
Strand-specific deep sequencing of the transcriptome
- Ana P. Vivancos1,4,5,
- Marc Güell1,4,
- Juliane C. Dohm1,2,4,
- Luis Serrano1,3,6 and
- Heinz Himmelbauer1,6
- 1 Centre for Genomic Regulation (CRG), UPF, 08003 Barcelona, Spain;
- 2 Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany;
- 3 Institució Catalana de Recerca i Estudis Avancats (ICREA), 08010 Barcelona, Spain
-
↵4 These authors contributed equally to this work.
Abstract
Several studies support that antisense-mediated regulation may affect a large proportion of genes. Using the Illumina next-generation sequencing platform, we developed DSSS (direct strand specific sequencing), a strand-specific protocol for transcriptome sequencing. We tested DSSS with RNA from two samples, prokaryotic (Mycoplasma pneumoniae) as well as eukaryotic (Mus musculus), and obtained data containing strand-specific information, using single-read and paired-end sequencing. We validated our results by comparison with a strand-specific tiling array data set for strain M129 of the simple prokaryote M. pneumoniae, and by quantitative PCR (qPCR). The results of DSSS were very well supported by the results from tiling arrays and qPCR. Moreover, DSSS provided higher dynamic range and single-base resolution, thus enabling efficient antisense detection and the precise mapping of transcription start sites and untranslated regions. DSSS data for mouse confirmed strand specificity of the protocol and the general applicability of the approach to studying eukaryotic transcription. We propose DSSS as a simple and efficient strategy for strand-specific transcriptome sequencing and as a tool for genome annotation exploiting the increased read lengths that next-generation sequencing technology now is capable to deliver.
Recent advances in high-throughput transcript sequencing have changed our views of gene expression and genomic organization (Morin et al. 2008; Mortazavi et al. 2008; Rosenkranz et al. 2008; Sultan et al. 2008; Maher et al. 2009; Tang et al. 2009; Wang et al. 2009), and precise prediction of transcript boundaries revealed exciting snapshots of the transcriptome (Nagalakshmi et al. 2008; Wilhelm et al. 2008), as well as intriguing aspects of gene regulation (Seila et al. 2008).
In mammals, antisense transcription has been shown to be a ubiquitous phenomenon (Katayama et al. 2005; He et al. 2008). The number of discovered antisense transcripts is growing steadily, and the definitive number is still unknown. Also, the effects of antisense transcription on their sense RNAs have not been clearly established yet (Carmichael 2003; Yelin et al. 2003). In prokaryotes, individual cases have been reported (Tomizawa and Itoh 1981; Wagner and Simons 1994; Guillier et al. 2006), and recently, it has been suggested that some antisense RNAs could be involved in gene regulation (Andre et al. 2008). Recent chip based reports in archaea (Koide et al. 2009) and in Listeria (Toledo-Arana et al. 2009) described extensive antisense transcription in such organisms.
Given that sequence reads generated on high-throughput next-generation sequencing instruments are getting progressively longer, sequencing libraries with longer inserts will massively increase the information content of the sequences. Longer sequences result in more precise mapping of reads, as the probability of matching to the genome is proportional to 1/4n, where n is the read length. In large genomes, this issue becomes crucial due to their higher complexity. In eukaryotic transcriptomes, alternative splicing of transcripts is the rule rather than the exception, with many genes exhibiting several transcript isoforms (Xing and Lee 2006).
Standard cDNA sequencing methods adapted to next-generation sequencing platforms do not generate strand information (e.g., Mortazavi et al. 2008). Accordingly, several approaches for strand-specific sequencing of RNA have been proposed for various next-generation sequencing platforms: Cloonan and Grimmond (2008) developed a protocol originally designed for Applied Biosystems SOLiD System sequencing; Lister et al. (2008) presented a method for the Illumina platform for very short RNA fragments based on Illumina's small RNA protocol; Croucher et al. (2009) proposed a method for Illumina sequencing library construction utilizing single stranded cDNA as a means to preserve information on strandedness; Ozsolak et al. (2009) proposed single-molecule direct RNA sequencing on the Helicos sequencing instrument; Parkhomchuk et al. (2009) enzymatically removed the second strand of the cDNA prior to sequencing on the Illumina platform; Wurtzel et al. (2010) developed a strand-specific method for the mapping of the 5′ ends of archaeal transcripts; Armour et al. (2009) used hexamers unable to prime cDNA synthesis from rRNA templates to generate cDNA in a strand-specific manner for Illumina sequencing; and Mamanova et al. (2010) devised a method whereby cDNA is generated from single molecules within an Illumina flow cell prior to cluster generation.
Here, we describe DSSS (direct strand specific sequencing), a new strand-specific protocol designed for transcriptome sequencing, using the Illumina next-generation sequencing platform. In this technology, fragmented RNA is modified with adapter sequences that are attached to both ends and that are complementary to oligonucleotides immobilized on a glass surface. By means of solid-phase PCR amplification, clusters each encompassing about 1000 identical molecules are generated. This is followed by parallelized sequencing-by-synthesis, using reversible terminator chemistry, that yields at least 60 million high-quality sequencing reads in a single instrument run (Bentley et al. 2008).
To assess the quality of DSSS results in one prokaryotic and one eukaryotic species, we have generated transcriptome data sets from Mycoplasma pneumoniae (strain M129), one of the smallest bacteria that can live outside a host cell, and from mouse (Mus musculus), respectively.
Results
Strand-specific deep sequencing
We have developed a protocol that allows strand-specific deep sequencing of prokaryotic and eukaryotic RNA samples. It is based on the small RNA sample preparation protocol from Illumina (Lu et al. 2007), which was developed to enable the investigation of 18- to 30-nucleotide (nt)-long RNA molecules and is strand-specific. The Illumina protocol encompasses sequential ligation of 5′ and 3′ adapters to RNA molecules, thus preserving strandness information. However, the Illumina protocol is limited in the lengths of RNA molecules that can be studied, as longer inserts tend to establish secondary structures that favor intramolecular ligation of the RNA. This introduces a bias against molecules that are prone to secondary structure formation, due to either their lengths or their base composition. We have experimentally determined that within the range of 50–100 nt the Illumina small RNA sample protocol becomes increasingly problematic and that it does not work at all with a molecule length >100 nt using standard conditions (Supplemental Fig. 1). We have adapted the small RNA Illumina protocol so that it enables the unbiased study of inserts >150 nt (Fig. 1).
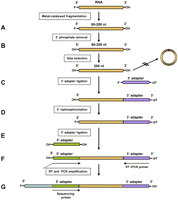
DSSS protocol workflow. (A) Fragmentation. RNA is fragmented to sizes in the range of 60–200 nt. (B) Dephosphorylation. 5′ phosphates are removed from RNA by treatment with alkaline phosphatase. (C) 3′ adapter ligation. Dephosphorylated 200-nt-long RNA fragments are selected by urea-PAGE. The 3′ adapter is ligated to the 3′ ends using T4 RNA ligase I. (D) Rephosphorylation. Fragments are rephosphorylated by treatment with T4 polynucleotide kinase as preparation for the next ligation step. (E) 5′ adapter ligation, preceded by removal of the nonligated 3′adapter by urea-PAGE size selection. (F) Reverse transcription (RT) and amplification of library. Molecules with 5′ and 3′ adapters were selected by urea-PAGE. First strand cDNA synthesis and PCR amplification were carried out with the indicated primers. (G) Sequencing.
In order to avoid intramolecular ligation of RNA molecules, we removed the 5′ phosphates and switched the order of adapter ligation in comparison to the Illumina protocol (Fig. 1B–E) and decided to use the 3′ adapter sequence to prime the first strand reverse transcription (Fig. 1F). 3′ biases are reduced due to the relatively short insert length (200 nt) after fragmentation (Fig. 1A). Size selection at each step of the protocol ensures homogeneous lengths of molecules in the population, thus minimizing the probability that the preference for shorter molecules during ligation steps or PCR amplification introduced biases.
We proceeded to establish DSSS for prokaryotic transcriptomes, exemplified by M. pneumoniae strain M129. Within its genome of 816 kbp, M129 contains 689 annotated protein-coding genes and 44 known genes that encode noncoding RNAs (Dandekar et al. 2000). We successfully mapped 100 million reads of 36 nt to the M129 genome (Supplemental Table 1) using MAQ (Li et al. 2008); mapping was limited to three mismatches, and quality values were taken into account (see Methods). Only 0.9% of the M129 genome sequence is included in repeats longer than 33 nt. Thus, mapping of 36-nt reads was assumed to provide unique positions in more than 99.1% of the genome.
The narrow size distribution of the sequenced fragments (200 ± 20 nt) allowed us to extend the reads in silico to 180 nt. The read extension is limited to the lowest library insert size that we observed, based on Agilent 2100 Bioanalyzer results (Supplemental Fig. 2), which show no insert population below 180 nt. Such a strategy is legitimate for transcript reads obtained from intronless species, where transcripts do not undergo splicing. Taking the data at face value, we observe 20% less coverage when mapping unextended reads, resulting in less coverage of 3′ regions, which are covered by library inserts (Supplemental Fig. 3a) but not by sequence reads (Supplemental Figs. 3b,c, 4). Due to the strand specificity of the protocol, the very last bases of any transcript are poorly covered. This bias may be overcome simply by generating longer reads, by paired-end (PE) sequencing, or by employing 454 Life Sciences (Roche) pyrosequencing.
In order to determine the coverage saturation, we estimated the ratio between the number of mapped positions within coding regions to the total length of all open reading frames (Fig. 2A). We estimated the coverage to be complete when the inclusion of additional read data did not significantly increase this parameter (Supplemental Fig. 5a). This conclusion was backed up by adding data from an M129 RNA biological replicate (one flowcell lane) (Supplemental Fig. 5b). To visualize genome-wide expression in M129, strand-specific, single-base resolution plots were prepared (Fig. 3; Supplemental Fig. 6). These resources provide a way to study the genomic landscape of gene expression. Base mapping clearly defined transcript boundaries, and no systematic error due to read extension could be detected (Fig. 3C). In addition, we observed even coverage along genes (Supplemental Fig. 3a). With unextended reads, overrepresentation of 5′ ends was detected. Underrepresentation of sequences at 3′ ends is a protocol-inherent feature in case of short-read, single-end sequencing (see above).
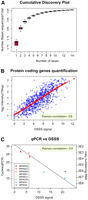
Comparative evaluation of deep sequencing. (A) Cumulative coverage of read data set, expressed as the ratio of sequenced bases located in annotated genes and the total number of bases in annotated genes. Exhaustive coverage is reached after 14 lanes (Supplemental Fig. 5). (B) Comparison of dynamic range of expression measurements (protein coding genes only) on tiling array and DSSS expressed on a log2 scale. (C) Validation of DSSS results using qPCR. DNase-treated RNA was reverse transcribed and subjected to SYBR green real-time PCR. Non-reverse-transcribed controls were included for each gene, as well as a genomic DNA dilution series. DSSS signal corresponds to the log2 transformed mean number of counts along the gene.
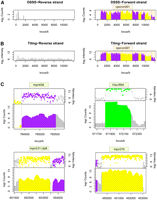
Visualization of the expression data collected from DSSS and tiling array data. Plots showing the transcription signals detected within genome coordinates 1–12,000 nt on the forward and reverse strand of the M. pneumoniae M129 genome. (A) DSSS deep sequencing data. (B) Tiling array data. Note the higher dynamic range and better signal/noise ratio of DSSS in comparison with tiling arrays. (C) Comparison of tiling array intensity signal and DSSS count (both on log2 scale) for Mycoplasma genes encoded on the forward or on the reverse strand. Transcript boundaries identified by DSSS do not show any systematic tendency compared with tiling arrays. mpn638: forward strand; 10sa RNA: forward strand; mpn531: reverse strand; mpn376: reverse strand. Purple/yellow, protein coding genes; green, RNA coding genes; gray, regions without annotated genes.
M129 transcriptome: Characterization and de novo gene discovery
We mapped 1.56 × 1010 sequenced bases to the M129 genome, counting the base content of extended reads. We estimated the expression of every transcribed base by counting the number of reads containing such a base. We detected 98% of the annotated open reading frames to be transcribed under the conditions used. Although the majority of the reads mapped to known gene predictions, 28% of reads were from intergenic regions, suggesting the existence of so far unknown transcripts in the Mycoplasma genome. Twenty-five percent of the reads mapping to intergenic regions overlap with transcriptional units on the opposite strand. Absence of correlation between both strands in DSSS (Supplemental Fig. 6), the existence of large sequence tracts devoid of any transcription (e.g., reverse strand of operon 001 in Fig. 3A), and the correlation with tiling arrays support the strand specificity of the protocol. Strand-specific DSSS data and low background in comparison to tiling arrays permitted the detection of antisense transcription (Fig. 4A,B). We report the details of two examples, one from the forward strand and another from the reverse strand (Fig. 4). These two examples are part of a larger data set where DSSS has been used in combination with tiling arrays to identify 117 new, mostly noncoding transcripts, 89 of them in antisense to known genes (Güell et al. 2009).
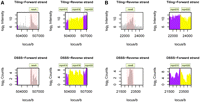
Visualization of antisense transcription. Detection of antisense transcription in Mycoplasma using tiling arrays or DSSS. Both technologies detect antisense transcription, but background is much reduced in DSSS. For the sake of clarity, annotated genes are displayed alternately colored purple or yellow. Signal recorded in regions lacking annotation is shown in gray. (A) M. pneumoniae M129 genome, interval 503,000–507,000 bp. Antisense transcription is detected on the forward strand. Reverse strand annotation at this position corresponds to open reading frame mpn420. (B) M129 interval 22,000–24,500 bp. Antisense transcription is detected on the reverse strand. Forward strand annotation at this position corresponds to open reading frames mpn019 and mpn020.
A case study: The ftsZ gene cluster
Detailed analysis of gene operons can help untangling gene function and regulation. Here, we provide an example of how DSSS could be used to characterize a gene operon of M. pneumoniae. ftsZ is the most conserved of all known bacterial cell division genes and codes for a homolog of tubulin that is involved in mechanical invagination of a dividing cell. ftsZ is located in the proximity of other genes forming the division/cell wall operon. In Escherichia coli and Bacillus subtilis, these operons are composed of 15 and 16 genes, respectively. In Mycoplasma genitalium and in M. pneumoniae, both lacking a cell wall, only six genes constitute the ftsZ operon (Fig. 5A). Benders et al. (2005) have previously provided an in-depth description of the ftsZ locus in the M129 strain, using standard molecular biology techniques. We compared DSSS to their published results. DSSS provided a precise quantification of the different transcript levels. Gene levels as determined by RT-PCR (Benders et al. 2005) were statistically equivalent to the DSSS-based gene expression measurement with the exception of mpn316 (Fig. 5B).
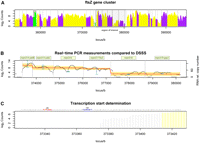
ftsZ gene cluster characterization. (A) Detection of transcription by DSSS in the interval between 355 kb and 395 kb on the forward strand of the M. pneumoniae M129 genome. Protein coding genes are alternately displayed in purple or yellow; genes encoding structural RNA are shown in green. Vertical gray lines separate annotated genes. The gray box at 373–380 kb highlights the ftsZ gene cluster. (B) Real-time PCR expression measurement compared to DSSS. Gene names are indicated in pale green boxes. Blue, transcript copy number estimated from RT-PCR (Benders et al. 2005); black, the DSSS signal; red, the average DSSS signal; orange, the confidence interval of gene expression (DSSS signal ± SD). Note that DSSS signal and RT-PCR measurements overlap in five of six genes. (C) Transcription start site determination with base-pair precision using DSSS. The first signal different from zero is used to determine the transcription start site. Blue, the predicted −10 region; red, −35 region. The result is in agreement with published results from Benders et al. (2005).
DSSS identified the transcription start site of the ftsZ operon (Fig. 5C), in agreement with previously performed primer extension and RNase protection assays (Benders et al. 2005). In a more general study, DSSS single base resolution provided a detailed map of the M. pneumoniae consensus promoter. The preferred −10 motif has been observed to be TAAAAT, and there is also a weak conservation of the TTGAXX and TTTAXX −35 motifs (Güell et al. 2009).
Statistical assessment and comparison with tiling arrays
DSSS displayed a high degree of reproducibility. Lanes from the same library in different flow cells (Illumina technical replicates) had a Pearson correlation of >0.95 (Supplemental Fig. 7). A biological replicate (RNA preparation from different culture of M129) showed a slightly lower Pearson correlation of 0.85–0.9 (Supplemental Fig. 7).
Tiling arrays are well established tools for transcriptome mapping (Carter et al. 2005; Mockler et al. 2005; Royce et al. 2005; Huber et al. 2006). We therefore compared the M129 DSSS data with an eight-base resolution tiling array data set that we had generated previously (Güell et al. 2009). The comparison with tiling array data revealed a strong overlap between both techniques (Fig. 3), with a Pearson correlation coefficient of 0.79 for protein coding genes (Fig. 2B). When considering genes that encode structural RNAs that have a much higher transcript number per cell compared with protein coding genes, this parameter was decreased to 0.74 due to the smaller dynamic range of array-based expression measurements.
Real-time PCR
To further validate DSSS with another standard technique, we performed real-time PCR on a set of eight genes (Supplemental Table 2), chosen according to their differing expression levels in the DSSS data set. We found an overall Pearson correlation between DSSS signal and normalized cycle threshold (Ct) values of 0.9 (Fig. 2C). Expression real-time PCR results not only back up the previous comparison to tiling arrays but also allow confirmation of DSSS results on those genes that, due to their high expression, are out of the dynamic range of tiling arrays.
Applying DSSS on eukaryotic samples
In order to test the DSSS protocol for eukaryotic transcriptomes, we performed paired-end (PE) Illumina sequencing of a mouse brain RNA sample. The sequencing of eukaryotic cDNA required an additional step before adapter ligation, i.e., removal of capped 5′ ends of RNA by the use of the decapping enzyme tobacco acid pyrophosphatase (Brock et al. 1992). The adapter sequences were redesigned to make them compatible with Illumina PE sequencing (Supplemental Table 3). From one PE flow cell run (seven lanes) on the Illumina Genome Analyzer (GA) IIx, we obtained 197 million good-quality reads in total (105 million sequences read 1; 92 million sequences read 2). Prior to sequencing, we had reduced the content of ribosomal RNA (28S, 18S, 5.8S, 5S) in the sample to 57.5% of the total read output.
Of all 148.5 million reads matching mouse RNA sequences, 127.2 million matched uniquely: 20,316 coding sequences were matched by 21.6 million reads, and 1171 noncoding RNA genes (including ribosomal RNA) were matched by the remainder of reads. We calculated the coverage along the normalized lengths of coding genes. The transcript sequences were well covered over the entire length (Fig. 6A). We noted a slightly decreased coverage of 3′ ends in the sequence data, which might be explained by the absence of library inserts containing stretches of polyA sequences. It has previously been shown that sequences from AT-rich regions are underrepresented in data sets generated on the Illumina GA platform (Dohm et al. 2008).
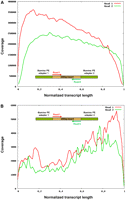
Strand specificity of eukaryotic DSSS. Coverage plot of reads obtained by paired-end DSSS of a mouse RNA Illumina library with an insert size of 200 nt. The reads were mapped against the reference sequences of the mouse transcriptome, transcript lengths were normalized, and the total number of reads in each of 100 bins per transcript was plotted. Only matches in coding sequences were taken into account. The reads keep the directionality information of the insert. Coverage observed is indicated for PE read 1 (red) and PE read 2 (green). (A) Sense reads; (B) antisense reads.
We determined the orientation of matching reads and found the fraction of reads in sense orientation to be 100% for 13,388 transcripts (9860 transcripts matched by 10 or more sense reads) and 99%–95% for 3056 transcripts. The number of transcripts with sense read fractions of 6%–94% was 2969. Reads mapping at the same position were counted only once per position.
In order to assess antisense background noise, we extracted the 1000 most highly expressed transcripts (according to the number of reads matching in sense orientation per number of transcript bases) and counted on average 2406.7 reads in sense orientation and 3.7 reads in antisense orientation, thus, on average, 648 times more sense than antisense reads. This factor can be considered as background rate, assuming that all antisense reads are attributed to background causing effects. Antisense reads were more frequently observed at the 3′ ends of genes (Fig. 6B). This might be explained by read-through from adjacent genes on opposite strands, arranged in tail-to-tail orientation.
Since the splicing process is directional, one would not expect to see exon junctions spanned by reads in antisense orientation. In order to analyze reads spanning exon junctions, we extracted exon information for 18,093 transcripts with more than one exon, resulting in 200,626 exon junctions in total. We considered reads spanning a junction with a minimum of four bases overlap to the right and to the left of the junction and found 95,285 junctions (from 13,044 different transcripts) spanned by 1,224,419 reads at distinct mapping positions. There were 94,611 exon junctions spanned in sense orientation only (by 1,215,075 reads in total) and 141 exon junctions spanned in antisense orientation only (by 231 reads in total). For the 1000 most highly expressed transcripts (see above), we counted 343,581 sense reads and 88 antisense reads spanning exon junctions, resulting in a factor of 3907 more sense than antisense coverage for exon junctions. In some cases, antisense reads spanning exon junctions were mapping artifacts (two mismatches in the four overlapping bases) or arose from inconsistencies between the annotation of the mouse genome and RefSeq mouse transcripts.
A more detailed characterization of the transcriptional activity within the mouse genome was performed by recording sense and antisense transcription within the boundaries of annotated mouse genes and 1000 bp upstream and downstream of these loci. Representative plots are shown in Supplemental Figure 8. We observed sense-coverage of exons in nonrepetitive regions consistent with gene annotation, and low background antisense-coverage (Fig. 7A; Supplemental Fig. 8a–g). Spikes of antisense transcription were observed within the loci of Glo1 and Snap25 (Fig. 7B; Supplemental Fig. 8d). Closer inspection of the data revealed the presence of processed pseudogenes (derived from Rps13 or Rpl21 encoding ribosomal proteins). Since the functional genes and the pseudogene sequences show high similarity, the observed signal most likely does not represent bona fide transcriptional activity. We detected antisense signals at locations of genes with overlapping annotation on different strands (Supplemental Fig. 8e–g).
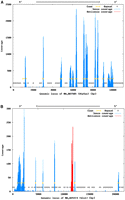
Analysis of mouse gene loci by DSSS. DSSS reads were mapped against the repeat-masked genomic mouse sequences plus 1 kbp upstream of and 1 kbp downstream of the gene locus. Exon annotation (yellow) is derived from the mouse genome annotation. The coverage by DSSS reads is shown in blue (sense orientation) or red (antisense orientation). (A) Atp5a1 locus; (B) Glo1 locus. The antisense signal in the Glo1 locus colocalizes with an Rps13 sequence (see text).
Taken together, the transcriptional patterns revealed by DSSS are highly consistent with the current annotation of the mouse genome.
Discussion
We have tested DSSS in two species, in a simple prokaryotic organism, and in a mammal. We have observed a high degree of strand specificity, single base resolution, and highly covered transcripts. Recent studies have reported exhaustive transcriptomic maps (Cloonan et al. 2008; He et al. 2008; Wilhelm et al. 2008); however, none of them addressed all these aspects in single data sets. Leading to deeper insights of the transcriptome, DSSS will help to escalate the understanding of various biological processes.
Tiling array–based transcriptome mapping correlated well with the Illumina data. However, we can suggest several advantages of DSSS in comparison with tiling arrays: DSSS provides a much higher dynamic range compared with tiling arrays. Transcriptomic experiments face the challenge of interrogating RNA species with abundance profiles differing by several orders of magnitude. We observed a much higher dynamic range using deep sequencing. Arrays spanned six to seven units (log2 scale), whereas sequencing easily reached 15 (Fig. 2B; Supplemental Fig. 9). Thus, DSSS may allow the study of transcripts being expressed at vastly different levels and even comparisons of different groups of RNAs (tRNA, rRNA, mRNA) (Supplemental Fig. 9) within a single experiment, which is currently not possible with array platforms due to their limited dynamic range or with mRNA-seq. The difference in abundance (in log2 scale) between the most abundant (mpns04) and the least abundant (mpn040) transcripts without considering ribosomal RNA was 6.3 in case of tiling arrays but 14.7 using DSSS, exemplifying the different quantitative space addressed in both of the approaches.
Key aspects of gene regulation models are cis- and trans-acting sequences. Presence of these motifs is linked to untranslated region (UTR) boundaries. Thus, it is essential to have an accurate description of such regions in order to characterize promoters and regulatory motifs. We are providing strand-specific data at single-base resolution (Fig. 5C), allowing a precise characterization of the transcripts.
Array experiments tend to be noisy due to cross-hybridization and background problems. Detection of transcripts with low levels of expression, or precise determination of UTR boundaries, could be affected by noise (Royce et al. 2005). Unfortunately, most of the transcribed species are present at levels just above background, in accordance with a transcriptional power-law distribution (Johnson et al. 2005). Therefore, when considering tiling array data, inherent background noise and signal distribution hamper the distinction between signal and background. Of course, this effect is increased in higher eukaryotes, where the percentage of coding DNA is much lower. DSSS provides a higher signal/noise ratio than arrays (Fig. 4; Supplemental Fig. 10). This effect is more apparent in regions that contain lowly expressed transcriptional units. Tiling array background is ubiquitous, whereas no signal is detected in untranscribed regions by DSSS (e.g., reverse strand panels in Fig. 3).
Transcriptome characterization using DSSS provides more accurate mapping data than tiling arrays at decreased experimental costs compared with custom arrays, which require specific design. In addition, tiling arrays are limited to species with known genome sequences, while DSSS is not. Taking advantage of the reasonable coverage reached with a single lane (70%–75%; Fig. 2A), DSSS could be used for addressing differential gene expression in small genomes when comparing two different samples. For Mycoplasma, we obtained an average of 15,303 bases mapped per protein coding gene in each of 15 independently run flowcell lanes. We consider this number large enough to eliminate secondary effects introduced during sample preparation and to allow quantitative comparisons between different samples. Therefore, DSSS provides a tool to interrogate differential expression for all species of transcripts. Arrays are clearly limited by the quality and completeness of the annotations on which they are based. Again, economic aspects and scientific performance of DSSS could outperform current array technologies.
Mapping DSSS reads to mouse transcripts showed strong strand specificity with very low background noise. This was further supported by analyzing reads spanning exon-junctions and by mapping to genomic loci. Cases with coverage by reads in antisense orientation were found to be attributed to overlapping genes on opposite strands or to inserted pseudogenes. Thus, read data generated by DSSS can be used for detailed tracking of eukaryotic gene structure and is providing a basis for comprehensive studies on antisense transcription and regulation.
The DSSS protocol can be easily implemented in any laboratory. Insert sizes of 200 nt make the method compatible with the generation of long sequence reads on the Illumina platform, thus avoiding the need for read extension, which is limited to genomes harboring intronless genes. Since DSSS, so far, supports long inserts up to 350 nt (data not shown), specificity will even increase further, as soon as read length is increased. Present-day Illumina standard runs on the GA IIx yield 75-nt reads, but read length is expected to be increased to 100–150 nt in the near future. In addition, the insert size that our method is able to provide makes it suitable for 454 Life Sciences (Roche) pyrosequencing, which presently supports read lengths of ∼500 nt.
In summary, DSSS is a simple and fast strand-specific sequencing method that is as accurate as standard conventional quantitative molecular biology protocols and that allows the detection of all coding and noncoding RNA transcripts. DSSS opens the challenging landscape of antisense transcription, providing an efficient and robust tool to contribute to this still unexplored field. We propose this method for fast and straightforward transcript sequencing.
Methods
Bacterial strains and culture conditions, and mouse tissue
M. pneumoniae strain M129 was grown in 75 cm2 tissue culture flasks with 50 mL of modified Hayflick medium (18.4 g of PPLO broth, 29.8 g of HEPES, 10 g glucose, 5 mL of 0.5% phenol red, 35 mL of 2 N NaOH per liter; horse serum and penicillin were included to a final concentration of 20% and 100 U/mL, respectively) for 96 h at 37°C. Mouse brain was dissected from adult male C57BL/6J mice (Harlan) after cervical dislocation.
Mycoplasma RNA isolation and depletion of ribosomal RNA
After growth, surface-attached mycoplasma were washed once with phosphate-buffered saline (PBS; 0.15 M NaCl, 10 mM sodium phosphate at pH 7.4) and immediately lysed in the cultivation flask by adding 1.5 mL of Qiazol lysis reagent from the Qiagen miRNeasy kit per flask. This isolation method is used for RNA extraction and preserves most of the small RNAs. Content in rRNA was reduced using the Ribominus Transcriptome Isolation kit (Invitrogen), with one round of rRNA depletion. Ten micrograms of total RNA was annealed with 400 pmol biotin-labeled probes targeting the 16S and 23S rRNA by incubation for 5 min at 37°C. Streptavidin-coated magnetic beads were added and allowed to bind for 15 min at 37°C with occasional gentle mixing. Finally, we placed the reaction for 1 min in a magnetic stand and collected the supernatant, containing the rRNA-depleted fraction. The RNA was precipitated by addition of 0.1 vol of 3 M sodium acetate (pH 5.2), 3 μg of glycogen (Ambion), and 2.5 vol of 100% ethanol, with 30 min incubation at −80°C and spinning for 25 min at 13,200 rpm. The dried pellet was dissolved in 10 μL of RNase free water.
Mouse RNA isolation and depletion of ribosomal RNA
Mouse RNA was isolated from brain tissue using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Two rounds of rRNA depletion using the Ribominus Eukaryote kit for RNA-seq (Invitrogen) were performed. Eight micrograms total mouse RNA was annealed with 800 pmol biotin-labeled probes targeting the 5S, 5.8S, 18S, and 28S rRNA by incubation for 5 min at 75°C, followed by a gentle ramp (0.1°C/ sec) to 37°C in a PCR thermocycler. Thereafter, treatment was as described above for mycoplasma RNA.
Fragmentation of rRNA depleted total RNA
RNA was fragmented to sizes between 60 and 200 nt using the RNA Fragmentation Reagents kit (Ambion), based on metal-catalyzed heat fragmentation. We added 1.1 μL of 10× fragmentation reagent (buffered zinc solution) to 10 μL of RNA and incubated for 5 min at 70°C. The reaction was terminated by adding 1.1 μL of stop solution (containing a metal chelating agent) and chilling on ice. After ethanol precipitation, the pellet was dissolved in 16 μL of RNase free water.
Decapping of fragmented mouse RNA
Cap structures at mRNA 5′ ends were removed with tobacco acid pyrophosphatase (TAP). A 25-μL reaction containing 16 μL of fragmented RNA, 2.5 μL of 10× TAP buffer, and 5 U of TAP (Epicentre) was incubated at 37°C for 2 h.
RNA dephosphorylation
We dephosphorylated the 5′ ends of fragmented RNA (100 ng) by treating with 10 U of calf intestinal phosphatase (New England BioLabs) in standard buffering conditions (50 mM Tris-HCl at pH 7.9, 100 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol) in a final volume of 25 μL for 30 min at 37°C. Following phenol:chloroform extraction, the RNA was precipitated in the presence of 15 μg of glycogen and 62.5 μL of 100% ethanol by incubation of 30 min at −80°C, followed by centrifugation at 13,200 rpm for 25 min. The pellet was washed once with 750 μL of 70% ethanol, dried on ice for 5 min, and dissolved in 10 μL of RNase free water.
Size selection
Size selection of fragmented RNA was achieved by denaturing urea-PAGE in a precast 6% TBE-urea gel (Invitrogen) at 200 V for 1 h. After staining with ethidium bromide, we excised the band corresponding to 200 nt (mycoplasma sample) or to 150 or 200 nt (mouse samples) and eluted in 300 μL of SRA + 0.3 M NaCl buffer (Illumina small RNA sample preparation kit) by incubation for 4 h at room temperature with gentle agitation. We then precipitated the eluted material by addition of 3 μg of glycogen and 750 μL of 100% ethanol, followed by 30-min incubation at −80°C and spinning for 25 min at 13,200 rpm. The pellet was dissolved in 5.4 μL of RNase free water.
3′ adapter ligation
We ligated a 3′ adapter to the size-selected RNA fragments. The 3′ adapter is modified at its 3′ end with an idT (inverted deoxythimidine) moiety, which renders it blocked in order to prevent further ligation reactions with any phosphorylated 5′ end. For mycoplasma sample preparation, we used a 3′ adapter compatible with single-end sequencing, while a PE compatible adapter was ligated to the mouse RNA (Supplemental Table 3). DMSO was added to the reaction to minimize any secondary structure formation in the RNA molecules. The reaction was set up as follows: 5.4 μL of sample, 0.6 μL of 3′ adapter (100 μM), 1 μL of 10× T4 RNA ligase buffer (50 mM Tris-HCl at pH7.8, 10 mM MgCl2, 1 mM ATP, 10 mM DTT), 1 μL of DMSO (Sigma), 1 μL of RNase out (Invitrogen), 20 U of T4 RNA ligase 1 (Invitrogen). The ligation reaction was performed for 6 h at 20°C, followed by 15-min incubation at 65°C to heat inactivate the T4 RNA ligase.
Rephosphorylation of 5′ ends and fragment purification
RNA fragments with the ligated 3′ adapter were phosphorylated on the 5′ end as a prerequisite for attachment of a 5′ adapter in the next step of the protocol. We incubated the following reaction mixture for 30 min at 37°C: 10 μL of sample, 1 μL of 10× T4 RNA ligase buffer (as fresh ATP supply), 10 U of polynucleotide kinase (New England BioLabs), 3 μL of RNase free water. After addition of 2× loading dye and incubation for 5 min at 65°C, the reaction was loaded onto a denaturing urea-PAGE gel in order to separate the fragments with ligated 3′ adapter from nonligated adapter (band sizes: insert size + 23 nt for single end sequencing; insert size + 34 nt for paired end libraries). The procedure of size selection was as described above, and the pellet was finally dissolved in 4.7 μL of RNase free water.
5′ adapter ligation and fragment purification
We used a 5′ adapter compatible with single-end sequencing for mycoplasma sample preparation, while a PE compatible 5′ adapter was ligated to the mouse RNA (Supplemental Table 3). The ligation reaction with the 5′ adapter was set up as follows: 4.7 μL of sample, 1.3 μL of 5′ adapter (100 μM), 1 μL of 10× T4 RNA ligase buffer, 1 μL of DMSO, 1 μL of RNase out, 20 U of T4 RNA ligase. The ligation reaction was performed for 6 h at 20°C. A fragment consisting of insert RNA and ligated 5′ and 3′ adapters was purified by denaturing urea-PAGE, and the recovered fragment was dissolved in 4.5 μL of RNase free water.
First strand reverse transcription and PCR amplification
We annealed 0.5 μL (100 μM) of the RT primer (Supplemental Table 3) to the sample by incubation at 65°C for 10 min and allowing it to cool down to 48°C. Thereafter, the following was added to the reaction—2 μL of 5× first strand buffer (250 mM Tris-HCl at pH 8.3, 375 mM KCl, 15 mM MgCl2), 0.5 μL of 12.5 mM dNTP mix, 1 μL of 100 mM DTT, 0.5 μL of RNase out—and incubated 3 min at 48°C. Next, we added 200 U of SuperScript II reverse transcriptase (Invitrogen) and allowed first strand synthesis to proceed for 1 h at 44°C. The product of first strand synthesis was amplified in a PCR reaction containing 10 μL of ssDNA, 10 μL of 5× Phusion HF buffer, 0.5 μL of primer 1 (Supplemental Table 3), 0.5 μL of primer 2 (Supplemental Table 3), 0.5 μL of 25 mM dNTP mix, and 0.5 μL of Phusion High-Fidelity DNA polymerase (New England BioLabs). The PCR protocol used for the mycoplasma libraries was as follows: 30 sec, 98°C/17× (10 sec, 98°C/30 sec, 60°C/30 sec, 72°C)/10 min, 72°C/hold at 4°C. Mouse libraries were amplified with 12 PCR cycles.
Purification of the amplified cDNA product
We ran the amplified cDNA in a 6% TBE PAGE at 200 V for 30 min. After staining with ethidium bromide, we excised the band and (insert size + 70 nt for single reads; insert size + 111 nt for PE libraries) and eluted the amplified material in 100 μL of 1× gel elution buffer (Illumina) by gently mixing for 2 h at room temperature. Next, we precipitated the DNA by addition of 1 μg of glycogen, 10 μL of 3 M sodium acetate (pH 5.3), 325 μL of ice-cold absolute ethanol, followed by centrifugation for 20 min at 13,200 rpm at room temperature. We washed the pellet with 70% ethanol and dried it in a SpeedVac (Salvant) for 5 min at 37°C. Finally, we dissolved the sequencing library in 10 μL of resuspension buffer (Illumina). We ran a lab-on-a-chip (DNA 1000, Agilent) to check the size distribution of the library (Supplemental Fig. 2). The library has a sharp size distribution with 200-bp ± 20-bp-long inserts; 180 bp is the shortest observed insert size (Supplemental Fig. 2). Mouse libraries showed similar profiles with 150 nt and 200 nt insert length.
Library quantification
The mouse library was quantified by TaqMan qPCR (Quail et al. 2008). For the mycoplasma sample, we redesigned oligonucleotides: primerF, 5′-AATGATACGGCGACCACCG-3′; primerR, 5′-CAAGCAGAAGACGGCATACG-3′; and probe, 5′-CAGAGTTCTACAGTCCGACGAT-3′.
Sample sequencing and data processing
We loaded samples at a concentration of 3.5–4 pM (mycoplasma) or 10 pM (mouse PE reads), respectively, per flow cell lane and generated clusters in the Illumina cluster station as recommended by the manufacturer. Following annealing of the Illumina sequencing primer (Supplemental Table 3), we performed sequencing runs on the Illumina GA II (35–40 cycle recipes) and on the GA IIx sequencing instrument (2 × 54 cycle recipe). Image analysis and base calling were performed using Illumina pipeline v1.0, v1.3.2 (GA II), and v1.4.0 (GA IIx), respectively. Run statistics using these pipeline versions and ELAND as standard alignment tool are displayed in Supplemental Table 1.
Real-time PCR
We performed SYBR green real-time RT-PCR of eight M. pneumoniae genes (Supplemental Table 2). Primers were designed using Primer3 (http://primer3.sourceforge.net), with an annealing temperature of 60°C and lengths of amplification products of 60–150 bp. We treated 4.2 μg of total RNA with 2 U of Turbo DNase (Ambion) in 1× Turbo DNase buffer in a 50-μL reaction. The reaction was allowed to proceed for 30 min at 37°C. We stopped the reaction by addition of 0.1 vol of DNase inactivation reagent (Ambion) and incubation for 5 min at room temperature, followed by centrifugation (10,000g; 1.5 min). The supernatant contained the DNase-treated total RNA. We synthesized cDNA with the first strand cDNA synthesis kit for RT-PCR (AMV) from Roche according to the manufacturer's instructions. All real-time PCR reactions were performed in triplicate. A 15-μL reaction volume contained the following: 7.5 μL of 2× Power SYBR green PCR master mix (Applied Biosystems), 1 μL of premixed primer pair (5 pmol of each primer), and 32 ng of cDNA. We also performed real-time reactions for non-reverse-transcribed RNA, diluted cDNA (dilutions 1:10 and 1:1000), and Nanodrop-quantified genomic DNA with the following numbers of genome copies: 0/300/3,000/30,000/300,000/3,000,000. Ct values for the serial dilution of genomic DNA allowed us to perform absolute quantification of the transcript levels for each of the genes.
Mapping and analysis of mycoplasma data
Fastq files were processed and mapped to the M. pneumoniae M129 reference genome (NC_000912) with MAQ (Li et al. 2008). We collected a DSSS score for each position of the genome (log2 of the number of times a particular base was sequenced). Average expression of genes was determined by calculating the average log2 expression value for the bases contained within a transcriptional unit, or open reading frame. The ShortReads Bioconductor package (http://www.bioconductor.org) was used to load data into the R software (http://www.r-project.org). R scripting was used for further data processing and display.
Mapping and analysis of mouse data
We analyzed data from seven lanes of one Illumina flow cell with PE reads (insert size, 200 nt) of 54-nt length on either side. For sequence quality reasons, the reads were trimmed to 50 bases (read 1, left end) and 42 bases (read 2, right end), respectively. Reads containing one or more “dots” in the sequence as well as reads that did not pass the filtering of the Illumina basecalling pipeline were discarded, resulting in 105,066,337 (read 1) and 92,126,147 (read 2) reads, respectively. The mouse RNA reference sequences were downloaded from the NCBI ftp server at ftp://ftp.ncbi.nih.gov/refseq/M_musculus/mRNA_Prot/mouse.rna.fna.gz. Exon information for the transcripts was downloaded from the NCBI ftp server at ftp://ftp.ncbi.nih.gov/refseq/M_musculus/mRNA_Prot/mouse.rna.gbff.gz. Genomic loci of genes were downloaded from the UCSC pages at http://genome.ucsc.edu/cgi-bin/hgTables using the following settings: Mammal, Mouse, Juli 2007 (NCBI37/mm9), Gene and Gene Prediction Tracks, RefSeq Genes, refGene. Fasta records were obtained for CDS exons, introns, and the surrounding region 1000 bp upstream/downstream of each locus. Mapping of the sequencing reads against RNA or genomic reference sequences was performed using GenomeMapper (http://1001genomes.org/downloads/genomemapper.html), permitting one insertion or deletion and two additional mismatches between reads and reference sequences (default parameters except for choosing -M 2 and setting the output option -e).
Analyses were performed using Perl v5.8.9 and Unix shell commands. Data plots were generated with Gnuplot v4.2.
Acknowledgments
M.G. is supported by an FPU fellowship. J.C.D. is supported by the GABI FUTURE grant 0315069A of the German Ministry of Education and Research (BMBF). We thank Fundación Marcelino Botín for funding. We thank Ester Castillo for experimental help; and Mònica Bayés, Matthew Ingham, Debayan Datta, and Fan Lai for insightful discussions and comments on the manuscript.
Author contributions: The original idea was formulated by A.P.V. and M.G. A.P.V. developed and implemented the DSSS sequencing protocol. M.G. performed mapping and downstream analysis of Mycoplasma Illumina and tiling array data, and J.C.D analyzed mouse Illumina data. All authors contributed to the preparation of the manuscript. H.H. and L.S. supervised the project.
Footnotes
-
↵5 Current address: Vall d'Hebron Institute of Oncology, Vall d'Hebron University Hospital, 08035 Barcelona, Spain.
-
↵6 Corresponding authors.
E-mail luis.serrano{at}crg.es.
E-mail heinz.himmelbauer{at}crg.es.
-
[Supplemental material is available online at http://www.genome.org. The tiling array CEL files from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE14014, and the Illumina fastq files have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA009091.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.094318.109.
- Received March 25, 2009.
- Accepted April 29, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








