
Interactome mapping suggests new mechanistic details underlying Alzheimer's disease
- 1 Institute for Research in Biomedicine, Joint IRB-BSC Program in Computational Biology, 08028 Barcelona, Spain;
- 2 Max-Planck Institute for Molecular Genetics, 14195 Berlin, Germany;
- 3 Institució Catalana de Recerca i Estudis Avançats (ICREA), 08010 Barcelona, Spain
-
↵4 These authors contributed equally to this work.
Abstract
Recent advances toward the characterization of Alzheimer's disease (AD) have permitted the identification of a dozen of genetic risk factors, although many more remain undiscovered. In parallel, works in the field of network biology have shown a strong link between protein connectivity and disease. In this manuscript, we demonstrate that AD-related genes are indeed highly interconnected and, based on this observation, we set up an interaction discovery strategy to unveil novel AD causative and susceptibility genes. In total, we report 200 high-confidence protein–protein interactions between eight confirmed AD-related genes and 66 candidates. Of these, 31 are located in chromosomal regions containing susceptibility loci related to the etiology of late-onset AD, and 17 show dysregulated expression patterns in AD patients, which makes them very good candidates for further functional studies. Interestingly, we also identified four novel direct interactions among well-characterized AD causative/susceptibility genes (i.e., APP, A2M, APOE, PSEN1, and PSEN2), which support the suggested link between plaque formation and inflammatory processes and provide insights into the intracellular regulation of APP cleavage. Finally, we contextualize the discovered relationships, integrating them with all the interaction data reported in the literature, building the most complete interactome associated to AD. This general view facilitates the analyses of global properties of the network, such as its functional modularity, and triggers many hypotheses on the molecular mechanisms implicated in AD. For instance, our analyses suggest a putative role for PDCD4 as a neuronal death regulator and ECSIT as a molecular link between oxidative stress, inflammation, and mitochondrial dysfunction in AD.
Alzheimer's disease (AD) is a devastating neurodegenerative disorder characterized neuropathologically by the extracellular accumulation of amyloid-beta (Aβ) plaques, and the intracellular accumulation of hyperphosphorylated tau protein in the form of neurofibrillary tangles (NFTs). Unfortunately, and despite the recent advances in characterization of the disease (Bettens et al. 2010; Querfurth and LaFerla 2010), current medical treatments for AD are purely symptomatic and hardly effective (Citron 2010). Thus, the complete understanding of the molecular mechanisms underlying AD is paramount for the development of novel therapies able to modify the biology of the disease and efficiently fight the increase of AD with age in our ever-increasing life expectancy.
Although highly heritable, AD is a genetically complex disorder associated with multiple genetic defects either mutational or of susceptibility, making genetic analysis difficult (Bertram and Tanzi 2008). It is well established that mutations in the genes encoding amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) can lead to altered production of Aβ, which is sufficient to cause rare, early-onset (∼50 yr of age) familial forms of AD (Selkoe and Podlisny 2002). However, the vast majority of disease cases are of late onset (>65 yr of age), and this sporadic form of AD is widely believed to be influenced by a combination of genes that probably affect a variety of pathways involved in the production, aggregation, and clearance of Aβ (Selkoe and Podlisny 2002). Indeed, the ɛ4 allele of apolipoprotein E (APOE) has been considered a key genetic factor to play a role in the multifactor pathogenesis of AD (Raber et al. 2004), which accounts for ∼50% of late-onset AD. In addition, several other genetic risk factors have been identified (e.g., A2M, SERPINA3, LRP1, IL1A, TNF, ACE, BACE1, BCHE, CST3, MTHFR, GSK3B, NOS3), although their susceptibility implication in AD still remains unclear (Bertram et al. 2007). These genes probably converge on common pathogenic mechanisms that lead to disease predisposition and age of onset but, unfortunately, current strategies for genome association studies have not been able to identify candidate loci effectively, probably due to the highly complex disease traits (Bertram and Tanzi 2009).
Recent studies have shown that causative/susceptibility genes for many disease phenotypes often work together within the same biological module (Oti and Brunner 2007), be it a protein complex, a pathway, or a protein interaction sub-network, highlighting a strong link between protein connectivity and disease (Zanzoni et al. 2009; Pujol et al. 2010). Indeed, the number of interactions observed between disease-causing genes in several pathologies is often much higher than what would be expected by chance, and the discovery of unexpected relationships between apparently unrelated genes has emerged as a powerful tool for the identification of novel genes involved in complex diseases such as breast cancer (Pujana et al. 2007), Huntington (Goehler et al. 2004), schizophrenia (Camargo et al. 2007), or cerebral ataxias (Lim et al. 2006). In the particular case of AD, computational analyses showed that the integration of genetic information with physical and functional interaction data can be useful for prioritizing candidate genes (Krauthammer et al. 2004; Chen et al. 2006; Liu et al. 2006).
In this manuscript, we first explore whether, in the light of recent data, well-established AD-related genes are indeed highly interconnected. Based on the obtained results we set up an interaction discovery strategy to unveil and validate novel AD causative and susceptibility genes. Finally, we contextualize the discovered relationships in the global disease-associated network and formulate novel hypotheses that provide insights on the molecular mechanisms implicated in AD.
Results and Discussion
Identification of novel AD-related genes through interaction discovery experiments
It has been described that, in complex diseases, causative and susceptibility genes tend to be highly interconnected (Oti and Brunner 2007). This observation was shown to be true for the four major causative genes identified for AD (Chen et al. 2006; Liu et al. 2006). Based on this observation, we included in our set all 12 well-established AD causative/susceptibility genes (Table 1), that we name “seeds,” and checked the interconnectivity between them to see whether it was still significantly higher than expected. To do so, we computed the minimal distance between any pair of seed genes in the frame of the charted human interactome (see Methods). This measure, known as “shortest path length,” quantifies the connectivity degree between two given nodes in a network. We found that the shortest path between seed genes is 3.2, meaning that, on average, we need roughly three links (i.e., two intermediate proteins) to physically connect any two gene products within this set. To assess the statistical significance of this figure, we compared this result to two different reference distributions: one consisting of randomly picked sets of 12 proteins in the human interaction space (RND1, average shortest path = 4.7) and the second one, to avoid functional biases, composed by randomly picked disease-causing proteins belonging to different disorder classes (RND2, average shortest path = 4.9). In both cases, the average distance among AD-related genes was significantly shorter than that of the reference distributions (P-valueRND1 = 6.8 × 10−18; P-valueRND2 = 3.5 × 10−23), indicating that AD seed genes are indeed more interconnected than one would expect by chance.
Alzheimer's Disease genes
We next sought to exploit this finding to reveal novel genes that could be involved to the onset or progression of AD. This is, to identify those proteins that physically interact with AD seeds and that are located in susceptibility loci, as identified by genetic cross-linking experiments, or whose expression is dysregulated in AD patients. Accordingly, we defined an interaction discovery strategy to identify potential interactors of the already known AD-related genes (Table 1) in an adult brain (Fig. 1). From our initial list of 12 seed genes, we had to discard three (ACE, MPO, and SORL1) for which the open reading frames (ORFs) were not available. After converting the nine seeds into bait plasmids (see Methods for details), we carried out 45 yeast two-hybrid (Y2H) screens against an adult brain cDNA prey library (five replicates for each of the nine baits), which yielded 191 interactions between 151 distinct cDNA clones or preys. DNA sequence verification and a systematic BLAST search showed that 72 of the isolated potential interactors (i.e., preys) contained the downstream gene in frame with the GAL4 activation domain, while the remaining 119 clones showed plasmids with out-of-frame sequences or sequences from non-protein-encoding regions, which were discarded. We retested all of the 72 positive interactions by cotransformant pairwise Y2H arrays, and validated 32 of them, indicating that they were indeed specific interactions. Finally, as they were observed by two independent Y2H screenings, we considered them as high-confidence interactions. Most of the identified preys interacted with a single bait, while only two were observed up to four times as independent clones interacting with three different baits (ST13 and UMPS prey genes).
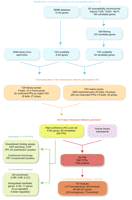
Flow strategy of the approach. Five major steps: (1) identification of potential and causative genes in AD; (2) characterization of the network by a Y2H screening; (3) generation of the AD protein interaction network; (4) experimental and computational assessment of the network coherence; and (5) functional module analysis of the generated AD-PIN.
Gene linkage analyses and genome-wide association studies have suggested that several chromosomal regions contain susceptibility loci involved in the etiology of late-onset Alzheimer's disease (LOAD) and familial AD with unknown genetic cause, confirming that additional AD genes remain to be identified (Lambert et al. 2006). As annotated in the Online Mendelian Inheritance in Man database (OMIM) (McKusick 2007), an association to AD has been demonstrated for four chromosome loci (7q36, 10q24, 19p13.2, and 20p), but very few associations have been unequivocally established with specific genes in these regions. Accordingly, to identify genes in these chromosomal loci potentially implicated in AD disease mechanisms, we decided to profit from our observation that AD causative and susceptibility genes tend to be physically connected. After discarding the 20p region, since it corresponds to an entire chromosome arm, we identified the 185 candidate genes within the three remaining loci and prioritized them according to their coexpression with known AD genes across a compendium of normal tissues and cell types. We estimated coexpression in terms of correlation coefficients computed using an expectation-maximization (EM) algorithm, and forced it to always consider all the brain related tissues to obtain the most relevant correlation for AD (see Fig. 1 and Methods). This procedure filtered out 60 candidate genes that did not coexpress with any of the known AD-related genes. With the aim of maximizing the use of genes suitable for Y2H screens, we discarded genes annotated as transcription factors (37 in total) from the original candidate gene list, as early studies indicated that they could behave as self-activators and trigger the expression of the reporter genes in the absence of a direct interaction with the prey protein, although this observation has been recently challenged. We also rejected genes encoding proteins that were highly glycosylated (one), extracellular (five), or containing several known/predicted transmembrane regions (29), as these might fold improperly, as well as nine genes for which the ORFs were unavailable. Finally, we ended up with 2809 binary interactions to be tested involving nine seed and 44 candidate genes, with the hope of finding clusters of interacting proteins involving known and candidate genes that could unveil novel AD causative or susceptibility elements.
We performed systematic matrix-based Y2H screens, by both cotransformation and mating approaches, to test pairwise interactions between seed and candidate genes (seed–seed, seed–candidate, and candidate–candidate combinations). We generated 53 preys (nine seeds and 44 candidates) and 43 baits (nine seeds and 34 candidate genes) from our gene selection list (see Methods), since we could not convert 10 of the prey vectors into baits. We excluded from the analyses six bait plasmids (one seed and five candidates) that resulted to be self-activating in the presence of empty prey clones. Out of the remaining 2050 pairwise protein interactions that we examined, we identified 246 nonredundant interactions between 19 baits and 52 prey proteins. Interestingly, we did not identify common protein–protein interactions to both matrix and library screens, revealing the advantage of performing pairwise and pool screens in parallel. All the detected interactions and putative susceptibility genes are reported in Supplemental File 1.
Based on the outcome derived from the Y2H screens, we generated a high-confidence (HC) interaction core set containing all the confirmed library interactions and those matrix interactions that were able to activate at least two reporter genes, as they required a more solid transcriptional activation. The definitive HC protein–protein interaction network comprises 200 nonredundant interactions among 74 genes: eight seeds (no HC interactions found involving PLAU), 27 library-identified, and 39 matrix-verified candidates (Fig. 1). Only four of these interactions had been reported previously, three between seed proteins (A2M–APOE, A2M–APP, and APP–PSEN1) and one involving one candidate (PSEN1–CDK5), meaning that the vast majority are entirely novel (Table 2). In addition, our screens did not recapitulate two other seed–seed interactions that had been found in other studies. This low overlap between different interactome networks is a well-known effect, and it is mainly attributed to the limited sampling of the interactome space and the detection capabilities of the different techniques (Russell and Aloy 2008; Venkatesan et al. 2009). In our case, this is particularly pronounced since we specifically chose our candidate genes to maximize the number of novel interactions added to the AD-related network and for which little interaction information was known (i.e., picking genes in susceptibility regions for which no direct proof of their implication in AD had been reported).
Overlap between the HC set of interactions and those previously reported
Although it is well-documented that different interaction discovery techniques are able to identify interactions of a different nature (i.e., binary/multimeric, transient/dedicated, etc.) (Venkatesan et al. 2009) we sought to validate some of our interactions derived from Y2H screens with complementary strategies previously employed in the identification of interactions involving AD proteins (Xia et al. 1997; Hughes et al. 1998). To this end, we randomly selected a subset of genes and protein interactions from our HC set, hoping that the results obtained would represent the general trends of the whole experiment (Fig. 2A). We first tested 11 HC protein–protein interactions, involving 11 genes, with GST pull-down assays (see Methods), and were able to validate seven positive interactions (Fig. 2B). We also tested 17 HC interactions, involving 12 proteins, by coimmunoprecipitation (co-IP) binding experiments in SH-SY5Y neuroblastoma cells, using specific antibodies against endogenously expressed proteins. Strikingly, all the tested interactions resulted positive, and most in the respective reverse co-IP experiment as well, hence confirming the interaction specificity (Fig. 2C). Overall, using both co-IP and GST-affinity binding methods, we could confirm 21 out of the 24 protein interactions we selected from our Y2H core set, yielding a verification rate of 87%. Of the four interactions analyzed by both techniques, one was positive by co-IP although negative in the pull-down assay (PSEN1-CDC37), while the remaining three interactions were confirmed as positive by both methods (IFIT3–CDC37; NOS3–CDC37; PSEN1–ECSIT). In addition, we also tested ten of the detected interactions that were not included in the HC set (seven in pull-downs, four in co-IPs), and we could only validate four of them. This verification rate of 40% indicates that, indeed, there are some real interactions among the 155 that we flagged as low-confidence, but in a much lower proportion than the ones contained in the HC set.
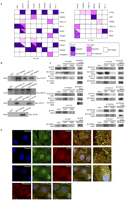
Validation of Y2H interactions by downstream binding assays. In vitro binding experiments: (A) schematic diagrams showing the interactions examined by co-IP or pull-down experiments, for the high-confidence (HC) and low-confidence (LC) sets, respectively; (B) by GST pull-down, blotting with anti-MYC antibody to detect the bound partner; (C) by coimmunoprecipitation (CoIP), blotting with a specific antibody for the bound partner, respectively. (Input) Cell lysate, used as loading control; (IP) immunoprecipitated protein; (IB) immunoblotted protein; (NIgG) nonimmune rabbit or mouse immunoglobulins, used as negative IP controls. When using NIgG as IP agent, no precipitation lines were detected against IB antibodies, indicating that CoIPs were protein-specific. Furthermore, reverse CoIPs using IB antibodies as IP agents, followed by IP antibodies for blotting, led to the same results in almost all cases, hence confirming the interactions. Expected molecular weights are also indicated. (D) In vivo colocalization of interacting partners by double immunofluorescence staining using confocal microscopy. (Upper panel) Double immunofluorescence confocal micrographs, labeled with a rabbit anti-ECSIT antibody and a secondary Alexa488-labeled anti-rabbit IgG (visualized in green pseudocolor), and with a mouse anti-PSEN2 visualized in red pseudocolor with a secondary Alexa568 labeled anti-mouse IgG (Invitrogen). Colocalized immunolabeling (merged window) appears as yellow staining in some areas (a framed area is also displayed in greater detail, see white arrows). Nuclei are displayed in blue by Hoechst staining. (Second panel) Double immunofluorescence of ECSIT and APOE following the same procedure. APOE was labeled with a mouse anti-APOE and an Alexa568 (visualized in red pseudocolor). (Third panel) Double immunofluorescence of GCDH labeled with a rabbit anti-GCDH and an Alexa488 (visualized in green pseudocolor), and NOS3 labeled with a mouse anti-NOS3 and an Alexa568 (visualized in red pseudocolor). (Bottom panel) Mitochondria staining with MitoTracker Deep Red stain (visualized in red pseudocolor). Merging with GCDH and NOS3 labeling, respectively, appears as yellow staining, indicating their mitochondrial localization.
Additionally, we analyzed the in vivo colocalization of proteins involving nine HC interactions in mammalian cells, where endogenously expressed proteins were labeled by double immunofluorescence staining and visualized using confocal microscopy (see Methods). We were able to detect the colocalization of three interactions: ECSIT–PSEN2, ECSIT–APOE, and GCDH-–NOS3, which were also validated by co-IP assays. Moreover, we also analyzed the subcellular distribution of the interactions by a double immunofluorescence staining of the respective partners in addition to the mitochondrial marker and, as expected, the GCDH–NOS3 interactions did both present a mitochondrial localization (Fig. 2D).
While the accuracy achieved is very high (i.e., very few false-positive interactions), the coverage is indeed low, meaning that we expect many more interactions involving the tested proteins than the ones detected in this study. It would be tempting to attribute this limited coverage to the intrinsic properties of many AD-related proteins which, due to their transmembrane or secreted nature, are experimentally difficult to handle. To check whether this was the case, we compared the number of interactions detected for each of the nine seed proteins employed (three secreted, three transmembrane, and three intracellular), and found no significant difference (10.3, 11.7, and 10.3 interactions per protein on average, respectively), although the use of different Y2H setups specially designed to deal with transmembrane proteins might indeed increase the coverage (Snider et al. 2010). Thus, in this particular study, failure to detect interactions is likely to be the result of the high stringency applied to our Y2H assays, particularly designed to minimize false-positives, although this criterion might penalize detection of some weak or transient interactions.
Functional and gene expression analysis of the obtained AD-related interactions
The first analysis that we applied to our HC set of interactions was to look for enrichment of particular functional terms as defined in the Gene Ontology (GO) database (Ashburner et al. 2000) (see Methods). In total, we identified 14 significantly enriched terms (adjusted P-value < 0.05) comprising three biological processes, five molecular functions, and six cellular component terms (see Table 3). Some of these are related to AD seed proteins (e.g., redox signaling or cytoskeletal proteins) and are consistent with current knowledge of the biological functions and compartments implicated in AD (Reddy 2009). However, more interestingly, we also found significant enrichments for certain unexpected functions or subcellular localizations that are not associated to known AD genes (e.g., regulation of apoptosis or actin binding activities).
Functional enrichment in the HC set; list of GO annotations enriched in the HC set
We also checked whether the genes present in our HC set had been found to be related to the AD phenotype based on microarray data (Fig. 3; Blalock et al. 2004). In this study, Blalock and colleagues analyzed hippocampal gene expression of nine controls and 22 individuals suffering from AD of varying severity (incipient, moderate, and severe) and tested the correlation of each gene expression with MMSE (mini-mental status examination) and NFT (neurofibrillary tangles) scores across all 31 subjects regardless of the diagnosis (see Methods). We found that 17 of the 66 genes in our HC interactors are either up- or down-regulated in AD subjects compared to control, which represents a 25.7% of the total. This figure is comparable to the one observed for the known seed genes and their direct interactors, where 26 out of 97 genes (26.8%) are dysregulated, and significantly higher than expected by chance when comparing the percentage with altered expression in AD within the human genome (2508/24210; P-value < 3 × 10−4 in a two-sided Fisher's test) and the genes present in the microarray employed by Blalock et al. (2004), although statistically insignificant (2508/13,030; P-value < 0.12). Curiously, the fraction of AD down-regulated genes in our HC gene set (0.53) is slightly higher than the fraction detected in the original study (0.43), but doubles the one found among the AD-related genes and interactors curated from the literature (0.23), which suggest a small bias toward the study of up-regulated genes in directed experiments (P-value < 0.057 in a two-sided Fisher's test). In any case, the discovery of direct interactions between dysregulated genes in AD subjects and well-characterized seeds could certainly trigger further functional studies to investigate their potential role in the disease phenotype.
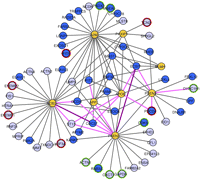
The HC interaction network. Visual representation of the relationships between AD seeds and HC interactors. Seeds are depicted in pale orange whereas HC matrix and library interactors are colored in dark and light blue, respectively. The AD dysregulated interactors are highlighted in red (up-regulated) and green (down-regulated). Dark violet lines denote those interactions confirmed by pull-down and coimmunoprecipitation experiments, whereas lilac lines represent interactions confirmed only by coimmunoprecipitation. Candidate–candidate interactions are removed for clarity.
Finally, we investigated whether any candidate from the HC set was listed in the AlzGene database (November 2010 download) (Bertram et al. 2007). We found that six out of the 58 genes that we identified as direct interactors of the AD-seed proteins are present in the AlzGene database, three candidates (CYP2C8, CDK5, LIPF) and three coming from the library screens (EFEMP1, GAPDH, TCN2). In addition, there are two more (DYNC1H1, EID1) that are located next to the linked regions identified by recent GWAS experiments (Bertram and Tanzi 2009). Interestingly, none of the candidates that do not interact with seeds is present in AlzGene, which reinforces the message that the genes identified in our interaction discovery pipeline might certainly play a role in AD.
Direct interaction partners among AD seed proteins
To date, the experimental difficulties associated to the majority of AD proteins has resulted in a very limited number of literature reported interactions among AD seed proteins, a number that is further reduced for direct interactions (Xia et al. 1997; Krimbou et al. 1998; Hesse et al. 1999). Interestingly, we identified seven direct interactions among AD causative genes, four of which correspond to novel interactions that might provide new insights into the molecular pathways underlying this disorder (Fig. 3). For instance, we found an interaction between alpha2-macroglobulin (A2M) and APP, which corroborates the finding that A2M is a strong and specific interactor of Aβ peptide in AD plasma (Mettenburg et al. 2002). But, more interestingly, we also found A2M to interact with PAXIP1, a gene that was identified by linkage and association studies as a novel locus for AD at 7q36 in a Dutch population-based sample (Rademakers et al. 2005). However, its functional role in AD still remains unclear, and this novel interaction with A2M can be used as a starting point for further investigations. In addition, we also detected an interaction between A2M and NOS3, found in close proximity to amyloid plaques (Probst et al. 1982), which supports the suggested link between plaque formation and inflammatory processes (Luth et al. 2001). However, the most interesting link is between APOE and PSEN1, which had not been reported to date. PSEN1 is primarily localized to the endoplasmic reticulum and it is required for efficient proteolysis of APP within their transmembrane domains. Mutations in PSEN1 increase the production of β-amyloid, strongly supporting the hypothesis that mutant PSEN1 interacts with APP in a way that enhances the intramembranous proteolysis (Vetrivel et al. 2006). Therefore, our direct evidence of APOE and PSEN1 binding could provide insights into the intracellular pathogenic role of APOE as a regulator of PSEN1 in APP cleavage. Furthermore, we also detected an interaction between PSEN1 and PSEN2, previously suggested to intimately cooperate as part of the gamma-secretase complex in APP cleavage. The direct binding of APP with both PSEN1, which we confirmed by co-IP, and PSEN2 had been previously suggested (Xia et al. 1997); however, there was not reported evidence of a direct PSEN1-PSEN2 binding (Haass and De Strooper 1999).
Collectively, these four novel interactions detected among central proteins in AD strengthen our initial observation that, as for other neurodegenerative disorders (Lim et al. 2006), AD causative and susceptibility proteins are, directly or indirectly, highly interconnected.
Interactome network associated to AD
To contextualize the 200 novel AD-related interactions between 74 proteins that our study has revealed, we integrated them with all the interaction data reported in the literature to build the most complete interactome associated to AD with the data that is currently available (see Fig. 1 and Methods). This network view will permit the undertaking of functional analyses that reflect the global properties of the network, and not only single proteins or interactions. We thus retrieved from the databases all the proteins identified as direct interactors of the group of seeds considered in our study and merged them with our HC set of interactions, making a total of 403 interactions between 183 proteins. Additionally, we further extended this initial network to the next level (i.e., we included all the direct interactors, the initial set and the interconnections among them), obtaining a network of 5881 interactions among 1704 proteins, which we call the AD protein interaction network (AD-PIN). The general topology of the AD-PIN shows a path length of 3.7 and a characteristic node degree distribution that approximates a power law (γ = 1.65, R2 = 0.911).
We next studied the structure of the AD-PIN to detect the presence of potential functional modules, defined as groups of proteins that are densely interconnected and that are functionally homogenous (i.e., functional annotation shared by the maximum number of module proteins). To identify these modules we used the MCL algorithm (van Dongen 2000), since it has proved to be more robust and tolerant to noise than other modules detection methods (Brohee and van Helden 2006; Vlasblom and Wodak 2009).
With this procedure, we identified 172 modules in the AD-PIN, of which 117 showed a high degree of functional homogeneity, roughly containing 55% of the proteins in the network. Additionally, we found that 68 of them were significantly enriched for one or more GO biological process annotations, the most frequent ones being related to signal transduction, transcription regulation, proteolysis, apoptosis, protein transport, and oxidative stress. If we look for the positioning of the seeds and proteins in our HC set in the AD-PIN, we find that most of them have been grouped into 38 distinct modules (69% and 80%, respectively), 24 of which are homogenous for, at least, one GO annotation (Fig. 4). If we compare these figures to the results obtained without including our 200 newly discovered interactions, we see that the number of clusters has risen from 146 to 172, and the number of homogeneous and enriched groups have also increased in nine and 14 modules, respectively. Perhaps more relevant to AD is the fact that the functional modules containing the seeds now include 13 new proteins, which are serious candidates to play a role in AD. All the AD-PIN modularity data is reported in Supplemental File 2.
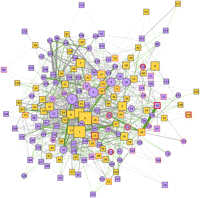
The modular structure of the AD-PIN. Representation of the network modules identified in the AD-PIN by the MCL algorithm. Functionally homogeneous modules are depicted as square nodes, and nonhomogeneous modules as circle nodes. Homogeneous modules that are enriched are in pale orange. Modules containing HC interactors have a thin red border, while those modules including AD seeds have a thick red one. Node labels correspond to the module identifiers provided in Supplemental File 2. The thickness of the edges is proportional to the number of interactions connecting two given clusters (max: 22; avg: 1.61; edges with at least two interactions: 257, 26.96%). Green edges (20.56% of the total, connecting 116 clusters) indicate if two clusters have at least one enriched/most abundant GO term in common.
Globally, the integration of our HC set of interactions into the larger AD-PIN, together with the analyses and visualization of the functional modules, have issued many hypotheses that might trigger novel lines of research. In the following paragraphs, we present some of the most interesting ideas that spanned out of the novel interactions reported and the AD-PIN analyses. These potential roles are mainly sustained on literature references and would indeed need further experimental validation.
Putative role of PDCD4 as neuronal death regulator in AD
Amongst the most promising interactions, there are two that relate the seed proteins PSEN2 and APOE with the programmed cell death 4 (PDCD4) candidate gene (on chromosomal region 10q24), which encodes a protein localized to the nucleus under normal growth conditions, but it can also shuttle to the cytoplasm (see Fig. 2B showing the experimental validation by co-IP). It is thought to be involved in apoptosis, although the specific role has not yet been determined (Lankat-Buttgereit and Goke 2003). Expression of this gene is modulated by cytokines in natural killer and T cells, inhibiting protein translation. In addition, PDCD4 has been found to inhibit AP-1-mediated transactivation and to induce expression of the cyclin-dependent kinase inhibitor p21. As a result, loss of PDCD4 confers growth advantages to the cells by several means (Talotta et al. 2009).
In the constructed AD-PIN, PDCD4 is present in a network module functionally homogeneous and enriched for “translation elongation,” which is consistent with its ability to inhibit protein translation (Yang et al. 2003). Notably, this gene is up-regulated in AD human brain tissues and thereby our observations suggest that PDCD4 could play a role in Aβ neurotoxicity in conjunction with APOE and PSEN2 (see Fig. 5).
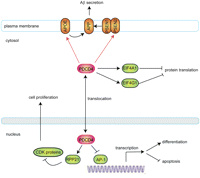
Putative role of PDCD4 as neuronal death regulator in AD. The AD candidate gene PDCD4 undergoes a complex regulation by cytokines, which results in the inhibition of protein translation. This gene is up-regulated in AD human brain tissues and thus the novel associations with AD seeds suggest that PDCD4 could play a role in Aβ neurotoxicity in conjunction with APOE and PSEN2. Literature reported interactions are depicted with black lines, while novel interactions are depicted with red lines. AD seeds are displayed as orange ellipsoids, candidates as red, and literature interactors as green ellipsoids.
Hypothetical role of ECSIT as molecular link between oxidative stress, inflammation, and mitochondrial dysfunction in AD
Chronic Aβ exposure increases protein oxidation in cultured neurons and in AD brains, indicating that mitochondria play a critical role in Aβ cytotoxicity and thereby in the pathogenesis of AD (Cumming et al. 2007). In the AD-PIN network we detected several modules linking redox signaling and immune responses. The most interesting one includes the candidate gene ECSIT (evolutionarily conserved signaling intermediate in Toll pathway), located in the susceptibility region 19p13.2 and presenting 13 interaction partners within the two modules (see Fig. 2 and Supplemental File 1). Based on this data, we hypothesize that ECSIT might constitute a molecular link between mitochondrial processes and AD lesions.
The ECSIT gene is an adapter protein involved in NFKB activation and also plays a role in the BMP signaling pathway required for normal embryonic development (Kopp et al. 1999). Although ECSIT acts as a cytoplasmic signaling protein in these two pathways, an N-terminal targeting signal directs ECSIT to mitochondria as well (Vogel et al. 2007). In fact, cell knockdowns present a disturbed mitochondrial function that supports a role for ECSIT in linking assembly of oxidative phosphorylation complexes to inflammatory response (Vogel et al. 2007).
Our AD interaction map shows the association of ECSIT with the mitochondrial proteins Lon protease homolog (LONP1), required for intramitochondrial proteolysis as a cellular response to oxidative stress, and glutaryl-CoA dehydrogenase (GCDH), involved in redox signaling, which also interacts with the AD seed NOS3 (Fig. 2D). In addition, we observe ECSIT interactions with other endoplasmic reticulum redox proteins, like the lysyl-oxidase homolog 4 (LOXL4) and the CYP2C18 (cytochrome P450 2C18), involved in an NADPH-dependent electron transport pathway. Although an altered ECSIT gene expression has not been reported in AD patients to date, its expression is significantly up-regulated in Huntington's patients (Borovecki et al. 2005), and we found it to physically interact with genes altered in AD brain, namely peroxiredoxin-2 (PRDX2) and interferon-induced protein with tetratricopeptide repeats 5 (IFIT5) (see Supplemental File 1). This gives further support to the hypothesis that ECSIT might modulate the energetic requirements upon inflammatory response by regulating the rate of complex I synthesis (Vogel et al. 2007).
Most interestingly, we observed a novel association of ECSIT with the AD gene APOE (Fig. 2C,D), which was shown to bind Aβ in its oxidized form (Strittmatter et al. 1993). In AD affected neurons, APOE is proteolysed and associates with neurofibrillary tangle-like structures and mitochondria, although it still remains unclear how the fragments associate and cause mitochondrial dysfunction (Nakamura et al. 2009). The physical interaction of APOE and ECSIT could thus highlight an association mechanism that would place APOE on the mitochondrial membrane for further cleavage in AD affected cells. The additional interactions of ECSIT with cleaving enzymes PSEN1 and PSEN2 (Fig. 2C,D) proves that ECSIT is involved in several pathways which are functionally connected, supporting the hypothesis that points ECSIT as a molecular link among oxidative stress, inflammation, and mitochondrial dysfunction in AD (Fig. 6).
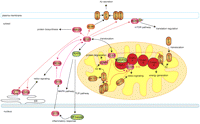
Potential contribution of new molecular mechanisms to mitochondrial dysfunction in AD. The AD candidate gene ECSIT, which is involved in TLR and BMP signaling pathways and also in the assembly of mitochondrial redox complexes, shows additional interactions with several AD causative and candidate genes that are functionally connected. Thus ECSIT can constitute a molecular link among oxidative stress, inflammation, and mitochondrial dysfunction in AD. Literature reported interactions are depicted with black lines, while novel interactions are depicted with red lines. AD seeds are displayed as orange ellipsoids, candidates as red, and literature interactors as green ellipsoids. (ETC) Electronic transport chain.
Concluding remarks
Network and systems biology strategies offer a global perspective to explore the molecular mechanisms underlying complex diseases beyond individual genes and proteins. In this work, we have shown how a combination of interaction discovery experiments and the computational analyses of diverse biological data can provide further evidence for potential causative/susceptibility genes related to Alzheimer's disease, suggesting novel hypotheses as to their molecular functioning. However, to be most valuable, these functional hints coming from global analyses will need to be individually validated. The finding that causative genes are often highly interconnected, even in complex heterogeneous disorders, places network biology strategies in a privileged position to complement genome-wide association studies and next generation sequencing techniques in the quest for novel genes associated to human pathologies. We anticipate that large international efforts, such as the ongoing initiatives to chart disease-related interaction maps (Charbonnier et al. 2008), will soon permit the generation of the basic wiring inherent to most physiopathological processes and refine systems biology models to the point where they can be effectively applied to biomedicine.
Methods
AD-related genes and chromosomal region selection
We extracted all the disease-related proteins from the OMIM Morbid Map database (January 2008) and picked as AD genes those loci with the “(3)” tag, which showed evidence that at least one mutation is known to be associated with AD (Table 1). We then identified four chromosomal regions, namely 7q36 (MIM:609636), 10q24 (MIM:605526), 19p13.2 (MIM:608907), and 20p (MIM:607116) for which the association with the AD phenotype was confirmed but that did not contain any gene directly related to AD. We discarded the region 20p, since it comprised the whole short arm of chromosome 20.
Connectivity assessment
We built a human interactome fetching the most recent available data (September 2009) from DIP, IntAct, and MINT databases (Salwinski et al. 2004; Aranda et al. 2010; Ceol et al. 2010). We selected experimentally verified direct interactions and added those interactions described as binary according to the associated detection methods (Rual et al. 2005). We further extended the interactome including the HPRD data set (Keshava Prasad et al. 2009), obtaining a human binary interactome consisting of 22,194 interactions between 8347 proteins.
We then evaluated the interconnectivity of AD-related genes in terms of average shortest path length. To assess the statistical significance of the connectivity measure, we defined two reference distributions: 10,000 instances of size equal to 12 (the number of AD-related genes) consisting of randomly picked proteins from (1) the human binary interactome and (2) disease-associated proteins belonging to distinct disorder classes (Goh et al. 2007) and present in the human binary interactome. We compared the AD genes average shortest path length and the random set average shortest path length using the Mann-Whitney U test.
Correlation in gene expression profiles
We used the microarray data from Su et al. (2004), a compendium of gene expression profiles from 73 normal tissue and cell types. We applied a mixture model in order to obtain correlation coefficients that are robust under the presence of noise. We fit the model using the expectation maximization (EM) algorithm (Dempster et al. 1977). We defined two genes as coexpressed if their EM correlation coefficient was >0.5 and the probability of noise <0.5.
Y2H cotransformation screens
We individually transferred the ORFs corresponding to the selected AD genes into Y2H destination vectors by Gateway recombinational cloning (ProQuest System, Invitrogen Inc.). We cloned the seed genes into pDEST32 to generate bait plasmids. Seed and candidate genes were cloned into pDEST22 to obtain prey plasmids.
We pairwise cotransformed bait and prey plasmids into a MaV203 yeast strain in a 96-well array format. We plated cotransformed cells onto selective SD2 (lacking Leu and Trp amino acids) agar media and incubated them for 48 h at 30°C. After a colony replica clean plating, we then replicated cotransformant arrays onto different selective media agar plates to detect colony growth. To assay the activation of the HIS3 reporter gene, SD3 (lacking Leu, Trp, His) agar plates were supplemented with 12–100 mM 3-aminotriazole (3AT, Sigma-Aldrich), 50 mM 3AT being the optimal concentration for positive HIS3 activation colonies. Similarly, we assayed the activation of the URA3 reporter gene by plating onto SD3 (lacking Leu, Trp, uracil) media or SD2 supplemented with 5-Fluoroorotic acid (5FOA, Sigma-Aldrich) for negative colony selection. Double reporter HIS3/URA3 activation was evaluated by SD4 (lacking Leu, Trp, His, uracil) agar plates. We tested the lacZ reporter gene by the beta-galactosidase assay on a nylon membrane placed onto a SD2 agar plate.
Y2H mating screens
We individually transformed bait and prey clones into MATα or MATa yeast haploid strains, respectively, in a 96-well array format. We cultured the single transformants into appropriate selective liquid medium (lacking Trp for the baits or Leu for the preys) to ensure the selection of transformants. We mated 43 MATa yeast cells individually expressing baits against 44 MATα prey-expressing cells in a pairwise format. We subsequently incubated the mates onto YPD (yeast rich media) plates for 48 h at 30°C. We then replated the coexpressing colonies onto SD2 (without Leu or Trp) agar medium and transferred the mated cells onto SD3 and SD4 agar plates to assess the activation of HIS3 and URA3 reporter genes. lacZ reporter gene was evaluated by a beta-galactosidase assay on a nylon membrane placed on a SD2 agar plate.
Y2H library screens
We performed a Y2H library screen using an adult human brain cDNA prey library (ProQuest, Invitrogen). We transformed yeast cells expressing individual baits (generated from the seeds) with the cDNA prey library and screened them onto selective agar media to check HIS3 and URA3 reporter gene activation. After 7 d incubation at 30°C, we picked up positive growing colonies and cultured them in prey selective liquid medium (lacking Trp). In each screen, we typically tested 6 × 104 auxotrophic transformants on selective plates, obtaining 1–15 positive colonies in average. We extracted the prey plasmid DNAs from the cultures and we subsequently carried out the bacterial transformation of each plasmid in order to enable DNA sequencing and subsequent gene identification by BLAST search.
We further tested the preys we identified by the library together with their respective baits in cotransformation assays for activation of reporter gene expression, in a similar procedure as explained above.
In vitro pull-down assays
For GST pull-down assays (PD), we selected genes that yielded detectable protein overexpression in COS-7 mammalian cells. As several genes encode for membrane proteins, we observed limited overexpression, which enabled us to only test 12 genes involved in 11 protein–protein interactions. We transferred each partner gene into a GST- or MYC-expression vector using the Gateway system (Invitrogen, Inc.), and transfected GST-fused plasmids into COS7 mammalian cells using Lipofectamine 2000 following the manufacturer's instructions. We cultured cells in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/mL penicillin and 10–6 mg/mL streptomycin). All reagents were purchased from Invitrogen Inc.
Two days after transfection, we harvested and lysed cells with lysis buffer (0.2% NP-40, 0.05% Triton X-100, 50 mM Tris-HCl at pH 7.85, 50 mM NaCl, 5 mM MgCl2, 50 μM ZnCl2, 0.5 mM EDTA, 10% glycerol, and complete protease inhibitor cocktail [Roche]). We cleared whole cell lysates by centrifugation for 20 min at 16,000g at 4°C and we purified the soluble protein complexes using glutathione Sepharose 4B beads (GE Healthcare). We then extensively washed the beads three times with lysis buffer. We eluted and analyzed bound proteins by SDS-PAGE and Western blotting.
We detected MYC- and GST-tagged proteins using mouse anti-MYC monoclonal antibody (mAb) (Invitrogen, cat. 13-2500) and rabbit polyclonal antibody (pAb) (Invitrogen, cat. 71-7500) or mouse mAb anti-GST (Invitrogen, cat. 13-6700).
Coimmunoprecipitation (Co-IP) assays
For co-IP assays, we selected those interactions involving proteins with both commercially available and compatible specific antibodies.
We cultured SH-SY5Y human neuroblastoma cells in DMEM plus F12 (1:1) supplemented with 10% FBS, 2 mM sodium pyruvate, and 2 mM nonessential amino acids (NEAA). After lysis with a mild buffer (0.5% Triton X-100, 50 mM HEPES at pH 7.50, 150 mM NaCl, 1 mM MgCl2, 1 mM EGTA, and complete protease inhibitor cocktail [Roche]), we cleared whole cell lysates by centrifugation for 20 min at 16,000g at 4°C.We then precleared the lysates by adding protein A/G Sepharose beads (GE Healthcare) (10% of the total lysate). After 30 min of rotation at 4°C, we removed the beads by centrifugation at 16,000g at 4°C for 10 min. We added the appropriate antibody (1ug) to the lysate. After incubation for 1 h at 4°C on a rotating plate, we added 30 μL of protein A/G bead slurry and incubated under rotation at 4°C overnight. We collected and washed the beads extensively three times with lysis buffer, we eluted the complex and after SDS-PAGE separation, we detected the binding partner of the precipitated protein using the corresponding specific antibody.
Commercially available antibodies were mouse anti-PSEN1 mAb (cat. ab15456), mouse anti-NOS3 mAb (cat. ab2801), mouse anti-APOE mAb (cat.ab1906), mouse anti-IFIT3 mAb (cat. ab76818), mouse anti-PSEN2 mAb (cat. ab15549), rabbit anti-APP pAb (cat.ab15272), rabbit anti-ECSIT pAb (cat. ab21288), rabbit anti-CDC37 pAb (cat. ab61773), rabbit anti-GCDH pAb (cat. ab75324), rabbit anti-PDCD4 pAb (cat. ab45124), and rabbit anti-ST13 pAb (cat. ab73917). We purchased those antibodies from Abcam Inc.
Double immunofluorescence and confocal microscopy
COS-7 cell monolayers were harvested at 24 h post-infection, fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) and permeabilized in 0.1% (v/v) Triton X-100 in PBS. Cells were blocked with 1% BSA in PBS (PBS-BSA) and reacted with a protein specific polyclonal rabbit antibody (1:200 in PBS-BSA) and Alexa Fluor 488-labeled goat anti-rabbit IgG (Invitrogen), and a protein mouse monoclonal antibody (1:200 in PBS-BSA) and Alexa Fluor 568-labeled goat anti-mouse IgG antibody (Invitrogen). Samples were treated with Hoechst stain (Invitrogen) for nuclei staining and with MitoTracker Deep Red stain (Invitrogen) for mitochondrial staining. They were subsequently mounted on slides. Samples were analyzed using a Leica TCS SP2 confocal microscope.
Gene Ontology annotation
We used the human GO annotation extracted from the Entrez gene2go file (NCBI, September 2009) and assessed the statistical significance of GO term enrichment using the Fisher's exact test. We adjusted the P-values for multiple testing, applying the Bonferroni correction.
Overrepresentation of AD-dysregulated genes
We downloaded the lists of AD-dysregulated genes from the The Molecular Signatures Database (Subramanian et al. 2005; http://www.broadinstitute.org/gsea/msigdb/). We assessed the overrepresentation of AD-dysegulated genes in the AD genes interactor sets using the Fisher's exact test.
Identification of functional modules within the AD-PIN
We applied the MCL algorithm (van Dongen 2000) to identify the cluster representing putative functional modules. Since the granularity of the clustering depends on one parameter, the inflation coefficient, I, we ran MCL on the AD-PIN exploring a wide range of I (from 0.1 to 10.0 by steps of 0.1).
We chose the value of I that maximized the number of functionally homogenous clusters, i.e., modules, containing at least three proteins. We evaluated the functional relatedness of modules in terms of GO homogeneity (Goh et al. 2007), GH, defined as the maximum fraction of proteins in the same module that have the same GO terms from the biological process branch. For the GH computation, we required that 50% of the proteins be present in the module to be annotated with at least one GO term.
We then assessed the statistical significance of each homogeneous module, comparing its GH to the mean GH of a reference distribution obtained by computing the GH for 10,000 randomly generated sets of the same size of the module. We picked proteins for the randomization from the human binary interactome.
Acknowledgments
We thank Roland A. Pache for useful discussions, David Rossell for help implementing the EM correlation algorithm, and Guillermo Suñé for assistance with the in vitro binding assays.
Footnotes
-
↵5 Corresponding author.
E-mail patrick.aloy{at}irbbarcelona.org.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.114280.110.
- Received August 18, 2010.
- Accepted December 1, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








