9-m9R and 10 μg/mL sgRNA complexed with 9R at various weight ratios. (C) Increasing the Cas9-m9R concentration increases the mutation frequency. The cells were simultaneously treated with various concentrations of Cas9-m9R and 10 μg/mL sgRNA complexed with 9R at a weight ratio of 1:5; this process was repeated three times. (D) The effect of exposure time on the mutation frequency. The cells were sequentially treated with 1.5 μM Cas9-m9R and 10 μg/mL sgRNA complexed with 9R at a weight ratio of 1:5 three times every day for the indicated exposure time per day. The exposure time was controlled by changing the media. (E) Incubating the cells at 30°C instead of 37°C mildly increases the mutation frequencies. Here, the cells were simultaneously treated three times with 1 μM Cas9-m9R and 10 μg/mL sgRNA complexed with 9R at a weight ratio of 1:5.
Application of Cas9-m9R and sgRNA:9R to several human cell types
We also tested whether this protein–RNA delivery method is applicable to other cell types. The T7E1 assay showed that treating HEK293T, HeLa, and NCCIT (a human pluripotent embryonal carcinoma cell line) cells with Cas9-m9R and sgRNA:9R resulted in mutation frequencies of 16%, 5.5%, and 2.7% at the CCR5 locus (Fig. 6A). Because genome editing in primary cells and stem cells has possible therapeutic applications, we also tested our method in these cell types. The T7E1 assay revealed that the treatment of human dermal fibroblasts and human embryonic stem cells (H9) with Cas9-m9R and sgRNA:9R led to mutation frequencies of 8.4% and 2.3%, respectively, at the CCR5 locus (Fig. 6B).
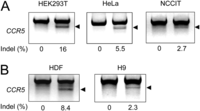
Endogenous gene disruption through CPP-mediated delivery of Cas9 protein and guide RNA in several cell lines. Cell lines such as HEK293T (human embryonic kidney cells), HeLa (human cervical cancer cell line), and NCCIT (human embryonal carcinoma cell line) cells (A), and primary cells such as human dermal fibroblasts (HDF) and human embryonic stem cells (B), were treated with Cas9-m9R and sgRNA:9R once per day for 3 d. The gene disruption efficiencies were determined using the T7E1 assay 3 d after the first treatment. The arrow indicates the expected position of DNA bands cleaved by T7E1.
Reduced off-target cleavage effects of Cas9-m9R and sgRNA:9R
When we checked the off-target effects of Cas9-m9R and sgRNA:9R, again using the sgRNA targeting the CCR5 gene, we did not observe any detectable off-target mutations (Supplemental Fig. 6). However, no off-target mutations have been reported in previous studies using this sgRNA (Cho et al. 2013a, 2014). Thus, we next evaluated off-target effects using sgRNAs that do have reported off-target activities (Cho et al. 2014; Fu et al. 2014). The off-target mutation frequencies in cells treated with Cas9-m9R and sgRNA:9R were drastically lower than those treated with plasmids, whereas the on-target mutation frequencies were comparable between the two groups; the ratio of on- to off-target activities was higher for direct delivery versus plasmid transfection by 2.2-fold (4.7/2.1), 4.1-fold (22/5.3), and more than 3.8-fold (more than 56/15) for the three sgRNAs tested (Fig. 7). This reduction in off-target effects associated with our CPP-mediated direct RGEN delivery is in line with the finding that direct ZFN protein delivery reduced off-target mutations (Gaj et al. 2012). We also compared gene disruption of the CCR5 and ABCC11 genes after CPP-mediated RGEN delivery versus plasmid-mediated delivery. The T7E1 assay revealed that the mutation frequencies associated with the two methods were similar or were only slightly lower in the CPP-based experiments (Supplemental Fig. 7). However, given that (1) the efficiency (on-target effect) and specificity (off-target effect) are strongly affected by Cas9 protein and guide RNA concentrations (Fu et al. 2013; Hsu et al. 2013; Pattanayak et al. 2013), and (2) it is challenging to exactly control these concentrations so that they are the same in the CPP-mediated direct delivery and plasmid-mediated expression systems, it is difficult to compare the efficiency and specificity of RGENs between the two methods directly in a quantitative manner.
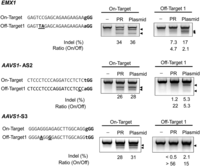
Reduced off-target effects of Cas9-m9R and sgRNA:9R. HEK293T cells were treated with Cas9-m9R and sgRNA:9R once per day for 3 d. Three days after the first treatment, the mutation frequencies at on- and off-target sites were determined using the T7E1 assay. The arrows indicate the expected position of DNA bands cleaved by T7E1. The protospacer adjacent motif is shown in bold. Mismatched bases are underlined.
Generation of mutant clones via CPP-mediated delivery of Cas9 protein and sgRNA
We next determined whether clones containing RGEN-induced mutations can be generated via this CPP-mediated RGEN delivery. To facilitate the selection of mutant clones (i.e., to reduce the number of clones that must be screened to obtain mutant clones), we used a surrogate magnetic reporter that expresses H-2Kk in the presence of programmable nuclease activity at the target sequence as previously described (Kim et al. 2013; Ramakrishna et al. 2014). After transfection of the reporter, HEK293T cells were treated with Cas9-m9R and sgRNA:9R. An H-2Kk–expressing cell population that was enriched with cells containing RGEN-induced mutations (Ramakrishna et al. 2014) was magnetically separated and seeded into 96-well plates at an average density of 0.25 cells per well. In 16 d, single cell-derived clones were selected via individual microscopic evaluation. Genomic DNA was isolated from the clones and subjected to the T7E1 assay and sequencing, which revealed that 8.3% (7/84) of clones contained RGEN-induced mutations in the CCR5 gene and that four out of the seven mutant clones had biallelic mutations (Supplemental Fig. 8).
Discussion
Here we showed the generation of endogenous gene disruptions by CPP-mediated direct delivery of Cas9 protein and sgRNA in several human cell types. This is the first study to show that CPP can enable direct delivery of Cas9 protein and sgRNA into cultured mammalian cells. CPP-mediated delivery of TALEN protein was reported while our manuscript was under review (Liu et al. 2014). In addition, a similar study in review at the same time reported highly efficient genome editing through electroporation-mediated delivery of purified (unmodified) Cas9 protein and sgRNA complex into human cells (Kim et al. 2014).
Genome editing using CPP-mediated direct RGEN delivery has several advantages. First, our approach circumvents potential problems associated with plasmid-based methods, which include uncontrolled integration of the plasmid DNA into the host genome and unwanted immune responses (Hemmi et al. 2000; Wagner 2001) caused by plasmid bacterial sequences. Furthermore, Cas9-m9R and sgRNA:9R do not contain the antibiotic-resistant genes that are included in plasmids, which are potentially unsafe if taken up by pathogenic bacteria in humans or animals. Second, our approach involves a simple, cloning-free process that is readily customized for genome editing at loci of interest. To deliver ZFNs and TALENs as protein, de novo protein synthesis for each protein is required. However, because Cas9 is common among all RGENs, additional subclonings and optimization of protein synthesis are not required for newly designed RGENs. As with restriction enzymes, a large amount of CPP-conjugated Cas9 could be manufactured by companies or nonprofit organizations and distributed to users. In the case of sgRNA, the generation of plasmids encoding sgRNA requires a subcloning step, whereas sgRNA can be prepared from templates generated by simple annealing of custom-ordered oligonucleotides without subcloning. Third, CPP-mediated delivery reduces off-target effects relative to plasmid-mediated delivery. This finding may be attributable to a reduction in RGEN working time caused by rapid degradation of directly delivered proteins and RNA; compatible with this idea, a previous report showed rapid ZFN degradation when ZFN proteins were delivered directly (Gaj et al. 2012). Recently, other methods to reduce RGEN-associated off-target effects, such as use of truncated sgRNA (Fu et al. 2014) and paired nickases (Ran et al. 2013; Shen et al. 2014), have been reported. Because these approaches reduce off-target effects by different mechanisms, combining our delivery method with these two methods may reduce off-target effects further.
In addition to these advantages, our approach has potentially important implications. Given that CPPs can enable protein and RNA delivery into cells in vivo (Nakase et al. 2013; Regberg et al. 2013) and are under clinical trials (Vasconcelos et al. 2013), our successful gene disruption in human cells in vitro using this approach might represent a major step toward potential development of “in vivo genome editing” using RGENs, potentially applicable to human patients. Furthermore, if advanced CPPs are developed, this approach can be improved as well.
During isolation of genomic DNA, cell lysates could potentially be mixed with the previously applied Cas9-m9R and sgRNA:9R. Given that mixing cell lysates with nucleases can lead to indel formation (Budman and Chu 2005), it needs to be determined whether functional Cas9 protein and guide RNA enter live cells. The generation of mutant clones via Cas9-m9R and sgRNA:9R treatment suggests that these factors disrupt endogenous genes in live cells after cell entry rather than by acting in the cell lysates.
In summary, we modified Cas9 protein and sgRNA by conjugating and complexing them, respectively, with CPPs, which enabled targeted gene disruption via the delivery of these two components into several human cell types without the use of any additional tools. We envision that this plasmid-free, CPP-mediated direct delivery system will facilitate RGEN-based genome editing and potentially will be applicable to in vivo genome editing.
Methods
Cell culture and transfection
HEK293T (human embryonic kidney cell line), HeLa (human cervical cancer cell line), NCCIT (human embryonal carcinoma cell line), and human dermal fibroblast cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum and a penicillin/streptomycin mix (100 U/mL and 100 μg/mL, respectively). The H9 (human embryonic stem cell line; WiCell Research Institute) cells were cultured in DMEM/F12 medium supplemented with 20% serum replacement, 1% nonessential amino acids, 1% penicillin–streptomycin, 0.1 mM β-mercaptoethanol, and 4 ng/mL basic fibroblast growth factor (all from Gibco) in the presence of mitomycin C–treated mouse embryonic fibroblasts as feeder cells. HEK293T cells were transfected using polyethyleneimine (linear; MW, ∼25,000; Polysciences)
Expression and purification of Cas9 protein
A sequence encoding Cas9 with a cysteine at the C terminus was prepared by PCR amplification using a previously described Cas9 plasmid (Cho et al. 2013a) as the template and then cloned into the pET28-(a) vector (Novagen, Merk Millipore), which includes a sequence encoding a His-tag at the N terminus. To induce Cas9 protein expression, E. coli BL21 cells transformed with the pET28-(a) vector encoding Cas9 were cultured overnight at 30°C in the presence of 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG; Promega). Cells were collected by centrifugation and lysed by sonication (40% duty, 10-sec pulse, 30-sec rest, for a total of 10 min, on ice) in a lysis buffer (20 mM Tris-Cl at pH 8.0, 300 mM NaCl, 20 mM imidazole, 1× protease inhibitor cocktail, 1 mg/mL lysozyme). After centrifugation at 20,000g for 20 min at 4°C, the Cas9 protein was purified from the soluble fraction by using a column containing Ni-NTA agarose resin (Qiagen) and an AKTA prime instrument (AKTA prime; GE Healthcare) at 4°C. The column-bound protein was eluted with an elution buffer (20 mM Tris-Cl at pH 8.0, 300 mM NaCl, 250 mM imidazole, 1× protease inhibitor cocktail) and dialyzed against a storage buffer (50 mM Tris-HCl at pH 8.0, 200 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 20% glycerol). The purity of eluted proteins was analyzed using SDS-PAGE, and the protein concentration was quantitated using the Bradford assay (Bio-Rad).
In vitro DNA cleavage assay
Purified Cas9 protein (50 nM) was incubated with supercoiled plasmid DNA (300 ng) containing the target sequence and sgRNA (50 nM) in NEB buffer 3 in a reaction volume of 20 μL for 1 h at 37°C. Digested DNA was analyzed by electrophoresis on a 0.8% agarose gel.
sgRNA preparation
RNA was in vitro transcribed through run-off reactions by T7 RNA polymerase. Templates for sgRNA transcription were generated by annealing and extension of two complementary oligonucleotides (Supplemental Table 1). Transcribed RNA was resolved on an 8% denaturing urea-PAGE gel. RNA was recovered in nuclease-free water followed by phenol:chloroform extraction, chloroform extraction, and ethanol precipitation. Purified RNA was quantified by spectrometry.
Conjugation of m9R to the Cas9 protein
The conjugation was carried out by drop wise mixing of 1 mg Cas9 protein (2 mg/mL) with 50 μg 4-maleimidobutyryl-GGGRRRRRRRRRLLLL (for brevity, m9R; 2 mg/mL; Peptron) in PBS (pH 7.4) followed by incubation on a rotator for 2 h at room temperature. To remove unconjugated m9R, the samples were dialyzed against DPBS (pH 7.4) for 24 h at 4°C using 50-kDa molecular-weight cutoff membranes. Cas9-m9R protein was collected from the dialysis membrane, and the protein concentration was determined using the Bradford assay (Bio- Rad).
Preparation and characterization of the sgRNA:9R complex
sgRNA (1 μg) in 1 μL of deionized water was gently added to the C3G9R4LC peptide (for brevity, 9R) in sgRNA:peptide weight ratios that ranged from 1:2.5 to 1:40 in 100 μL of DPBS (pH 7.4). This mixture was incubated for 30 min at room temperature and diluted 10-fold using RNase-free deionized water. The hydrodynamic diameter (size) and zeta-potential (charge) of the formed nanoparticles were measured using DLS (Zetasizer-nano analyzer ZS; Malvern Instruments). For the electrophoretic mobility shift assay, sgRNA (1 μg) samples complexed with the 9R peptide at various weight ratios were loaded onto 2% agarose gels containing EtBr and subjected to electrophoresis. For the EtBr exclusion assay, 1.5 μg EtBr in 100 μL of DPBS (pH 7.4) was added to sgRNA (1 μg) complexed with the 9R peptide at various weight ratios and incubated for 15 min. The fluorescence was determined using a UV/fluorescence reader (SpectraMax M2; Molecular Devices) at an excitation wavelength of 544 nm and emission wavelength of 590 nm and normalized to that of free sgRNA. The surface morphology of a sgRNA:9R complex at a weight ratio of 1:5 was determined using a scanning electron microscope (Nova Nano SEM 450; FEI) at a voltage of 10 kV.
Episomal reporter assay
Flow cytometric reporter plasmids containing an RGEN target sequence from the CCR5 gene were constructed as previously described (Kim et al. 2011). The episomal reporter assay was performed as previously described (Kim et al. 2011; Cho et al. 2013a; Ramakrishna et al. 2013). Briefly, at 24 h after reporter plasmid transfection, HEK293T cells were treated with Cas9-m9R and sgRNA:9R as described above. The cells were analyzed using a flow cytometer (FACSAria II; BD Biosciences). Cells transfected with reporters alone were used as an analysis control (Kim et al. 2011).
Cas9-m9R protein and sgRNA:9R treatments
One day after plating, cells were washed with Opti-MEM and treated with Cas9-m9R and sgRNA:9R either sequentially or simultaneously. The sgRNA:9R complex was formed by incubation of 10 μg of sgRNA and 30–50 μg of 9R peptide in 250 μL (for sequential treatment) or 100 μL (for simultaneous treatment) of Opti-MEM medium at room temperature for 30 min. For sequential treatment, cells were treated with the sgRNA:9R complex for 4 h at 37°C, washed with Opti-MEM medium, and treated with 250 μL Cas9-m9R (2 μM) for 2 h at 37°C. For simultaneous treatment, 150 μL Cas9-m9R (2 μM) protein was mixed with 100 μL sgRNA:9R (10:50 μg) complex, and the mixture was applied to the cells for 4 h at 37°C. Cells were simultaneously treated with Cas9-m9R and sgRNA:9R unless otherwise specified. After either treatment, cells were incubated at 30°C in serum-containing complete media until additional treatments the following day. Cells were treated three times and analyzed 3 d after the first treatment unless otherwise specified.
T7E1 assay
The T7E1 assay was performed as previously described (Kim et al. 2009, 2011; Ramakrishna et al. 2013). Briefly, after the isolation of genomic DNA using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions, the region of DNA containing the nuclease target site was nested PCR-amplified using primers listed in Supplemental Table 1. The amplicons were denatured by heating and annealed to form heteroduplex DNA, which was treated with 5 units of T7E1 (New England Biolabs) for 15–20 min at 37°C, and analyzed using 2% agarose gel electrophoresis. Mutation frequencies were calculated as previously described based on the band intensities using Image J software and the following equation (Guschin et al. 2010): mutation frequency (%) = 100 × [1 −(1 − fraction cleaved)1/2], where the fraction cleaved is the total relative density of the cleavage bands divided by the sum of the relative density of the cleavage bands and uncut bands.
Sequencing analysis
PCR products were cloned into the T-Blunt vector using a T-Blunt PCR Cloning kit (SolGent). Cloned products were sequenced using the T7 promoter primer (5′-TAATACGACTCACTATAGGG-3′).
Clonal analysis
Beginning 1 d after transfection with a magnetic reporter (Kim et al. 2013), HEK293T cells were treated daily with Cas9-m9R (2 μM) and sgRNA:9R (1:5) for 3 d. The day after the last treatment, the H-2Kk+ cells were magnetically separated as previously described (Kim et al. 2013) and seeded into 96-well plates at an average density of 0.25 cells/well. Sixteen days after the cell seeding, each well was microscopically evaluated, and single cell-derived round colonies were selected. Each selected colony was individually trypsinized and replated into 24-well plates. Four days after the subculture, genomic DNA was isolated from each clone and subjected to the T7E1 assay and sequencing as described above.
Mass spectrometry
The proteins were diluted in distilled water and concentrated using Centricon (Millipore) according to the manufacturer’s instructions. The samples were mixed with DHB matrix solution (10 mg/mL; 1:2, v/v) and dropped on to the MALDI plate. Mass analysis of the proteins was performed using an Ultraflextreme MALDI-TOF/TOF mass spectrometer (Bruker Daltonics) equipped with a HM2 TUVO high-mass detection system (CovalX) at the Korea Basic Science Institute, Daejon. Using FlexControl 3.0, the instrument was operated in a linear mode supplemented with accelerating voltage by HM2. Calibration was performed using a C150 Calibration kit (CovalX). Obtained mass spectra were analyzed using FlexAnalysis 3.0.
Acknowledgments
We thank Ho Min Kim (Korea Advanced Institute of Science and Technology), Je-Min Choi (Hanyang University), and Sung-Rae Cho (Yonsei University) for their helpful discussions. We thank Young Hee Nam (Flow Cytometer Core Center, Yonsei Medical Research Center), Kun Ho Chung (S.-K.L. laboratory), Soo Jung Kim, and Young-Ho Chung (Korea Basic Science Institute) for their technical assistance. This work was supported in part by the National Research Foundation of Korea (2011-0013568, 2008-0062287, 2011-0019357, 2013M3A9B4076544) and Converging Research Center Program (2013K000275) funded by the Ministry of Science, ICT, and Future Planning, and the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (H10C1740).
Author contributions: S.R., H.K., and S.-K.L. designed the experiments. H.K. and S.R. wrote the manuscript. S.R. and all the other authors performed the experiments.
Footnotes
-
↵3 Corresponding author
E-mail hkim1{at}hanyang.ac.kr
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.171264.113.
Freely available online through the Genome Research Open Access option.
- Received December 17, 2013.
- Accepted March 25, 2014.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








