
Nucleotide composition-linked divergence of vertebrate core promoter architecture
- Simon J. van Heeringen1,3,
- Waseem Akhtar1,3,
- Ulrike G. Jacobi1,
- Robert C. Akkers1,
- Yutaka Suzuki2 and
- Gert Jan C. Veenstra1,4
- 1 Radboud University Nijmegen, Department of Molecular Biology, Faculty of Science, Nijmegen Centre for Molecular Life Sciences, 6500 HB Nijmegen, The Netherlands;
- 2 Department of Medical Genome Sciences, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8562, Japan
-
↵3 These authors contributed equally to this work.
Abstract
Transcription initiation involves the recruitment of basal transcription factors to the core promoter. A variety of core promoter elements exists; however for most of these motifs, the distribution across species is unknown. Here we report on the comparison of human and amphibian promoter sequences. We have used oligo-capping in combination with deep sequencing to determine transcription start sites in Xenopus tropicalis. To systematically predict regulatory elements, we have developed a de novo motif finding pipeline using an ensemble of computational tools. A comprehensive comparison of human and amphibian promoter sequences revealed both similarities and differences in core promoter architecture. Some of the differences stem from a highly divergent nucleotide composition of Xenopus and human promoters. Whereas the distribution of some core promoter motifs is conserved independently of species-specific nucleotide bias, the frequency of another class of motifs correlates with the single nucleotide frequencies. This class includes the well-known TATA box and SP1 motifs, which are more abundant in Xenopus and human promoters, respectively. While these motifs are enriched above the local nucleotide background in both organisms, their frequency varies in step with this background. These differences are likely adaptive as these motifs can recruit TFIID to either CpG island or sharply initiating promoters. Our results highlight both the conserved and diverged aspects of vertebrate transcription, most notably showing co-opted motif usage to recruit the transcriptional machinery to promoters with diverging nucleotide composition. This shows how sweeping changes in nucleotide composition are compatible with highly conserved mechanisms of transcription initiation.
An essential step in the regulation of gene expression is the initiation of transcription, which involves the recruitment of the basal transcription machinery to the core promoter. The core promoter is defined as the ∼100-bp sequence around the transcription start site (TSS) that is minimally required for the assembly of the core transcription machinery and initiation of transcription. A number of core promoter elements have been identified, and their contribution to basal transcription has been documented (Smale and Kadonaga 2003; Juven-Gershon et al. 2006). Each of these motifs is present in only a subset of core promoters, and there are many core promoters that do not feature any of these motifs. Among the most studied motifs are the TATA box (Wasylyk et al. 1980; Mathis and Chambon 1981), the initiator (Inr) (Smale and Baltimore 1989), TFIIB recognition elements (BREs) (Lagrange et al. 1998; Deng and Roberts 2005), and the downstream promoter element (DPE) (Kadonaga 2002).
Recent large-scale promoter analyses have shown that the Inr is the most prevalent motif in the Drosophila and mammalian promoters, whereas the TATA box is present in 10%–20% of the promoters, most of which represent tissue-specific promoters with precise TSSs (Gershenzon et al. 2006; Sandelin et al. 2007). These core promoter motifs are specifically recognized by the components of the basal transcription machinery. TATA box binding proteins and TFIIB bind to the TATA box and the BREs, respectively, whereas TAF subunits of TFIID interact with the Inr and the DPE (Smale and Kadonaga 2003; Jallow et al. 2004). These core promoter motifs work cooperatively and exhibit synergy with each other (Juven-Gershon et al. 2008). Other core promoter elements have also been identified, including the motif 10 element (MTE) (Lim et al. 2004) and the X core promoter element (XCPE) 1 and 2 (Tokusumi et al. 2007; Anish et al. 2009). Recent computational analyses have also identified a number of other sequence elements that cluster in promoters (FitzGerald et al. 2004; Xie et al. 2005; Carninci et al. 2006; FitzGerald et al. 2006; Gershenzon et al. 2006; Vardhanabhuti et al. 2007; Frith et al. 2008; Tharakaraman et al. 2008; Yokoyama et al. 2009).
The TATA box is the only known core promoter element that is conserved from yeast to human. The DPE and Inr elements are shared between human and fly, although the fly Inr has a stricter consensus than does the human Inr. On the other hand, the DCE and XCPE1 motifs have only been identified in human promoters, indicating that core promoter elements have different representations in different species. This raises the question how promoter sequences compare among vertebrates. Up to now, most genome-wide promoter studies have focused on mammalian promoters, human and mouse in particular. To gain more insight into vertebrate promoter architecture, we decided to systematically compare Xenopus tropicalis and human core promoters. The draft genome of the Western clawed frog, X. tropicalis, an important model organism for vertebrate development, has recently been published. Xenopus is phylogenetically well positioned to compare to other vertebrates, and its genome shows significant long-range synteny with the human genome (Hellsten et al. 2010).
To date there is no genome-wide data set of promoter sequences available for Xenopus; therefore, such a collection needs to be determined in order to perform a comparative analysis of the human and amphibian promoter motifs. In addition, a robust motif finding and comparison pipeline needs to be established. The identification of enriched sequence motifs from a set of sequences is computationally complex, and a variety of tools and techniques have been developed to deal with this problem (for reviews, see Das and Dai 2007; Zhang 2007). However, performance of these methods remains poor, especially when used on eukaryotic sequences (Tompa et al. 2005). It has been suggested that combining different computational techniques, rather than focusing on a single method, should improve the effectiveness of motif prediction (Hu et al. 2005). Indeed, several so-called ensemble methods have been shown to perform better than their individual components (Carlson et al. 2007; Wijaya et al. 2008).
Ensemble methods that combine different de novo methods generally identify multiple redundant motifs. These highly related motifs need to be clustered to remove this redundancy. Transcription factor binding elements such as core promoter motifs are commonly represented as matrices that reflect the frequency of each nucleotide at every position in the motif, the position frequency matrix (PFM). One relatively straightforward approach to combine the results of different methods in an ensemble approach is to cluster the PFMs; however, this demands a sensitive motif similarity metric. Various metrics have been proposed (Mahony et al. 2007), but one important aspect that most of these scoring systems do not take into account is the relative importance of the individual motif positions. Specifically, positions with nucleotide frequencies close to the background have a similar contribution to the score, as do well-conserved, important positions that show a preference toward a single nucleotide. We propose a similarity metric, the weighted information content (WIC) score, that incorporates the relative entropy or information content (IC) (Shannon 1948; Schneider and Stephens 1990) of the motif positions into the comparison. This metric compares favorably to existing methods.
In order to compare the core promoter structure between Xenopus and human, we have obtained a collection of X. tropicalis TSSs by TSS-seq, a deep-sequencing-adjusted method to determine the 5′ ends of capped transcripts (Tsuchihara et al. 2009) similar to the CAGE approach (FANTOM Consortium and Riken Omics Science Center 2009). To predict core promoter motifs using this TSS data set, we developed a de novo motif discovery pipeline that incorporates the new WIC motif similarity metric to cluster similar motifs. By using this pipeline, we have identified a number of sequence elements in Xenopus promoters, including motifs shared with mammals. Intriguingly, Xenopus promoters feature distinctly different nucleotide frequencies and sequence motifs around the TSS compared with those of human promoters. We highlight the different behavior of promoter motifs with respect to this nucleotide background and have identified several Xenopus-specific promoter motifs. The findings reported here reveal a nucleotide composition-linked plasticity of the core promoter architecture.
Results
Selection of TSSs
A key issue in the analysis of core promoter sequences is the reliability and the positional precision with which TSSs can be determined. By using RNA from oocytes and gastrula stage embryos, we obtained a high-quality collection of X. tropicalis TSSs by high-throughput sequencing of 5′ cap–specific transcripts (TSS-seq) (Tsuchihara et al. 2009). After oligo-capping, cDNA was synthesized, amplified, and sequenced using an Illumina Genome Analyzer. The reads were mapped to the Xenopus genome to obtain a data set of precise TSS coordinates. This resulted in a total of 2.5 million mapped positions.
As intra-exonic and other nonpromoter reads have been observed for CAGE data (Mercer et al. 2010), we used ChIP-sequencing to determine the genomic binding sites of the TATA-binding protein (TBP) for verification of the core promoter positions. TBP is a key factor in the assembly of the transcription preinitiation complex and is expected to bind to the core promoter. The location of the TBP reads relative to all annotated 5′ ends of genes is visualized in Supplemental Figure S1. This distribution clearly shows that the binding location of TBP is in the core promoter just upstream of the annotated 5′ end.
Figure 1, A and B, shows two examples of the TSS-seq data together with the ChIP-seq profile for TBP, as well as our previously published data for RNA polymerase II (RNAPII); the chromatin mark H3K4me3, associated with the TSS of actively transcribed genes; and RNA-seq (Akkers et al. 2009). Figure 1A shows a site of focused transcription initiation, while Figure 1B illustrates the dispersed initiation for the eif1ax gene. The expressed sequence tags (ESTs) at this locus support the broad pattern of TSSs uncovered by the TSS-seq reads (data not shown).
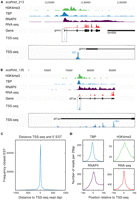
TSS-seq accurately defines transcription start sites. (A) Focused transcription initiation at the gmnn gene locus on scaffold_213, visualized using the UCSC Genome Browser. Shown are ChIP-seq profiles for (from top to bottom) H3K4me3 (green), TBP (blue), and RNAPII (purple), as well as RNA-seq data (red). The reads obtained by sequencing the 5′ end of oligo-capped transcripts (TSS-seq) are shown in the lower two tracks. The lower track shows a 90× magnification of the TSS-seq track. (B) Dispersed transcription initiation at the eif1ax gene locus on scaffold_135, visualized using the UCSC Genome Browser. (C) Histogram of the distance between the TSS-seq reads and the 5′ end of the closest EST, summarized for every TSS-seq read. (D) ChIP-seq profiles of TBP (blue, upper left), H3K4me3 (green, upper right), and RNAPII (purple, lower left), as well as RNA-seq (red, lower right), around the start position of TSS-seq reads.
As has been previously demonstrated, the oligo-cap method is a reliable method for TSS identification (Tsuchihara et al. 2009). This is further illustrated by the average distance between the TSS-seq reads and the 5′ end of the closest EST (Fig. 1C), as well as the profiles of the ChIP-seq and RNA-seq data, all of which are associated with the TSS of genes and, indeed, show a profile that peaks sharply around the TSS-seq reads, as expected (Fig. 1D). Finally, to obtain a high-confidence set of TSSs, covered by multiple TSS-seq reads, we filtered all positions with at least 20 overlapping TSS-seq reads. To exclude possible reads outside TSS regions, these positions were intersected with TBP and H3K4me3 peaks (Akkers et al. 2009). In total, this resulted in a collection of 4183 TSSs (Supplemental Table S1). For interspecies comparison between Xenopus and human, we obtained a comparable collection of 5561 human TSSs based on the CAGE data (Carninci et al. 2006).
Together these data indicate that we have obtained a robust, high-confidence set of TSSs in X. tropicalis, which can be used for promoter motif discovery and analysis.
Systematic motif prediction and comparison
De novo prediction of eukaryotic regulatory elements remains a computational challenge, and no single method achieves high all-around accuracy (Tompa et al. 2005). However, different computational tools often show complementary behavior, and ensemble approaches that incorporate several tools show improvement over single methods (Tompa et al. 2005; Carlson et al. 2007; Wijaya et al. 2008). Therefore, we chose to use a number of different motif prediction tools to obtain a comprehensive collection of Xenopus core promoter motifs.
To reduce the large motif redundancy resulting from predictions of different methods, we developed a motif similarity metric, the WIC, based upon the IC (Shannon 1948; Schneider and Stephens 1990). The WIC score is a function of both the similarity of the two positions in terms of IC, as well as the similarity to the background nucleotide frequency (see Equations 1 and 2 in Methods), and compares favorably to other similarity metrics in several different benchmarks (Supplemental Figs. S2, S3; Supplemental Methods).
We implemented the WIC score and an iterative clustering approach into a de novo motif discovery pipeline (Fig. 2). This pipeline uses the provided sequence data to predict, as well as validate, de novo motifs and is composed of the following steps:
-
Split the data into two sets: a prediction and a validation set. The first set of sequences is used to predict motifs, while the second data set is used to independently determine the significance of the predicted motifs.
-
Predict motifs using four different de novo motif prediction algorithms: Weeder (Pavesi et al. 2004), MDmodule (Liu et al. 2002), MotifSampler (Thijs et al. 2001), and MEME (Bailey et al. 2009).
-
Filter by significance: All predicted motifs are filtered using a hypergeometric enrichment test on the validation data set, compared to a random set of sequences generated by a first-order Markov model (similar dinucleotide frequency).
-
Filter by positional bias: As we expect core promoter motifs to be significantly enriched close to the TSS, all significant motifs from step 3 are filtered to select for motifs with a positional bias in the core promoter area compared with the upstream sequence, based on the clustering factor (CF) (similar to in FitzGerald et al. 2006).
-
Cluster similar motifs: All significant motifs are clustered using the WIC similarity metric, to provide a final set of nonredundant motifs.
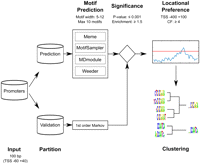
Schematic representation of the systematic de novo motif discovery pipeline. A set of input sequences is partitioned into two sets: a prediction set and a validation set. The prediction set is used as input for several different motif prediction algorithms. The validation set is used to produce a background set of random sequences generated with a first-order Markov model trained on the validation sequences. All predicted motifs are filtered for significance based on the hypergeometric distribution in the validation sequences compared with the random sequences. Only significant motifs with a positional bias, determined using the clustering factor, are kept. Subsequently this set of redundant motifs is clustered using an iterative procedure incorporating the new weighted information content (WIC) motif similarity score. To predict Xenopus promoter motifs, this pipeline was repeated 10 times.
Xenopus promoter elements
We proceeded to predict the core promoter elements in the Xenopus TSS data set using our comprehensive motif discovery pipeline (for a detailed description and parameters, see Methods). We first determined the CpG content of the promoters and divided them into CpG-rich and non-CpG subsets according to the overlap of the core promoter (−60 to +40 relative to the TSS), with CpG islands as predicted according to the method of Gardiner-Garden and Frommer (1987). The core promoter sequences of each subset, spanning −60 to +40 bp relative to the TSS, were subsequently used as input for the pipeline. All predicted motifs were further filtered for positional preference around the TSS. To obtain robust motifs, this procedure was repeated 10 times, and only motifs identified in at least half of the cases (five or more runs) were kept. All significant motifs were clustered, and a sequence logo (Schneider and Stephens 1990; Crooks et al. 2004) was generated for each cluster (Table 1; Supplemental Table S2). For comparison, we used the human TSSs determined by CAGE as the input for the same pipeline, which led to the identification of well-known promoter motifs (Supplemental Tables S3, S4). In addition, using the sequences of the TBP ChIP-sequencing peaks as input results in a similar set of well-defined promoter motifs (Supplemental Tables S5, S6).
Sequence motifs enriched in Xenopus promoters
We found 24 unique motifs that are enriched in the Xenopus core promoters (Table 1; Supplemental Table S2). In addition to the TATA box, several other well-known promoter elements were identified, some of which are known to be enriched in human but not Drosophila promoters (FitzGerald et al. 2004, 2006). These include the cAMP-response element (CRE), Ets, and Nrf-1 binding sites (Felinski et al. 2001; Scarpulla 2002; Buchwalter et al. 2004), as well as the YY1/Kozak consensus sequence, a motif that may act in both transcription and translation (Xi et al. 2007). The NF-Y motif, known to be enriched in human promoters (FitzGerald et al. 2004), also occurs in our Xenopus promoter data sets. A reverse complement of the SP1-like element (Zhao and Meng 2005) is also identified.
In addition to these known elements, we identified several unknown motifs with no unambiguous match to known promoter motifs. The relatively uncharacterized xt7 motif enriched in the Xenopus promoters was previously identified and called Clus1, because it clusters in human promoters (FitzGerald et al. 2004). It is a conserved motif, present in the promoters of many housekeeping genes (Wyrwicz et al. 2007), and shown to be important for the human HNRNPK and FBN1 promoters (Guo et al. 2008; Mikula et al. 2010). The identity of the protein that binds this element is unknown, although ZBED1 (zinc finger, BED-type containing 1) can bind to this element in the promoters of ribosomal genes (Yamashita et al. 2007). The motif is also identified in our analysis of human CAGE data (Supplemental Table S3).
One identified motif (xt17) resembles the upstream stimulatory factor (USF) binding motif (FitzGerald et al. 2004) but more closely matches the consensus of the helix-loop-helix transcription factor CBF1, a yeast protein involved in nucleosome positioning (Kent et al. 2004). Several newly identified motifs with a distinct consensus do not match any known motif (xt14, xt15, xt16, xt19, xt21, and xt22). Furthermore, several purine-rich motifs were identified (xt10, xt18, xt20, xt23, and xt24).
Some core promoter elements function in a specific orientation. To evaluate this, we analyzed the difference in abundance and positional distribution of the newly identified motifs between the plus strand and the minus strand. The motifs with a different distribution between the two strands are shown in Figure 3A. Actually, most motifs are not limited to a specific orientation (for all motifs, see Supplemental Fig. S4). Several novel Xenopus elements (xt15, xt16, x19, xt21, and xt22), however, are very specifically oriented, with almost no enrichment at the reverse orientation. The purine-rich motifs xt10 and xt24 also show an orientation-specific peak just downstream from the TSS.
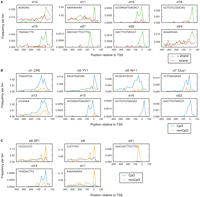
Differences in distribution of Xenopus promoter motifs. (A) Distribution of predicted Xenopus promoter motifs (Table 1) in the + and the − orientation. The matches in the region from −400 to +100 relative to the transcription start site (TSS) are binned at 20-bp resolution for the forward (green) and the reverse (red) orientation. Only motifs for which the distribution is different are shown (for all motifs, see Supplemental Fig. S4). (B) Predicted motifs containing a CpG dinucleotide are preferentially enriched in CpG-island promoters (blue) versus non-CpG promoters (orange). (C) Predicted motifs without a CpG dinucleotide preferentially enriched in either CpG-island promoters (blue) or non-CpG promoters (orange).
To evaluate the motif distribution in CpG-island and non-CpG promoters, we analyzed the differences between those two classes of promoters (Fig. 3B,C; Supplemental Fig. S5). As expected, all motifs containing at least one CG dinucleotide are much more prevalent in CpG-island promoters (Fig. 3B). This includes the known motifs YY1, CRE, and Nrf-1, as well as the newly identified motifs xt7, xt13, xt15, xt16, and xt22. One motif does not contain a CpG (Fig. 3C) but is still more abundant in CpG promoters compared with non-CpG promoters (xt19). Four motifs are preferentially enriched in non-CpG promoter sequences, two C-rich motifs (xt8 and xt9), one motif (xt11) that consists of a stretch of As, and xt21, which is only present in eight promoters.
Known core promoter elements
Some well-known human core promoter elements with degenerate consensus sequences were not found in the Xenopus promoters by our analysis, including the human Inr (consensus YYAnWYY), the human upstream BRE (BREu; consensus SSRCGCC), the downstream BRE (BREd; consensus STDKKKK), the human DPE (consensus RGWYV), the XCPE1 (consensus DSGYGGRASM) and XCPE2 (consensus VCYCRTTRCMY), and the MTE (consensus CSARCSSAACGS). We looked specifically for these elements in the Xenopus promoters (Fig. 4).
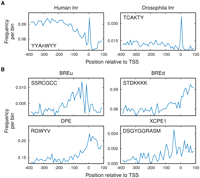
Distribution of known core promoter motifs in Xenopus promoters. The distribution of the positions of the motifs within a region from −400 to +100 relative to the TSS was determined by binning these positions at 10-bp resolution.
Both the human Inr and the stricter Inr element described as the Drosophila Inr are clearly present and precisely positioned at the TSS (Fig. 4A). The Drosophila Inr has a similar distribution in human promoters (Supplemental Fig. S6). As described for human promoters, the BREu and BREd elements are enriched upstream of and downstream from the TSS, respectively, in Xenopus (Fig. 4B). However, the frequency of BREu is much lower than that in human promoters (11% and 34%, respectively).
The DPE is present in Xenopus and enriched downstream from the TSS. The XCPE1 motif peaks broadly around the start site, but the frequency is relatively low. Less than 4% of the Xenopus promoters feature this element in the −60 to +40 region. This frequency and positional bias is similar to that observed previously in human promoters (Tokusumi et al. 2007). For XCPE2 and MTE, we did not find any clustering in Xenopus promoters (Supplemental Fig. S6).
It has been suggested that a downstream GC-rich sequence, also referred to as the gcg motif and similar to the YY1 motif, is the equivalent of the MTE in mammalian promoters (Juven-Gershon et al. 2008; Frith et al. 2008). The YY1 motif does indeed show clear enrichment downstream from the TSS of Xenopus promoters and is specifically positioned (peaking at approximately +10) (Fig. 3; Table 1).
Nucleotide frequencies differ between Xenopus and human promoters
To get more insight in the conservation of vertebrate core promoter evolution, we wanted to compare the motif distribution in the Xenopus and human promoters. One complicating matter in this comparative analysis is the substantially different sequence composition of warm- and cold-blooded vertebrates. Although the overall GC percentage of Xenopus and human genomes is similar (40.1% vs. 40.9%), GC-rich isochores are very scarce in Xenopus compared with mammals and birds, possibly due to the difference in body temperature (Costantini et al. 2009). Indeed, the relative nucleotide frequencies of the Xenopus and human promoter sets are clearly different (Fig. 5A). Additionally, the relative frequencies of most dinucleotides differ between the Xenopus and human promoters, although the shape of the dinucleotide distribution patterns around the TSS is mostly similar (Supplemental Fig. S7). As expected, this has an effect on the DNA stability, as predicted by the calculated 11-bp melting temperature (Supplemental Fig. S8).
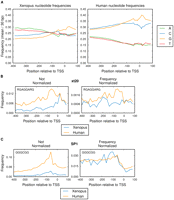
Normalization by nucleotide frequencies. (A) The nucleotide frequency (mean nucleotide fraction in a 30-bp window) is plotted for Xenopus (left panel) and human (right panel) promoters (A, green; C, blue; G, yellow; T, red). (B) The effect of nucleotide frequency normalization on the distribution of the xt20 motif. The distribution of the positions of the motif within a region from −400 to +100 relative to the TSS was determined by binning these positions at 20-bp resolution. Shown is the unnormalized (left panel) and normalized (right panel) distribution in Xenopus (blue) and human (orange) promoters. The frequencies in the right panel are normalized, based on the motif consensus, using the nucleotide frequencies in that bin. (C) The effect of nucleotide frequency normalization on the distribution of the SP1 motif.
The different promoter nucleotide frequencies between the two organisms have a large influence on motif distribution. While all predicted Xenopus motifs are overrepresented compared with the randomly generated sequences with a similar dinucleotide composition, we wondered how exactly the nucleotide background would influence the spatial motif distribution around the TSS. As the single nucleotide frequencies are significantly different between the human and Xenopus promoters (Fig. 5A), we can use these frequencies to normalize the motif distributions (for details, see Methods). For every bin, the motif frequency is normalized to the local frequency of the nucleotides present in the motif. This allows us to determine to what extent the positional bias of motifs follows the positional bias of their nucleotide frequencies.
The effect of this normalization is shown for two examples, xt20 (consensus RGAGGARG) (Fig. 5B) and the well-known human promoter motif SP1 (consensus GGGCGG) (Fig. 5C). While xt20 has a higher frequency in human promoters, it also strongly peaks around the TSS in the Xenopus promoters. The normalized frequencies, on the other hand, are much more similar, with less apparent positional bias around the TSS. The SP1 motif, known to be bound by the SP1 family of transcriptional activators (Zhao and Meng 2005), is highly enriched in the human promoters compared with Xenopus. Strikingly, this difference disappears when taking the nucleotide frequencies into account, suggesting that these relative differences are reflective of the general sequence composition characteristics of promoters in both species with no apparent selection against these trends.
To further facilitate this analysis, sequence motifs can be classified according to their distribution before and after normalization (Table 2). Promoter-enriched motifs that have a similar frequency and distribution in both species before normalization can be considered “conserved.” Some of these motifs show differences after normalization for nucleotide composition; other motifs in this group have a similar frequency also after normalization. In both cases, the positional biases toward the core promoter and the frequency are similar regardless of a changed nucleotide composition. A second group of motifs has different motif frequencies before normalization but is similar after correcting for the nucleotide background. Examples are the xt20 and SP1 motifs shown in Figure 5, B and C. These motifs seem to have changed their frequencies along with the nucleotide composition. Such a change in motif frequency could be adaptive, as their relative, normalized enrichment is similar between species, even though the nucleotide background is widely different. Finally, the last group is composed of motifs that are selectively enriched in one of the species both before and after normalization, and represents truly species-specific motif enrichment independent of nucleotide composition trends.
Motif classes
Comparison of Xenopus and human promoter elements
To extend this analysis, we implemented the nucleotide frequency normalization in a comprehensive comparison to identify the elements preferentially enriched in either the Xenopus or human core promoters. We combined 1794 human promoter sequence motifs predicted in several promoter studies (FitzGerald et al. 2004; Xie et al. 2005; Vardhanabhuti et al. 2007; Tharakaraman et al. 2008; Yokoyama et al. 2009) with the Xenopus motifs predicted in this study, and selected all positionally enriched motifs.
For a robust comparison between species, we also checked the positional enrichment of these motifs in completely independent validation data sets for Xenopus and human to ensure that the analysis is not biased toward TSS-seq or CAGE. For this purpose, we predicted TSSs based on a strict selection of spliced ESTs that overlap with the 5′ exons of known genes and share the same orientation relative to genomic sequence (Ohler et al. 2002; for details, see Supplemental Methods and Supplemental Fig. S9). By using this approach, we obtained 3867 X. tropicalis TSSs (Supplemental Table S7A) and a set of 6761 human TSSs (Supplemental Table S7B). These TSSs cluster just upstream of the 5′ end of annotated genes (Supplemental Fig. S9B) and are generally positioned within 20 bp of the actual start sites as determined by primer extension and analysis of the nearest TSS-seq read in 1997 promoters that are present in both promoter collections (Supplemental Fig. S9).
We selected all motifs with a positional bias in either the TSS-seq (Xenopus) or CAGE (human) data, with a consistent positional bias in the corresponding EST TSS data set. For all positionally enriched elements identified in the first step, the frequency in the core promoters was determined (Supplemental Table S8). We then selected the motifs that were more abundant (at least twofold difference) in either Xenopus or human, resulting in a set of 898 differential motifs (adaptive or species-specific) (cf. Table 2) versus 966 conserved motifs with a comparable promoter distribution between species (difference less than twofold). The differential motifs were clustered to obtain a set of nonredundant motifs. Finally, we normalized the motif frequencies of all clustered nonredundant motifs on the basis of the single nucleotide frequencies in the core promoter, and checked for preferential enrichment using the normalized frequency (Supplemental Table S9). This resulted in a set of 12 nonredundant motifs, preferentially enriched in either the Xenopus or human core promoters (Fig. 6C).
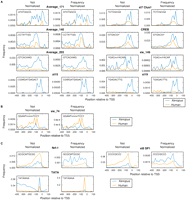
Comparison of Xenopus and human promoter elements. (A) Distribution of motifs specifically enriched in Xenopus promoters relative to human promoters. The distribution of the positions of the motifs within a region from −400 to +100 relative to the TSS was determined by binning these positions at 20-bp resolution in Xenopus (blue) and human (orange) promoters. The left panel for each motif shows the frequency per bin; the right panel shows the normalized frequency, based on the motif consensus, using the nucleotide frequencies in that bin. (B) Distribution of motifs specifically enriched in human promoters relative to Xenopus promoters. (C) Distribution of motifs that have a different species-preferential enrichment before and after nucleotide frequency normalization.
Eight motifs are preferentially enriched in Xenopus, showing relatively low enrichment in the human promoters, independent of nucleotide composition bias (Fig. 6A). These include the Clus1 motif (xt7) and the two novel motifs xt15 and xt19 identified in this study. The CRE motif also shows positional enrichment in human promoters but occurs with higher frequency in Xenopus promoters. This group includes four motifs predicted in human promoter studies (Average_140, Average_171, Average_203, xie_149), which, however, show much stronger enrichment in Xenopus promoters.
One motif appears to be preferentially enriched in human promoters independent of nucleotide composition bias (Fig. 6B): Motif 74 predicted by Xie et al. (2005).
The Nrf-1 and xt8 (a SP1 reverse complement) motifs and the TATA box (Fig. 6C) are special cases as they are partially conserved and partially adaptive with regard to nucleotide bias. Nrf-1 and xt8 are more prevalent in the human promoters but are more enriched in the Xenopus promoters after normalization. For the TATA box, the opposite differential enrichment before and after normalization is observed; this core promoter motif is more abundant in Xenopus promoters. For all three motifs, the Xenopus–human frequency differences are inverted after normalization, which is observed in both the TSS-seq/CAGE and the EST TSS data sets (cf. Fig. 6C and Supplemental Fig. S10). This shows not only that their frequency has changed significantly along with the nucleotide background but also that this change would have been larger had these motifs not been partially retained against the nucleotide composition trends. The frequency of the TATA box is higher in the more AT-rich Xenopus promoters, while the SP1 frequency is higher in the human promoters, which have a higher GC content (Figs. 5A, 6C). These results show a remarkable degree of plasticity in the well-known promoter motifs between vertebrates.
Discussion
In this study, we have analyzed the genome-wide core promoter architecture of Xenopus based on a set of approximately 4000 TSSs, which were obtained by oligo-capping in combination with high-throughput sequencing. We defined a high-confidence set of TSSs by combining this data set with an experimental ChIP-seq data set of the transcription initiation factor TBP. To be able to systematically predict eukaryotic motifs, we developed a motif prediction pipeline (Fig. 2). This pipeline uses an ensemble of different complementary motif prediction tools to avoid being dependent on a single computational approach (Tompa et al. 2005). To compare and cluster motifs, we developed a motif similarity metric based on the IC, the WIC score. This similarity metric compared favorably to current similarity metrics and performs well in an iterative motif clustering approach (Supplemental Figs. S2, S3).
A search for motifs enriched in the sequence surrounding the TSSs in Xenopus led to the identification of 24 significantly enriched motifs (Table 1; Supplemental Table S2). Though most of these are also present in the human promoters, there are some that are specific to Xenopus, indicative of both similarities and differences between the two species.
Some of the known core promoter motifs and the known CRE, Ets, Nrf-1, YY1, and NF-Y promoter motifs are also found in this study. All these positionally enriched elements are shared with mammals but not flies, most likely reflecting similarities in mechanisms of transcriptional regulation in vertebrates. The YY1 element plays a dual regulatory role in promoters (Xi et al. 2007). It can function in transcriptional regulation by recruiting YY1, but if present in the plus-orientation downstream from the TSS, it can act either as a Kozak consensus site in the transcribed mRNA for translation or as a binding site for YY1 (Xi et al. 2007). The Clus1 element, xt7, identified previously (FitzGerald et al. 2004; Wyrwicz et al. 2007; Yamashita et al. 2007; Guo et al. 2008; Mikula et al. 2010) is one of the most well-positioned motifs identified in the Xenopus promoters. The exact identity of this motif remains unclear, but the highly specific positioning relative to the TBP binding peak could indicate that it is bound by an important element in the core transcriptional machinery, warranting further investigation. The mammalian Inr (consensus YYAnWYY) (Smale and Baltimore 1989) is enriched in the Xenopus promoters; however, the more strict Drosophila Inr (consensus TCAGTY) Ohler et al. 2002) shows a much stronger positional enrichment in the Xenopus promoters.
It is known that CpG and non-CpG island promoters are structurally and functionally different from each other. CpG island promoters are associated with housekeeping genes, show a broad distribution of start sites, and seem to be particularly rapidly evolving in mammals (Carninci et al. 2006). There is also evidence for functional differences in the requirements of the basal transcription factors between the CpG and non-CpG island promoters (Denissov et al. 2007). Although the CpG dinucleotide content of the Xenopus promoters, and of the whole genome in general, is lower than that of homeothermic vertebrates (Costantini et al. 2009), we find that promoter elements are differentially enriched in these two classes of promoters. The C- or A-rich motifs are specific to the non-CpG promoters, whereas all predicted motifs with a CpG are enriched in the CpG island-containing promoters.
The single nucleotide frequencies are markedly different between the Xenopus and human promoters (Fig. 5A). The Xenopus promoter region is relatively AT-rich, whereas the human promoters are more GC-rich. In Xenopus, at and around the TSS, only G is enriched relative to the other nucleotides. The nucleotide composition has a significant impact on the distribution of some motifs. The TATA box has a higher frequency in Xenopus compared with the human promoters. However, when normalized, it shows significantly higher enrichment in the human promoters (Fig. 6C). The opposite is true for the SP1 motif. The strong positional enrichment around the TSS in the human promoters seems to be mostly a product of the nucleotide background, as the normalized motif frequency shows no such peak. This motif is bound by the transcriptional activator SP1 (Zhao and Meng 2005). SP1 has been shown to interact directly with several TFIID components, including TBP, and is essential for TFIID recruitment in the absence of a TATA box (Emili et al. 1994; Gill et al. 1994; Kaufmann and Smale 1994; Chiang and Roeder 1995). In addition, SP1 motifs not only recruit TBP via SP1 but also keep CpG islands methylation-free (Brandeis et al. 1994; Macleod et al. 1994). Nonmethylated CpG islands recruit Cfp1, which allows H3K4me3 deposition and promoter activity (Thomson et al. 2010). This raises the interesting possibility that the evolution of GC-rich promoters in some vertebrates, which seems to be particularly rapid in mammals (Carninci et al. 2006), may have driven the increased use of functional GC-rich motifs, such as SP1, to recruit the transcription machinery to these promoters, accompanied by a correspondingly reduced usage of the TATA box to recruit TFIID.
We identified two Xenopus-specific motifs (xt15, consensus GCGWGATGAGACT; xt19, consensus TGAGACTTG) in this study. These novel motifs match no known TF binding sites. Further investigation should clarify the role of these motifs and whether they can function as bona fide core promoter elements. In addition, two motifs were identified (xt7/Clus1 and CREB) that show a stronger enrichment in the Xenopus promoters compared with the human promoters.
In conclusion, this report represents the first analysis of a large set of amphibian core promoters and a first comparison with human core promoter elements. Although Xenopus promoters differ in nucleotide composition compared with human promoters, most of the known core promoter motifs are shared, indicating a similar vertebrate core promoter architecture. However, the distribution of some motifs is different. Motifs such as SP1 and the TATA box seem to be adapted to the local nucleotide background, whereas other motifs are strongly conserved, despite different nucleotide background frequencies. This may indicate that while there is a functionally conserved set of essential core transcription factors, the motif frequencies reflect adaptive changes in factor usage in response to a changing nucleotide composition. This allows for extensive cis-regulatory plasticity in the presence of a highly conserved transcription machinery.
Methods
Animal procedures
X. tropicalis embryos were obtained by natural mating, dejellied in 3% cysteine, and collected at the indicated Nieuwkoop-Faber stages.
RNA isolation and TSS-seq
X. tropicalis oocytes and embryos of stages 10–12 were collected, and total RNA was isolated using TRIzol and the QIAGEN RNeasy Kit. The oligo-capping and sequencing were performed as has been previously described (Tsuchihara et al. 2009). Briefly, 50 μg of purified total RNA was dephosphorylated with bacterial alkaline phosphatase, ligated with oligo-RNA (5′-AAUGAUACGGCGACCACCGAGAUCUACACUCUUUCCCUACACGACGCUCUUCCGAUCUGG-3′) using T4 RNA ligase, and cDNA was then synthesized using a random hexamer primer (5′-CAAGCAGAAGACGGCATACGANNNNNNC-3′) with Super Script II (Invitrogen). The cDNA was amplified by 20 cycles of PCR, and massive parallel sequencing was executed with a Genome Analyzer (Illumina) to obtain reads with 36 nt length. All reads were mapped to the X. tropicalis genome (Joint Genome Institute, assembly version 4.1) (Hellsten et al. 2010) using ELAND. All unmapped reads, or reads mapping to multiple positions, were discarded. To call TSSs, all positions with at least 20 overlapping reads were filtered to overlap with either a TBP peak or a H3K4me3 peak (Akkers et al. 2009).
TBP ChIP-sequencing
Embryos were collected at stage 12. Chromatin harvesting and ChIP using the α-TBP antibody (SL33) were performed as described previously (Jallow et al. 2004) with minor modifications: 12.5 μL of Prot A/G beads (Santa Cruz) were used, and during reversal of cross-linking, proteinase K was omitted from the buffer. Sequencing samples were prepared according to the manufacturer's protocol (Illumina). Briefly, adapter sequences were linked to the generated ChIP sample; the library was size selected (300 bp) and amplified by PCR. The subsequent sequencing was carried out on a Genome Analyzer (Illumina). All 35-bp reads were mapped to the X. tropicalis genome, Joint Genome Institute, assembly version 4.1 (Hellsten et al. 2010), using ELAND (GAPipeline, version 1.4, Illumina) allowing one mismatch. Peaks were called using MACS (Zhang et al. 2008) with a P-value cutoff of 10−7.
Data availability
The TSS-seq and ChIP-seq data have been submitted to the NCBI Gene Expression Omnibus (GEO) (Edgar and Barrett 2006) and are accessible through GEO Series accession no. GSE21482 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21482). Visualization tracks are available at http://www.ncmls.nl/gertjanveenstra.
WIC motif similarity score
The WIC score reflects the comparison of two motif columns and is based on two terms. The first term is an indication of how informative the motif positions are; the second is a measure of their difference. A position with a strong preference for a specific nucleotide will likely be more important for binding of the transcription factor to the DNA and therefore will be more informative. The score can be summarized as follows: WIC = Information − Difference. The WIC score will be higher for more informative positions, compared with positions that are not informative. Similarly, the WIC score will be lower for different positions and higher for more similar positions. The first term is based on the IC, while the second term expresses the differences between two positions similar to the IC, as detailed in the formulas below.
The IC of a specific motif position is defined as follows: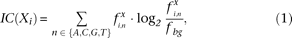 where Xi is the IC of position i of motif X,
where Xi is the IC of position i of motif X, 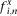 is the frequency of nucleotide n at position i, and fbg is the background frequency (0.25).
is the frequency of nucleotide n at position i, and fbg is the background frequency (0.25).
The WIC score of position i in motif X compared with position j of motif Y is defined as follows: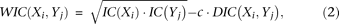 where c is a scaling constant and is a differential IC (DIC) defined in equation 3. The constant c is set to 2.5. This value was based on optimal performance in benchmarks of JASPAR data (Supplemental Fig. S3B) and was confirmed
using other benchmarks (Supplemental Fig. S3C,D).
where c is a scaling constant and is a differential IC (DIC) defined in equation 3. The constant c is set to 2.5. This value was based on optimal performance in benchmarks of JASPAR data (Supplemental Fig. S3B) and was confirmed
using other benchmarks (Supplemental Fig. S3C,D).
The DIC of position i in motif X and position j in motif Y is defined as follows: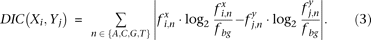 The WIC score of all individual positions in the alignment is summed to determine the total WIC score of two aligned motifs.
To calculate the maximum WIC score of two motifs, all possible scores of all alignments were calculated, and the maximum scoring
alignment was kept. Optionally an empirical P-value can be calculated based on the maximum WIC score and the length of the motif. This was done according to the method
of Sandelin and Wasserman (2004), based on simulated PFMs. Ten-thousand random PFMs were generated using the JASPAR website (http://jaspar.cgb.ki.se/).
The WIC score of all individual positions in the alignment is summed to determine the total WIC score of two aligned motifs.
To calculate the maximum WIC score of two motifs, all possible scores of all alignments were calculated, and the maximum scoring
alignment was kept. Optionally an empirical P-value can be calculated based on the maximum WIC score and the length of the motif. This was done according to the method
of Sandelin and Wasserman (2004), based on simulated PFMs. Ten-thousand random PFMs were generated using the JASPAR website (http://jaspar.cgb.ki.se/).
Motif clustering
Similar motifs were clustered using an iterative procedure. Pairwise comparisons were performed for all motifs using the WIC score. The two most similar motifs were merged, and an average motif was computed, weighted using the column frequencies of the PFMs. The pairwise scores of this new average motif to all other motifs were calculated, and the two most similar motifs are again merged. This procedure was repeated until the best-scoring alignment did not reach a predefined threshold (WIC, P ≤ 0.05).
Motif prediction on the Xenopus TSS data set
Motifs were predicted separately for CpG and non-CpG promoters and subsequently combined. The CpG and non-CpG sets of Xenopus promoters (−60 to +40 around the TSS) were split randomly in a prediction and a validation subset (each containing 50% of the sequences). The former subset was used to predict motifs using four de novo motif prediction tools: MEME (Bailey et al. 2009), MotifSampler (Thijs et al. 2001), Weeder (Pavesi et al. 2004), and MDmodule (Liu et al. 2002). Weeder performed generally well in a benchmark study (Tompa et al. 2005), while MEME and MotifSampler showed complementary behavior (Tompa et al. 2005). MEME, MDmodule, and MotifSampler were each used to predict 10 motifs for each of the widths between 5 and 12. We used the “medium” analysis setting for Weeder and the “zoops” distribution for MEME. Where possible we specified strand-specific motif prediction using only the plus strand relative to the promoter orientation. All other parameters were according to the default settings. The significance of the predicted motifs was determined by scanning the validation set, the remaining 50% of the promoter sequences not used for motif prediction, and a background set of random sequences generated according to a first-order Markov model, matching the dinucleotide frequency of the promoter sequences. P-values were calculated using the hypergeometric distribution with the Benjamini-Hochberg multiple testing correction (Benjamini and Hochberg 1995). Motifs with a P-value ≤ 0.001 and an absolute enrichment of at least 1.5-fold or greater compared with background were determined as significant. All significant motifs were passed through another level of filtering by looking at their enrichment compared with the surrounding sequences. To this end, the positions of these motifs were determined in the complete TSS promoter data set from −400 to +100 relative to the TSS. To determine if sequence motifs peak in the promoter region, a CF, similar to FitzGerald et al. (2006), was calculated. A local background mean (xmean) and standard deviation (σ) was calculated for the bins of length 20 between positions −400 and −250 relative to the TSS. The CF is calculated using the maximum bin value (xmax) between positions −250 and +50: CF = (xmax − xmean)/σ. The CF values were used to determine if a sequence motif is clustering in the promoter-proximal region. Only motifs with CF ≥ 4 were kept for further analysis. This whole prediction pipeline was repeated 10 times, and only motifs that were identified at least five times were kept.
Frequency of known motifs
The consensus sequences for known promoter elements were obtained for the human and Drosophila Inr (Juven-Gershon and Kadonaga 2010), BREu and BREd (Deng and Roberts 2006), DPE (Burke and Kadonaga 1996), XCPE1 (Tokusumi et al. 2007), XCPE2 (Anish et al. 2009), and MTE (Lim et al. 2004). These consensus sequences were converted to weight matrices, and the frequency was determined by scanning the whole set of core promoters (−60 to +40 around the TSS) with a strict cutoff of 0.95 of the score of the best possible match.
Nucleotide frequency normalization
First, the frequency of each motif was determined from −400 to +100 relative to the TSS and binned at 20-bp resolution. For
each of these 20-bp bins, the mean single nucleotide frequency was calculated. Subsequently, the motif frequency per bin was
normalized depending on the motif consensus.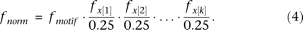
fmotif is the motif frequency for a specific bin, fx[1] is the nucleotide frequency (in that specific bin) of the nucleotide in position 1 of the motif consensus, fx[2] is the nucleotide frequency of the nucleotide in position 2 of the motif consensus, etc., and k is the motif length. If the consensus was a degenerate symbol, the sum of the frequencies of the individual nucleotides was used. For instance, the frequency of S (the IUPAC symbol for either a G or a C) is the frequency of G plus the frequency of C.
Comparison of human and Xenopus promoters
All human promoter motifs predicted in five studies (FitzGerald et al. 2004; Xie et al. 2005; Vardhanabhuti et al. 2007; Tharakaraman et al. 2008; Yokoyama et al. 2009) were retrieved and combined with the 24 Xenopus motifs determined in this study. The CF for each of these motifs was calculated for the human and Xenopus promoters, both in the predicted TSS set, as well as the validation set. All motifs with a consistent CF ≥ 4 in two independent TSS collections (Xenopus: TSS-seq and EST; human: CAGE and EST) were kept for further analysis. For each positionally enriched motif, the motif frequency was calculated for the human and Xenopus promoters in the region between −150 and +50 relative to the TSS (Supplemental Table S7). All motifs with a frequency of at least 1% in either the Xenopus or human promoters and at least a twofold difference in frequency between the Xenopus and human promoters were clustered. For all the clustered motifs, we determined the frequency, the nucleotide normalized frequency, and the CF (Supplemental Table S8).
Acknowledgments
This work was supported by NWO-ALW (Netherlands Organization for Scientific Research-Research Council for Earth and Life Sciences, grant no. 864.03.002) and NIH (National Institutes of Health, grant no. R01HD054356) with grants to G.J.C.V. and by the Higher Education Commission of Pakistan with a PhD fellowship to W.A. We thank R. Jurgelenaite and A.G. Uren for critical reading of this manuscript, R. Engels for help with frogs and animal care, and H.G. Stunnenberg, K.J. Françoijs, and E.M. Janssen-Megens for ChIP-sequencing support.
Footnotes
-
↵4 Corresponding author.
E-mail g.veenstra{at}ncmls.ru.nl.
-
[Supplemental material is available for this article. The sequence data from this study have been submitted to NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE21482.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.111724.110.
- Received June 14, 2010.
- Accepted December 29, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








