
Abstract
The generation of genome-wide data derived from methylated DNA immunoprecipitation followed by sequencing (MeDIP-seq) has become a major tool for epigenetic studies in health and disease. The computational analysis of such data, however, still falls short on accuracy, sensitivity, and speed. We propose a time-efficient statistical method that is able to cope with the inherent complexity of MeDIP-seq data with similar performance compared with existing methods. In order to demonstrate the computational approach, we have analyzed alterations in DNA methylation during the differentiation of human embryonic stem cells (hESCs) to definitive endoderm. We show improved correlation of normalized MeDIP-seq data in comparison to available whole-genome bisulfite sequencing data, and investigated the effect of differential methylation on gene expression. Furthermore, we analyzed the interplay between DNA methylation, histone modifications, and transcription factor binding and show that in contrast to de novo methylation, demethylation is mainly associated with regions of low CpG densities.
DNA methylation is an epigenetic mechanism involved in transcriptional regulation during embryonic development (Meissner et al. 2008) and reprogramming of somatic cells into induced pluripotent stem cells (Chan et al. 2009; Deng et al. 2009). Results from literature have shown severe effects of aberrant methylation, for example, its association with cancer (Jones and Baylin 2007; Irizarry et al. 2009). Furthermore, distinct genome-wide methylation patterns distinguish different cell-types (Eckhardt et al. 2006; Rakyan et al. 2008). Sequencing-based DNA methylation data are an emerging technology for analyzing epigenetic modifications (Laird 2010). Methylated DNA immunoprecipitation (MeDIP) depends on the use of an antibody specific for methylated cytosines in order to immunocapture methylated genomic fragments (Weber et al. 2005), which can be detected either by tiling arrays (MeDIP-Chip) or by next-generation sequencing (MeDIP-seq). Methylation profiles obtained by the MeDIP approach are not base pair–specific but reflect methylation levels on a resolution restricted by the size of the sonicated DNA fragments after amplification and size selection. In contrast, bisulfite sequencing detects cytosine methylation on a base-pair level. Although whole-genome single-base resolution maps have been generated (Lister et al. 2008, 2009), such techniques cannot yet be cost-effectively applied to screen large sets of sequences or samples. Reduced representation bisulfite sequencing (RRBS) was introduced in order to address this issue by selecting only some regions of the genome for sequencing. Here, reduced representation is achieved by the size-fractionation of DNA fragments after restriction enzyme digestion (Meissner et al. 2008; Laird 2010). In contrast to bisulfite sequencing, MeDIP-seq–derived methylation data are of far lower resolution, and therefore, it remains difficult to discriminate between CpG and non-CpG methylation when single-end short reads are considered. However, MeDIP-seq covers nearly as many CpGs per sample genome as does the more expensive whole-genome shotgun bisulfite sequencing (WGSBS) approach (Laird 2010). An advantage of the MeDIP approach is the generation of unbiased, cost-effective, and full-genome methylation levels without the limitations associated with methylation-sensitive restriction enzymes. The current bottleneck resulting from the advancing technology development in DNA methylation is the computational analysis of the large-scale sequencing data (Laird 2010). It has been previously shown that MeDIP-derived data need to be corrected for local CpG densities in order to compute unbiased methylation levels (Down et al. 2008; Pelizzola et al. 2008). This effect is caused by a varying efficiency of antibody binding and immunoprecipitation dependent on the local density of the methylated CpG sites. Although there are computational methods available for analyzing whole-genome methylation data (Down et al. 2008; Pelizzola et al. 2008), in particular the analysis of MeDIP-seq data remains disproportionally time-consuming. Moreover, important features for the design of MeDIP-seq experiments have not yet been addressed, including quality control metrics and identification of differential methylation. In particular, the number of sequencing reads necessary for obtaining a sufficient coverage of the methylome has to be estimated and the enrichment of CpG-rich short reads relative to the genomic background has to be analyzed in order to provide a quality measure for antibody binding and immunoprecipitation. Finally, there is a need for MeDIP-seq–specific methods that identify events of differential methylation. Here, we present MEDIPS, a comprehensive approach for normalizing and analyzing MeDIP-seq data that is based on the valuable concept of coupling factors presented by Down et al. (2008) but that outperforms computation time by orders of magnitude. As a proof of performance, we processed the available MeDIP-seq sperm data from Down et al. (2008), compared our results to benchmark data from the HEP project (Eckhardt et al. 2006), and show comparable concordance to the results of Down et al. (2008). We further applied our computational analysis approach to the analysis of cellular differentiation of human embryonic stem cells (hESCs). Because hESCs can be induced to differentiate into a wide variety of cell types, these cells hold promise for cell replacement therapy (Altun et al. 2010). Differentiation of hESCs along the endodermal lineage is induced by treatment with Activin A, a member of the TGFβ family of ligands (D'Amour et al. 2005; Agarwal et al. 2008), resulting in definitive endoderm (DE). We derived DE cells from hESCs and analyzed the resulting transcriptome and methylome profiles of both cell types using the Illumina bead array platform and MeDIP-seq technologies. Analogous to the study of Lister et al. (2009), we identified a large number of demethylation events, emphasizing an important role of demethylation during the differentiation of hESCs. In addition, we identified de novo methylation events at key regulatory acting genomic regions (e.g. in the POU5F1 transcription factor [TF] promoter). The entire computational approach (MEDIPS), including data processing, quality control, normalization, statistical analysis of differential methylation, and methods for the simulation of read coverage and saturation has been made available as an R software library. MEDIPS is suitable for any arbitrary genome available via Bioconductor's annotation libraries (Gentleman et al. 2004). Our results show that MEDIPS is an efficient approach for a genome-wide methylation analysis that significantly reduces the imbalance of sequencing data generation and analysis and that can assist further studies aiming to understand and characterize the function of DNA methylation.
Results
MEDIPS—MeDIP-seq data normalization performance
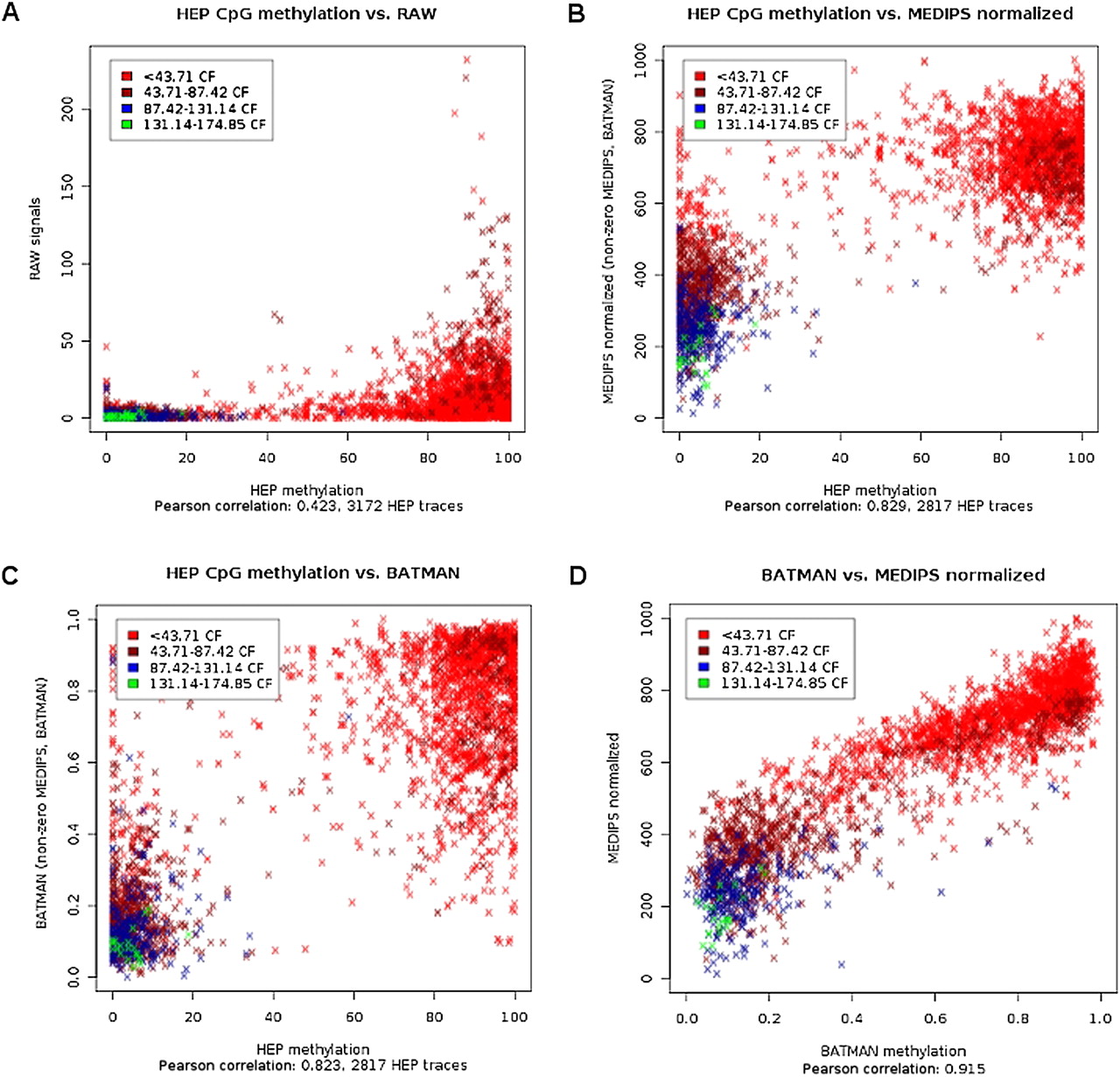
The rationale behind our normalization method is based upon the concept of coupling factors presented by Down et al. (2008). Based on a selected distance function for calculating coupling factors, we estimated the dependency between increasing total CpG density and increasing mean MeDIP-seq signals for the low range of coupling factors. Instead of transferring the identified normalization parameters to a computationally demanding Bayesian deconvolution process (Down et al. 2008), MEDIPS weighs the raw MeDIP-seq signals with respect to the estimated coupling factor–dependent normalization parameters. The main impact of this simplification is a significantly reduced run time for processing MeDIP-seq data by orders of magnitude. Moreover, CpG coupling factor–based normalization methods do not require an artificial reference experiment using fully methylated samples, as proposed by Pelizzola et al. (2008). For a detailed description of the normalization method performed by MEDIPS, see the Supplementary Methods. In order to test the performance of the MEDIPS procedure, we processed the MeDIP-seq data derived from a sperm sample published by Down et al. (2008). Additionally, we downloaded the normalized methylation values provided by Down et al. (2008) as well as the benchmark methylation data derived from bisulfite-sequencing of another sperm sample generated by the human epigenome project (HEP) (Eckhardt et al. 2006). Because we have mapped our short reads against the latest human genome build (hg19), we always transform genomic coordinates from other public sources to the hg19 build using UCSC's Batch Coordinate Conversion (liftOver) software (Rhead et al. 2010). The analysis revealed that both normalization methods improved the poor correlation of raw data from MeDIP-seq and bisulfite sequencing from a Pearson correlation of 0.42 (Fig. 1A) to 0.83 (MEDIPS) (Fig. 1B) and 0.82 (Batman) (Fig. 1C), respectively, and that both methods have a high correlation of 0.92 (Fig. 1D).
Normalization of MeDIP-seq data. We compared the normalization results of the MEDIPS method by processing publicly available MeDIP-seq data (Down et al. 2008) against bisulfite sequencing–derived methylation data from sperm samples (human epigenome project [HEP]) (Eckhardt et al. 2006). Each data point represents a genomic region analyzed by bisulfite sequencing (Eckhardt et al. 2006). The color code refers to four quantiles of the mean coupling factors (CpG densities) for these regions. Correlation plots show (A) raw MeDIP-seq signals (y-axis), (B) MEDIPS normalized signals (y-axis), and (C) Batman (Down et al. 2008) normalized (y-axis) signals against bisulfite data (x-axis) from the HEP project (Eckhardt et al. 2006). (D) Comparison of MEDIPS normalized against Batman (Down et al. 2008)-normalized MeDIP-seq data for the same genomic regions.
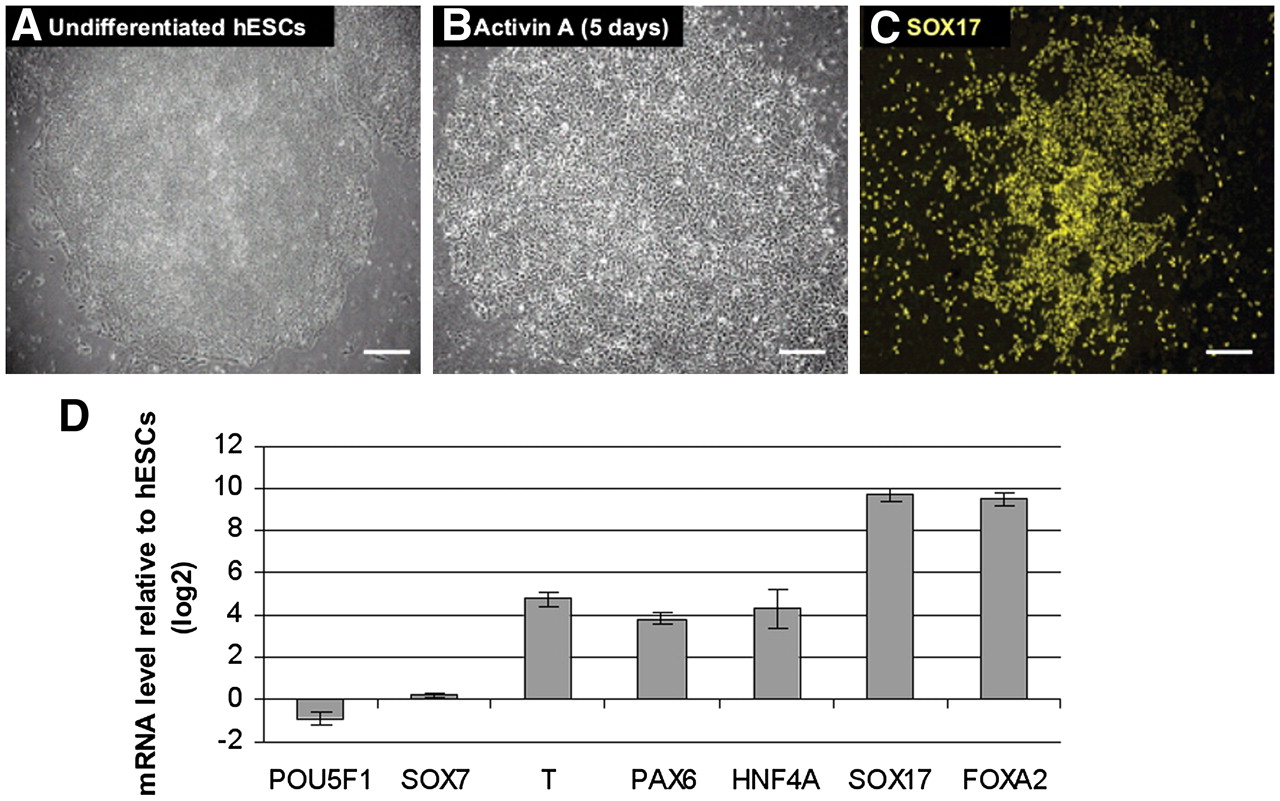
Differentiation of hESCs into DE
Upon treatment with Activin A (100 ng/mL) for 5 d, undifferentiated hESCs (H1, passage 53) changed morphology from typical, defined, tight colonies (Fig. 2A) into less dense, flatter cells (Fig. 2B; D'Amour et al. 2005). In order to confirm the differentiation into DE, we detected the expression of SOX17 using immunostaining (Fig. 2C) and investigated lineage-specific gene expression patterns by real-time RT-PCR (Fig. 2D). After 5 d of Activin A treatment, the majority of cells was devoid of the pluripotent marker POU5F1 but, however, showed expression of the TFs SOX17 and FOXA2, which are markers of DE. Importantly, there was a low level expression of the TF SOX7 (expressed in primitive endoderm but not in DE). This implies that the induction of SOX17 and FOXA2 expression was not a result of differentiation into primitive endoderm. PAX6 expression is detectable, demonstrating the presence of some ectodermal cells. Moreover, Brachyury (T) expression was also detected, which might imply a transition through the primitive streak stage of development. Furthermore, HNF4A is up-regulated and therefore indicative of early hepatic-like characteristics of the Activin A–treated cells.
Derivation of definitive endoderm from human ES cells. Phase contrast image of undifferentiated human ES cells (hESCs; A) and cells after 5 d of Activin A treatment (B). (C) Immunofluorescence labeling of differentiated cells showing SOX17 expression. Scale bars, 100 μm. (D) Effect of Activin A treatment on the gene expression of selected genes during differentiation of human ES cells. The ratios represent the mean of two independent biological replicates. Bars, SE between the biological replicates.
MEDIPS—MeDIP-seq quality control metrics
Based on the high-quality mapping hits of the generated short reads from hESCs, DE, and input (see Methods), we first performed saturation analyses resulting in genome-wide coverage saturation of 0.94 for hESCs and 0.96 for DE (see Supplemental Fig. 1A,B). Because the constellation of DNA fragments that have to be sequenced is much higher for the input samples than for the immunoprecipitated samples, the estimated saturation for the input sequences is lower (0.75) (see Supplemental Fig. 1C). Coverage analysis shows a good CpG coverage saturation of the approximately 28.2 million CpGs of the human genome. In the hESCs sample 22.4 million CpGs (79%), in the DE sample 23.2 million CpGs (82%), and in the input sample 25.4 million CpGs (90%) were covered at least once (see Supplemental Fig. 1D–F). The genome-wide Pearson correlation obtained when comparing MeDIP-seq data from the hESCs and DE samples is 0.9 (see Supplemental Fig. 2A,B). Moreover, we tested the enrichment of CpG-rich short reads derived from the immunoprecipitation step, and found a relative enrichment for CpG-rich short reads from the hESC sample (2.11) and DE sample (2.59) compared with the reference genome, whereas, as expected, the relative CpG enrichment is close to one (1.16) for the combined input samples (see Supplemental Table 1). Finally, the calibration curves clearly reveal the dependency between increasing MeDIP-seq signals and increasing local total CpG densities for the hESCs and DE samples resulting from immunoprecipitation, but not for the input sample (see Supplemental Fig. 3A–C). For a detailed description of the quality controls, see Supplementary Methods.
Comparing MeDIP-seq and WGSBS derived methylation profiles in hESCs
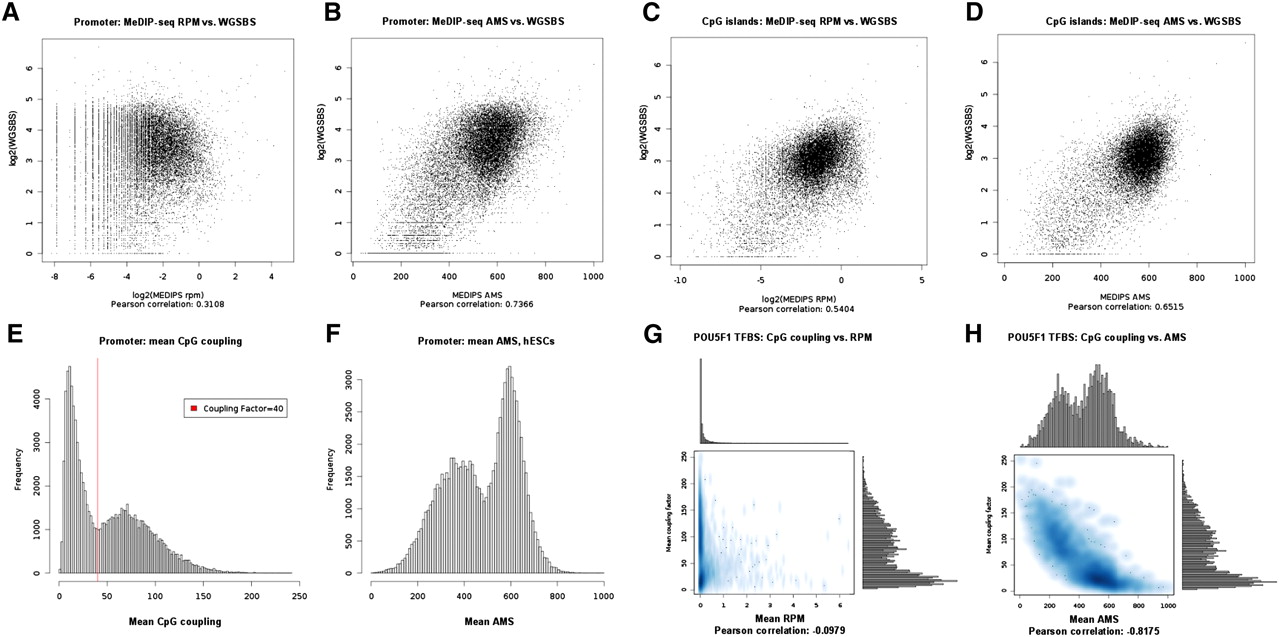
Recently, Lister et al. (2009) presented a genome-wide map of methylated cytosines in hESCs at base resolution generated from 1.16 billion short reads of a WGSBS approach. Moreover, they showed that 25% of all methylated cytosines in hESCs exist in a non-CpG context. Although MeDIP-derived methylation signals are not at a base resolution level, we were interested in comparing mean MeDIP-seq and mean WGSBS methylation values for defined regions of interest. We divided all the Ensembl (Birney et al. 2004) transcript proximal promoters (−1 kb to +0.5 kb around their transcription start sites [TSSs]) of chromosome 1 into 500-bp windows and calculated mean WGSBS-derived CpG methylation values and mean un-normalized (reads per million [rpm]) MeDIP-seq values from hESCs on that resolution. The scatterplot in Figure 3A shows a low correlation of the raw data of 0.31. Figure 3B shows that this correlation increases to 0.74 by normalizing the rpm MeDIP-seq signals into absolute methylation signals (ams) using MEDIPS. For CpG islands (Takai and Jones 2002), the correlation between mean rpm MeDIP-seq and mean WGSBS values is 0.54 (see Fig. 3C) and increases to 0.65 with MEDIPS normalized ams values (see Fig. 3D).
Promoter, CpG islands, and TFBS methylation and comparison to WGSBS. (A) We divided Ensembl (Birney et al. 2004) transcript promoters of chromosome 1 into 500-bp windows and show that mean WGSBS and mean reads per million (RPM) MeDIP-seq signals have a correlation of 0.31. (B) The WGSBS vs. MeDIP-seq correlation is increased to 0.74 after MEDIPS normalization of the MeDIP-seq signals into absolute methylation signals (AMS). For CpG islands, the correlation between mean rpm MeDIP-seq and mean WGSBS values is 0.54 (C) and is increased to 0.65 after MEDIPS normalization of the MeDIP-seq signals into ams (D). (E) DNA sequences underlying human promoters show a bimodal distribution of CpG densities (calculated as means of CpG coupling factors). By setting the coupling factor = 40, we define a threshold for discriminating between low (LCPs) and high (HCPs) CpG density promoters. (F) MEDIPS normalized ams reveal the bimodal promoter methylation distributions in hESCs. (G) POU5F1 binding sites (Lister et al. 2009) show low negative correlation (−0.10) between CpG density and un-normalized rpm values in hESCs. (H) MEDIPS normalized ams values reveal the negative correlation (−0.82) between CpG density and methylation present in POU5F1 binding sites. Interestingly, mean CpG coupling factors and mean normalized ams values indicate bimodal CpG density and methylation distributions of POU5F1 TFBSs.
Promoter methylation
We have, in particular, analyzed CpG density and methylation distributions in proximal promoter sequences (−1 kb to +0.5 kb around the TSSs) of 96,016 Ensembl (Birney et al. 2004) transcripts. Figure 3E shows the well-known bimodal CpG density distribution present in human promoters but calculated based on CpG coupling factors. By visual inspection of the plot, we define the coupling factor = 40 as threshold for discriminating between low CpG density (LCP; 48,021 transcripts) and high CpG density (HCP; 47,995 transcripts) promoters. Whereas bimodal promoter methylation is not obvious when considering non-normalized rpm MeDIP-seq signals from hESCs (see Supplemental Fig. 4A), MEDIPS-normalized ams MeDIP-seq values reveal the bimodal promoter methylation distribution present in hESCs (see Fig. 3F) and in DE (see Supplemental Fig. 4B). Consistent with previous findings (Koga et al. 2009), we observe distinct patterns of LCP and HCP methylation based on our MeDIP-seq data. For hESCs, a large fraction of LCPs (22,104, 46%) is highly methylated (mean ams ≥600), whereas only 3488 (7%) LCPs show low methylation levels (mean ams ≤400). For HCPs, this observation is reversed: 33,196 (69%) HCPs are lowly methylated, whereas only 189 (<1%) HCPs are highly methylated. For DE, a similar trend was observed (data not shown).
Methylation patterns of TF binding sites
We tested the ChIP-seq derived TF binding sites (TFBSs) of six TFs as presented by Lister et al. (2009) for mean CpG densities, rpm values, and ams values in hESCs. As shown in Figure 3G, a low negative correlation between CpG density and un-normalized rpm values is observed for POU5F1 TFBSs (Pearson correlation −0.10). An increased negative correlation is obtained (−0.82) when MEDIPS-normalized ams values are considered instead. Interestingly, mean CpG coupling factors and mean normalized ams values indicate bimodal CpG density and methylation distributions of POU5F1 TFBSs (see Fig. 3H). In addition to POU5F1, the binding sites of KLF4 and TAF1 show bimodal CpG density and ams distributions (see Supplemental Fig. 4C–F). In contrast, NANOG, SOX2, and EP300 binding sites cannot be distinguished into two groups of CpG densities or ams (see Supplemental Fig. 4G–L).
Identification of differentially methylated regions
Based on the MeDIP-seq data from hESCs, DE, and input, MEDIPS identified 62,142 distinct genome-wide regions that become demethylated during the differentiation of hESCs into DE (see Supplementary File 1). On the other hand, MEDIPS identified 10,435 genomic regions where de novo methylation occurs during the first step of differentiation along the endodermal linage (see Supplementary File 2). For a detailed description of the detection of differentially methylated regions (DMRs), see Methods. The comparatively higher number of demethylated regions compared with de novo methylated regions emphasizes the important role of demethylation during embryonic differentiation. As a comparison, Lister et al. (2009) identified approximately 6 million cytosines with higher levels of methylation in hESCs compared with differentiated fetal lung fibroblasts, and only 124,162 cytosines with higher levels of methylation in fetal lung fibroblasts compared with hESCs. From the 491 regions that are hypomethylated in hESCs compared with fetal lung fibroblasts (Lister et al. 2009), we also identified 62 regions (13%) where a de novo methylation event occurs during the differentiation into DE and only five regions (1%) that appear more methylated in hESCs compared with DE. Moreover, we tested the overlap between the identified genomic regions that become demethylated during differentiation of hESCs into DE and the partially methylated domains (8088 PMDs) identified by Lister et al. (2009) in fetal lung fibroblasts. From the 8028 PMDs remaining after transformation of the genomic coordinates to the hg19 genome build, 3067 (38.2%) overlap with at least one of the DMRs.
Genome-wide distribution of DMRs
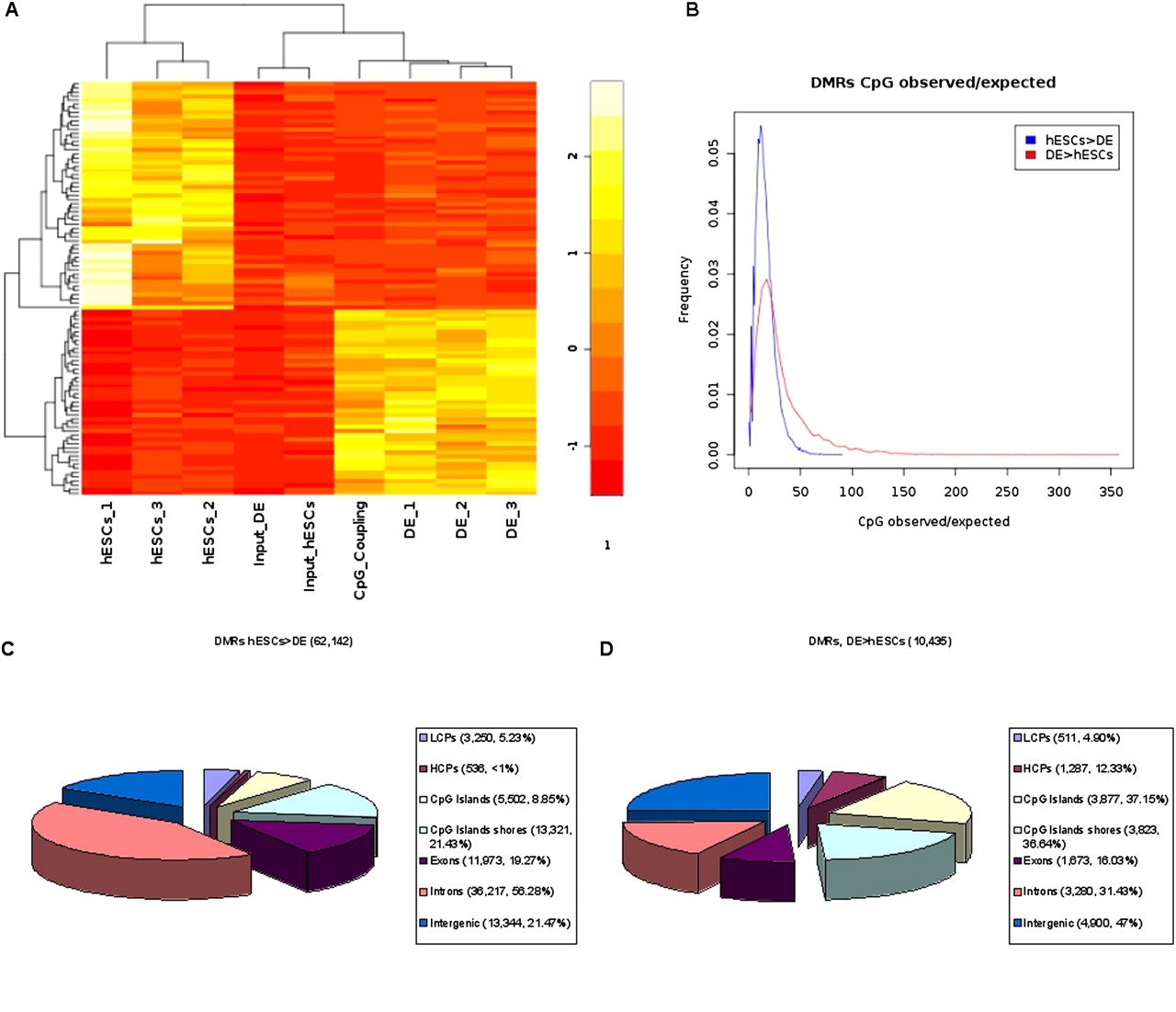
Figure 4A shows rpm MeDIP-seq values for the three biological replicates of hESCs, DE, and the input samples for a subset of the identified DMRs selected by highest variances between samples. This clustering approach clearly separates the hESCs, DE, and input samples into distinct groups. Additionally, the heatmap contains scaled CpG coupling factors of the DMRs. Interestingly, the DMRs that become demethylated during the differentiation of hESCs are associated with low CpG densities, and the DMRs that become de novo methylated are associated with higher CpG densities (see Fig. 4A). In addition to this observation, we calculated CpG observed/expected (obs/exp) (Gardiner-Garden and Frommer 1987) ratios as a measure for CpG density with respect to the amount of cytosines present in both strands of the DNA for both sets of DMRs, separately. Whereas the majority of demethylated regions are associated with very low CpG obs/exp ratios, de novo methylated regions tend to be associated with higher CpG obs/exp ratios, indicating higher densities of CpGs (see Fig. 4B). For the identified demethylated (Fig. 4C) and de novo (Fig. 4D) methylated regions, we tested if they overlap with HCPs (high CpG promoters) or LCPs (low CpG promoters), CpG islands (Takai and Jones 2002), the CpG island shores, exons, and introns, or if they are intergenically located. In addition, we have calculated the enrichment of DMRs with respect to selected regions of interest. Interestingly, a higher percentage of de novo methylated regions overlap with promoters (17.23%) or CpG islands (37.15%) compared with the percentage of demethylated regions (6.09% and 8.85%, respectively). For CpG islands, the DMR enrichment is 2.53 for demethylated regions and 11.20 for de novo methylated regions. We observed that less than 1% of all demethylation events occur within HCPs (enrichment of 0.49), whereas 12.33% of all de novo methylated regions overlap with HCPs (enrichment of 8.08). The percentage of demethylated regions that overlap with introns is considerably higher (56.28%) compared with the percentage of de novo methylated regions (31.43%). In total, a large fraction (78.53%) of all genome-wide demethylation events can be associated with transcript bodies or proximal promoters associated with 12,930 unique Ensembl (Birney et al. 2004) gene names (including miRNAs and others), whereas 53% of all de novo methylation events can be associated with the gene regions or proximal promoters of 4787 unique Ensembl genes.
Differentially methylated regions (DMRs). (A) Heatmap of 100 DMRs, selected by highest variances between samples, including mean rpm signals for the three biological replicates of hESCs, and DE cells, the input sample from hESCs, the input sample from DE, and scaled mean CpG coupling factors. Differential methylation was calculated based on the pooled sets for hESCs, DE, and input (see Methods and Supplementary Methods). (B) Distributions of CpG observed/expected (Gardiner-Garden and Frommer 1987) ratios for demethylated regions (hESCs > DE) and de novo methylated regions (DE > hESCs). The identified demethylated (C) and de novo (D) methylated regions were annotated for Ensembl (Birney et al. 2004) transcript promoters (−2 kb to +0.5 kb regions around their TSSs; divided into LCPs and HCPs), CpG islands (Takai and Jones 2002) and their shores (−0.5 kb from the start or +0.5 kb from the end of a CpG island), exons, introns, and intergenic regions (no overlap with promoters and transcript bodies). Regions can be associated to more than one annotation (e.g., exon and CpG island).
Differential methylation at TFBSs
We have tested the TFBSs of six TFs in hESCs as published by Lister et al. (2009) for overlap with regions identified as differentially methylated during endodermal differentiation of hESCs. In total, DMRs are not significantly enriched for any of the sets of TFBSs (data not shown). However, demethylations and de novo methylations occur within the genomic regions identified as binding regions of the TFs. For example, from the 3889 POU5F1 binding sites (Lister et al. 2009), there are 130 regions that become de novo and only 14 regions that become demethylated. Interestingly, although there are in total six times more DMRs that become demethylated than de novo methylated, the majority of DMRs that overlap with the TFBSs are associated with de novo methylation for all six TFs (see Supplemental Table 2). Binding regions of the class of TFs that show bimodal methylation distributions (these are POU5F1, KLF4, and TAF1) overlap more than twice as much with DMRs than TFBSs targeted by NANOG, SOX2, and EP300.
Enrichment analysis associates demethylation events to functional histone modifications
In order to further examine the identified DMRs, we performed overrepresentation analyses for the demethylated and de novo methylated regions, separately, using the statistical analysis software EpiGRAPH (for the full results, see Supplemental Tables 3, 4; Bock et al. 2009). Most interestingly, demethylation events are significantly enriched within regions associated with high signals of gene activating histone modifications (Table 1; Barski et al. 2007). On the other hand, events of demethylation are strongly under-represented within regions associated with higher signals of gene silencing histone modifications (Barski et al. 2007).
Overrepresented histone modifications in DMRs

[i] The table shows all histone modifications (Barski et al. 2007) that are highly significantly (Bonferroni-corrected) enriched (or under-represented, respectively) within the identified demethylated and de novo methylated regions. Statistical analysis was performed using EpiGRAPH (here, EpiGRAPHSs overlapRegionsCount annotation was considered) (Bock et al. 2009). + indicates associated with gene activation (Barski et al. 2007); −, associated with silent genes (Barski et al. 2007); controversial, no distinct effect (Barski et al. 2007); hESCs > DE, DMRs higher methylated in hESCs compared to DE; DE > hESCs, DMRs higher methylated in DE compared with hESCs; —, not associated to DMRs; ratio control/DMRs <1, overrepresented in DMRs; and ratio control/DMRs >1 underrepresented in DMRs.
Differential methylation and gene expression alterations
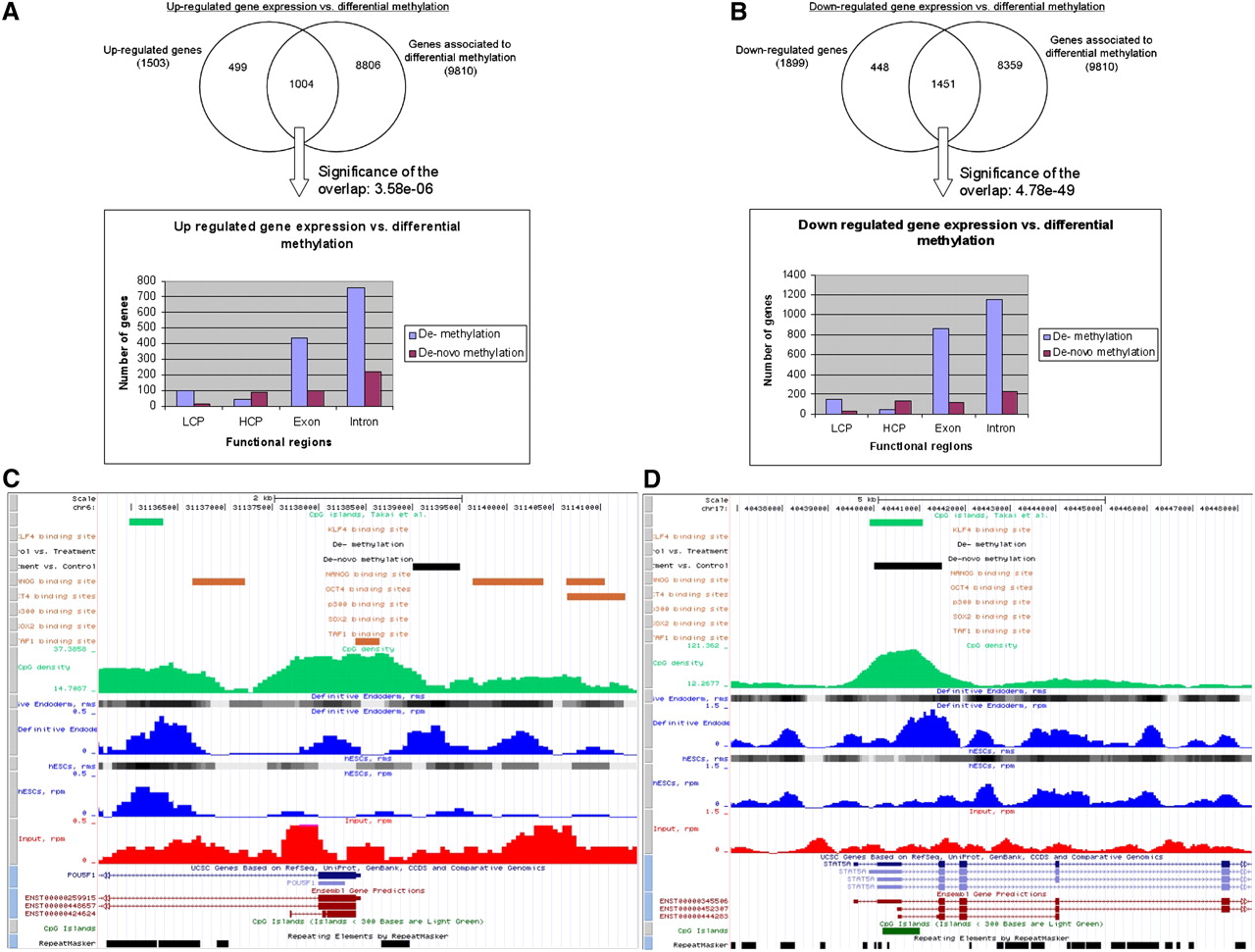
In order to analyze the interplay between DNA methylation and gene expression changes, we performed microarray-based gene expression analysis of hESCs and derived DE cells (see Methods); 2129 genes were significantly (P-value ≤ 0.01) down-regulated and 1661 genes up-regulated after differentiation (see Supplemental Table 5). In total, 15,947 genes were in common with Illumina arrays and with gene annotations matched to the identified DMRs. Based on these common genes, Figure 5 shows the overlap between genes that contain at least one identified DMR (demethylation or de novo methylation) in any of their associated transcript-exons, introns, or promoter regions with expression either up-regulated (Fig. 5A) or down-regulated (Fig. 5B) defined by a P-value of ≤0.01. In general, the events of differential methylation are significantly associated with up-regulated (P-value = 3.58 × 10−6) and down-regulated (P-value = 4.78 × 10−49) gene expression patterns (see Supplemental Table 5). However, the histograms in Figure 5, A and B, show similar location distributions over the tested gene-associated functional units of demethylation and de novo methylation events in both, up- and down-regulated genes. Although gene expression changes cannot, in general, be linked to distinct patterns of differential methylation, Figure 5 shows two examples of de novo methylation events located within the promoter regions of the TFs POU5F1 (Fig. 5C) and STAT5A (Fig. 5D), which appear along with down-regulation of gene expression in both cases.
Genetic and epigenetic dependencies. The figure shows the number of up-regulated (A) and down-regulated (B) genes with respect to the number of genes associated with differentially methylated regions (DMRs). For the genes that are differentially expressed and that contain a DMR, the histograms give an overview of the location of the respective demethylated and de novo methylated regions (LCP indicates low CpG density promotor; HCP, high CpG density promotor). (C) The promoter region of the down-regulated TF POU5F1, including an identified promoter de novo methylation event. (D) The promoter region of the down-regulated TF, STAT5A, including an identified de novo methylation event. Visualization of both regions was done with a local copy of the UCSC Genome Browser (Kuhn et al. 2009) (hg19). Included tracks are rpm (blue curves) and rms (gray blocks) values for hESCs and DE, rpm values for input (red curves), demethylated and de novo methylated regions (black blocks), CpG islands defined by UCSC (dark green blocks at the bottom) (Kuhn et al. 2009) and by Takai and Jones (2002) (light green blocks at the top), CpG densities along the chromosome (green curves, calculated by MEDIPS based on the CpG coupling factors), TFBSs of six TFs (orange blocks; genomic regions were transformed from hg18 to hg19 using UCSCs liftOver software; Rhead et al. 2010) (Lister et al. 2009), repeat masked regions (black boxes at the bottom), and RefSeq (Pruitt et al. 2007) and Ensembl (Birney et al. 2004) transcripts.
Discussion
Full-genome and base-specific methylomes of hESCs have been generated based on more than 1 billion short reads (Lister et al. 2009). Although MeDIP-seq data have far lower resolution, we have shown that MeDIP-seq enables the generation of full-genome methylation profiles based on about 20–30 million uniquely mapped short reads and thus reveals valuable information for whole-genome methylation analysis. A prerequisite is, however, a proper computational processing of the data, in particular a normalization procedure that takes into account the inherent sequence bias. In this work, we have developed and applied MEDIPS, a stand-alone computational tool, and showed that it is a cost- and time-effective software package for full-genome DNA methylation analysis. MeDIP-seq data are not base-specific, and therefore, it is still difficult to distinguish cytosine methylation within a CpG and non-CpG context based on single-end short reads. However, we have shown that for regions of interest, methylation profiles obtained by WGSBS can be correlated to normalized MeDIP-seq data. For biological material known to express cytosine methylation in non-CpG context, like for hESCs, correlation might even be improved when raw MeDIP-seq signals are calibrated with respect to a weighted combination of cytosine- and CpG-based coupling factors. The MEDIPS software package enables calculating coupling factors with respect to any arbitrary sequence pattern for immediate adapted calibration in future studies. Moreover, MeDIP-seq–derived methylation profiles enable the identification of regions showing differential methylation between samples on a full-genome level. Although in vivo liver development is specifically characterized by substantial demethylation, Brunner et al. (2009) reported some controversial observations on the number of demethylation events and the enrichment of demethylation and de novo methylation events at H3K27me3-bound regions and within LCPs when comparing in vitro and in vivo hepatic differentiation by a methyl-sensitive restriction enzyme–based sequencing approach. However, based on the normalized full-genome MeDIP-seq data of hESCs and DE, consistently with in vivo hepatic differentiation (Brunner et al. 2009), we observe high numbers of demethylation events and especially LCPs are specific targets for demethylation compared with de novo methylation. Although we compared our DMRs to histone modification signals obtained from human T cells (Barski et al. 2007), accordingly to the method of Meissner et al. (2008), we observed that H3K4 methylation events (activating mark) are associated with demethylation events in hESCs. CpG density and methylation analysis revealed two classes of TFs, namely, POU5F1, KLF4, and TAF1 on the one hand, and NANOG, SOX2, and EP300 on the other hand, thus suggesting distinct mechanisms in the interplay between TF binding and DNA methylation. Differential methylation is associated with differential gene expression in some key pluripotency regulating genes such as POU5F1. Similar cases can be found in the literature (Rakyan et al. 2008). However, we did not observe a general trend of gene-associated, region-specific methylation alterations that could explain up- and down-regulation of gene expression, thus implying the influence of additional factors in gene regulation. We have identified previously a core gene regulatory network of POU5F1 within the context of maintaining pluripotency in hESCs (Chavez et al. 2009), so that we were specifically interested in the effect of Activin A treatment on the induction of endodermal differentiation (D'Amour et al. 2005) by associating gene expression and DNA methylation for the members of this network. POU5F1 is a major factor in maintaining pluripotency and shows significant differential methylation in its promoter sequence (see Fig. 5C) associated to down-regulation of gene expression during differentiation. In contrast, direct target genes of POU5F1 show low or no promoter methylation differences, suggesting that expression of downstream genes is determined by promoter DNA methylation-independent regulation (see Supplemental Fig. 6). These observations indicate more complex dependencies in the interplay between gene regulation and DNA methylation during endoderm differentiation and of course gastrulation. Therefore, we propose that the effect of differential methylation on gene expression has to be further examined with respect to gene-specific locations of putative functional enhancer or silencer regions. Taken together, in our opinion and in line with that of D'Amour et al. (2005), we propose to further consider in vitro differentiation of hESCs along the endodermal lineage as a model for endodermal in vivo development.
Methods
Differentiation of hESCs cells into DE
hESCs (H1, passage 53) were treated with Activin A (100 ng/mL) for 5 d according to the method of D'Amour et al. (2005).
Library preparation
Genomic DNA was sonicated for 2 h as described previously (Parkhomchuk et al. 2009) to a size range of 100–400 bp. Fragmented DNA was purified using QIAquick spin columns and buffer QG (Qiagen) according to the manufacturer's protocol. Five micrograms of fragmented DNA was subjected to single end library preparation using the genomic DNA sample prep kit (Illumina) according to the manufacturer's instructions with the following modifications: 2.4 times increased amount of enzymes were used for end-repair and A-tailing. End repair was performed in the presence of 0.25 mM dNTPs Mix in a total volume of 317 μL; A-tailing was performed in a total volume of 88 μL. Adapters were ligated to the DNA fragments using 29 μL of Adapter oligo mix and two times excess concentration of ligase in a total reaction volume of 98 μL. The sequencing libraries were subjected to immunoprecipitation (see below). The amplification was performed after immunoprecipitation prior to gel-size selection. Twenty percent of the immunoprecipitated DNA or 40 ng of sheared DNA (input) was amplified using six amplification cycles in a total volume of 30 μL. Amplified libraries were size-selected on a 2% agarose gel to fragments of 150–400 bp (corresponding to insert sizes of 80–330 bp). Libraries were quantified on a Qubit fluorometer using the QuantIt dsHS Assay Kit (Invitrogen).
Immunoprecipitation of methylated DNA
MeDIP was adapted from a previously published protocol (Weber et al. 2005). Ten micrograms of monoclonal antibody against 5-methylcytidine (no. BI-MECY, Eurogentec) was coupled overnight with 40 μL of Dynabeads M-280 sheep anti-mouse IgG (Invitrogen) in 500 μL of 0.5% BSA/PBS and washed two times with 0.5% BSA/PBS and once with IP-buffer (10 mM sodium phosphate at pH 7.0, 140 mM NaCl, 0.25% Triton X100). The sequencing libraries were denatured for 1 min at 95°C. Four micrograms of library was immunoprecipitated for 4 h at 4°C with the 5-methylcytidine antibody coupled to Dynabeads in 230 μL of IP-buffer and then washed three times with 700 μL of IP-buffer. The beads were treated with 50 mM Tris-HCl (pH 8.0), 10 mM EDTA, 1% SDS for 15 min at 65°C and collected using a magnetic rack. The supernatant containing the methylated DNA (200 μL) was diluted with 200 μL of 10 mM Tris (pH 8.0), 1 mM EDTA and treated with proteinase K (0.2 μg/μL) for 2 h at 55°C, followed by phenol-chloroform-extraction and ethanol precipitation. The precipitated DNA was resuspended in 20 μL of 10 mM Tris (pH 8.5).
Illumina Genome Analyzer sequencing and data processing
After library quantification at a Qubit (Invitrogen), a 10 nmol stock solution of the amplified library was created. We loaded 12 pM of the stock solution onto the channels of a 1.4-mm flow cell, and cluster amplification was performed. Sequencing-by-synthesis was performed on an Illumina Genome Analyzer (GAIIx). After quality control of the first base incorporation (signal intensities, cluster density), the run was started. All MeDIP and input samples were subjected to a 36-bp single read sequencing. The raw data processing was done with the Illumina 1.4 pipeline version. After MAQ mapping (Li et al. 2008) of the generated reads against the human genome hg19 build downloaded from UCSC (Kuhn et al. 2009) (http://genome.ucsc.edu/), we obtained about 25.9 million unique high-quality (MAQ quality ≥ 10) mapping hits for pluripotent hESCs and about 32.6 million for DE. Additionally, we obtained about 22.6 million unique high-quality mapping hits from input samples of both conditions.
Identification of DMRs
Based on the MeDIP-seq data from hESCs, DE, and input, respectively, we calculated the short read coverage (extend value = 400) at genome-wide 50-bp bins using MEDIPS. In order to identify DMRs, MEDIPS calculates mean rpm (for hESCs, DE, and input) and mean rms (for hESCs and DE, only) values for overlapping genome-wide 500-bp windows where neighboring windows overlap by 250 bp. In order to estimate a minimal global background signal threshold, MEDIPS calculates the 0.9 rpm quantile (qt) of the input rpm signals of all overlapping 500-bp windows. Additionally, MEDIPS calculates P-values by comparing the rms signal distributions of the 50-bp bins of the hESCs and DE samples within each of the 500-bp windows. DMRs were identified by filtering for windows associated with a P-value ≤0.001, with a mean hESCs (or DE, respectively) rpm value ≥qt, with a local mean rpm hESCs/input (or DE/input, respectively) ratio ≥1.33, and with a mean rms hESCs/DE ratio ≤0.75 (or ≥1.33, respectively) (for a detailed description, see Supplementary Methods).
Gene expression analysis
Biotin-labeled cRNA was produced by means of a linear amplification kit (Ambion) using 500 ng of quality-checked DNase-free total RNA as input. Chip hybridizations, washing, Cy3-streptavidin staining, and scanning were performed on an Illumina BeadStation 500 platform (Illumina) using reagents and following protocols supplied by the manufacturer. cRNA samples were hybridized on Illumina human-8 BeadChips. We hybridized undifferentiated and Activin A–treated (DE differentiated) H1 cell line (passage 53) samples in biological triplicates. Raw data were obtained employing the manufacturer's software, BeadStudio 3.0.19.0. Subsequently, the raw data were imported into the Bioconductor environment (Gentleman et al. 2004), and quantile normalization was performed using the beadarray package (for boxplots of raw and normalized data, see Supplemental Fig. 5A,B; Dunning et al. 2007). In order to test for global gene expression similarities within biological replicates and between different treatments, pairwise Pearson correlation coefficients were calculated for all samples. Correlations within the groups are all >0.99, and correlations between the groups are from 0.92–0.93 (see Supplemental Fig. 5C). Finally, the dendrogram in Supplemental Figure 5D shows that the biological replicates of hESCs (control) and of DE (treatment) can be clearly separated into distinct groups.
Statistical analysis
For testing the enrichment of differentially expressed genes within the set of genes associated with DMRs, we used the hypergeometric distribution function phyper provided within the R framework (http://www.R-project.org). For identifying DMRs, the MEDIPS package calculates P-values (see above) using the default parameter settings of the ttest and wilcox.test functions (both two-sided) of the R framework. Differential gene expression was calculated using the limma (Wettenhall and Smyth 2004) package and by setting the level of significance to 0.01.
Acknowledgments
We thank Felix Dreher for extensive testing of the MEDIPS package and Thomas Down for help and feedback with installation of the Batman tool. Furthermore, we thank Stephan Beck and Jörn Walter for helpful discussions on whole-genome epigenetics. The work was funded in part by the German Ministry of Education and Research (BMBF) within its National Genome Research Network (01GS08111) and within the funding program Cell-based Regenerative Medicine (01GN0530) and by the Max Planck Society.
Author contributions: L.C. conceived the study, developed and implemented the MEDIPS package, did all data analysis, and drafted the manuscript. J.J. performed the differentiation of the hESCs into DE and the BeadArray gene expression experiments. C.G. performed the MeDIP experiments. J.D. assisted in implementing and optimizing the MEDIPS package. B.T. performed next-generation sequencing. R.H. and J.A. helped to conceive the study and drafted the manuscript. All authors have read and approved the final manuscript.
References
- ↵S Agarwal, KL Holton, R Lanza. 2008. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells 26: 1117–1127.
- ↵G Altun, JF Loring, LC Laurent. 2010. DNA methylation in embryonic stem cells. J Cell Biochem 109: 1–6.
- ↵A Barski, S Cuddapah, K Cui, TY Roh, DE Schones, Z Wang, G Wei, I Chepelev, K Zhao. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837.
- ↵E Birney, TD Andrews, P Bevan, M Caccamo, Y Chen, L Clarke, G Coates, J Cuff, V Curwen, T Cutts, . 2004. An overview of Ensembl. Genome Res 14: 925–928.
- ↵C Bock, K Halachev, J Buch, T Lengauer. 2009. EpiGRAPH: User-friendly software for statistical analysis and prediction of (epi)genomic data. Genome Biol 10: R14. doi: 10.1186/gb-2009-10-2-r14.
- ↵AL Brunner, DS Johnson, SW Kim, A Valouev, TE Reddy, NF Neff, E Anton, C Medina, L Nguyen, E Chiao, . 2009. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res 19: 1044–1056.
- ↵EM Chan, S Ratanasirintrawoot, IH Park, PD Manos, YH Loh, H Huo, JD Miller, O Hartung, J Rho, TA Ince, . 2009. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol 27: 1033–1037.
- ↵L Chavez, AS Bais, M Vingron, H Lehrach, J Adjaye, R Herwig. 2009. In silico identification of a core regulatory network of OCT4 in human embryonic stem cells using an integrated approach. BMC Genomics 10: 314.
- ↵KA D'Amour, AD Agulnick, S Eliazer, OG Kelly, E Kroon, EE Baetge. 2005. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 23: 1534–1541.
- ↵J Deng, R Shoemaker, B Xie, A Gore, EM LeProust, J Antosiewicz-Bourget, D Egli, N Maherali, IH Park, J Yu, . 2009. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol 27: 353–360.
- ↵TA Down, VK Rakyan, DJ Turner, P Flicek, H Li, E Kulesha, S Graf, N Johnson, J Herrero, EM Tomazou, . 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26: 779–785.
- ↵MJ Dunning, ML Smith, ME Ritchie, S Tavare. 2007. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics 23: 2183–2184.
- ↵F Eckhardt, J Lewin, R Cortese, VK Rakyan, J Attwood, M Burger, J Burton, TV Cox, R Davies, TA Down, . 2006. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38: 1378–1385.
- ↵M Gardiner-Garden, M Frommer. 1987. CpG islands in vertebrate genomes. J Mol Biol 196: 261–282.
- ↵RC Gentleman, VJ Carey, DM Bates, B Bolstad, M Dettling, S Dudoit, B Ellis, L Gautier, Y Ge, J Gentry, . 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80. doi: 10.1186/gb-2004-5-10-r80.
- ↵RA Irizarry, C Ladd-Acosta, B Wen, Z Wu, C Montano, P Onyango, H Cui, K Gabo, M Rongione, M Webster, . 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186.
- ↵PA Jones, SB Baylin. 2007. The epigenomics of cancer. Cell 128: 683–692.
- ↵Y Koga, M Pelizzola, E Cheng, M Krauthammer, M Sznol, S Ariyan, D Narayan, AM Molinaro, R Halaban, SM Weissman. 2009. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res 19: 1462–1470.
- ↵RM Kuhn, D Karolchik, AS Zweig, T Wang, KE Smith, KR Rosenbloom, B Rhead, BJ Raney, A Pohl, M Pheasant, . 2009. The UCSC Genome Browser Database: Update 2009. Nucleic Acids Res 37: D755–D761.
- ↵PW Laird. 2010. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 11: 191–203.
- ↵H Li, J Ruan, R Durbin. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858.
- ↵R Lister, RC O'Malley, J Tonti-Filippini, BD Gregory, CC Berry, AH Millar, JR Ecker. 2008. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133: 523–536.
- ↵R Lister, M Pelizzola, RH Dowen, RD Hawkins, G Hon, J Tonti-Filippini, JR Nery, L Lee, Z Ye, QM Ngo, . 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322.
- ↵A Meissner, TS Mikkelsen, H Gu, M Wernig, J Hanna, A Sivachenko, X Zhang, BE Bernstein, C Nusbaum, DB Jaffe, . 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770.
- ↵D Parkhomchuk, T Borodina, V Amstislavskiy, M Banaru, L Hallen, S Krobitsch, H Lehrach, A Soldatov. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37: e123. doi: 10.1093/nar/gkp596.
- ↵M Pelizzola, Y Koga, AE Urban, M Krauthammer, S Weissman, R Halaban, AM Molinaro. 2008. MEDME: An experimental and analytical methodology for the estimation of DNA methylation levels based on microarray derived MeDIP-enrichment. Genome Res 18: 1652–1659.
- ↵KD Pruitt, T Tatusova, DR Maglott. 2007. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–D65.
- ↵VK Rakyan, TA Down, NP Thorne, P Flicek, E Kulesha, S Graf, EM Tomazou, L Backdahl, N Johnson, M Herberth, . 2008. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 18: 1518–1529.
- ↵B Rhead, D Karolchik, RM Kuhn, AS Hinrichs, AS Zweig, PA Fujita, M Diekhans, KE Smith, KR Rosenbloom, BJ Raney, . 2010. The UCSC Genome Browser database: Update 2010. Nucleic Acids Res 38: D613–D619.
- ↵D Takai, PA Jones. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci 99: 3740–3745.
- ↵M Weber, JJ Davies, D Wittig, EJ Oakeley, M Haase, WL Lam, D Schubeler. 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37: 853–862.
- ↵JM Wettenhall, GK Smyth. 2004. limmaGUI: A graphical user interface for linear modeling of microarray data. Bioinformatics 20: 3705–3706.
Notes
[2] [Supplemental material is available at http://www.genome.org. The MeDIP-seq data from this study have been submitted to NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA012665. The bead array gene expression data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE21715. The MEDIPS software package, manual, data, and example data are available online at http://medips.molgen.mpg.de.]
[3] Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.110114.110.