
A positive but complex association between meiotic double-strand break hotspots and open chromatin in Saccharomyces cerevisiae
- 1 Department of Biology and the Carolina Center for Genome Sciences, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3280, USA;
- 2 Lineberger Comprehensive Cancer Center, University of North Carolina School of Medicine, Chapel Hill, North Carolina 27599-3280, USA
-
↵3 These authors contributed equally to this work.
Abstract
During meiosis, chromatin undergoes extensive changes to facilitate recombination, homolog pairing, and chromosome segregation. To investigate the relationship between chromatin organization and meiotic processes, we used formaldehyde-assisted isolation of regulatory elements (FAIRE) to map open chromatin during the transition from mitosis to meiosis in the budding yeast Saccharomyces cerevisiae. We found that meiosis-induced opening of chromatin is associated with meiotic DSB hotpots. The positive association between open chromatin and DSB hotspots is most prominent 3 h into meiosis, when the early meiotic genes DMC1 and HOP1 exhibit maximum transcription and the early recombination genes SPO11 and RAD51 are strongly up-regulated. While the degree of chromatin openness is positively associated with the occurrence of recombination hotspots, many hotspots occur outside of open chromatin. Of particular interest, many DSB hotspots that fell outside of meiotic open chromatin nonetheless occurred in chromatin that had recently been open during mitotic growth. Finally, we find evidence for meiosis-specific opening of chromatin at the regions adjacent to boundaries of subtelomeric sequences, which exhibit specific crossover control patterns hypothesized to be regulated by chromatin.
During meiosis, chromosomes undergo a highly orchestrated series of movements and reorganizations including pairing, synapsis, recombination, and two successive rounds of segregation (Zickler and Kleckner 1999). The end result is the basis for sexual reproduction: haploid gametes. Meiotic recombination contributes to haplotype diversity by generating new combinations of alleles not present in the parental chromosomes (Felsenstein 1974). Crossing over results in physical linkages between homologous chromosomes, which are critical for proper chromosome segregation in many organisms (Gerton and Hawley 2005). In this study, we explore how the location of recombination events and their precursors double-strand breaks (DSBs) are associated with how DNA is packaged into chromatin.
In Saccharomyces cerevisiae, recombination occurs during prophase I and is initiated by programmed DSBs catalyzed by the topoisomerase-related protein Spo11 (Keeney et al. 1997). Single-stranded DNA (ssDNA) filaments coated with Rad51 and Dmc1 catalyze strand invasion of homologous duplex DNA, resulting in four-strand intermediates known as joint molecules (Bishop et al. 1992; Schwacha and Kleckner 1994; Allers and Lichten 2001). After strand invasion, DSB repair proceeds and culminates in either crossover or noncrossover products (Szostak et al. 1983; Sun et al. 1991; McMahill et al. 2007). Regions of gene conversion may also be produced. It has been estimated from ssDNA mapping and high-resolution recombination maps that 90 of the 140–170 DSBs that occur during each S. cerevisiae meiosis are repaired as crossovers (Buhler et al. 2007; Mancera et al. 2008).
To a large degree, crossover distribution is governed by the initial placement of meiotic DSBs, which has been examined in S. cerevisiae using three different techniques. First, meiotic DSBs that accumulate in a rad50S background have been precisely mapped on chromosome III by Southern blot analysis (Baudat and Nicolas 1997). The rad50S strain is useful for studying DSB distribution because Spo11 remains covalently linked to the 5′ end of the DSBs, causing DSBs to accumulate instead of being converted rapidly into crossovers and noncrossovers (Gerton et al. 2000). However, rad50S mutants may have reduced DSB frequency and altered DSB placement relative to wild type (Buhler et al. 2007). More recently, methods that do not require the rad50S background were used to map DSBs (Blitzblau et al. 2007; Buhler et al. 2007). As a normal part of DSB processing following cleavage by Spo11, the 5′ ends of DSBs are resected, leaving 3′ single-stranded tails that participate in the subsequent strand-invasion process. These 3′ ssDNA tails can be used as a molecular tag to map DSBs. ssDNA was isolated from a dmc1Δ mutant, in which DSBs accumulate prior to arrest in late meiotic prophase (Bishop et al. 1992). Together, these studies provide an initial map of the DSB landscape and clearly demonstrate that DSBs occur preferentially in intergenic regions (particularly in transcriptionally active promoters), while also showing that chromosome ends and centromeres exhibit substantial DSB activity (Baudat and Nicolas 1997; Gerton et al. 2000; Blitzblau et al. 2007; Buhler et al. 2007).
In most organisms, neither DSBs nor crossovers occur with uniform frequency throughout the genome. Some regions, called hotspots, have very high rates of recombination (Lichten and Goldman 1995; Petes 2001). Conversely, coldspots are regions of the genome with lower than average rates of recombination (Petes 2001). The kinetics and location of meiotic recombination are likely to be influenced by chromatin organization (Wu and Lichten 1994; Fan and Petes 1996; Kirkpatrick et al. 1999b). The fundamental unit of chromatin, the nucleosome, is defined as ∼146 bp of DNA wrapped around a histone octamer (Luger et al. 1997). Modulation of nucleosome occupancy provides the cell with a mechanism to regulate access to DNA. Regions of DNA that are depleted of nucleosomes are known as “open” chromatin and are more accessible to DNA-binding proteins (Han and Grunstein 1988; Han et al. 1988).
During S. cerevisiae meiosis, hotspots at ARG4 and CYS3 show a meiosis-specific increase in sensitivity to micrococcal nuclease (MNase), a common assay for open chromatin (Ohta et al. 1994). Additionally, recombination rates can be artificially elevated at specific loci by inserting sequences that exclude nucleosomes, such as bacterial DNA or S. cerevisiae telomere fragments (Cao et al. 1990; Fan et al. 1995; Fan and Petes 1996). Recently, the distribution of a histone modification associated with transcriptionally active chromatin, H3 histones trimethylated at lysine 4 (H3K4me3), has been shown to be enriched at DSB sites (Borde et al. 2009).
Further evidence for a chromatin role comes from S. pombe. At the S. pombe ade6-M26 locus, MNase sensitivity increases prior to DSB initiation, and nuclease sensitivity patterns at ade6-M26 change during meiosis (Hirota et al. 2007, 2008). A study that examined three distinct S. pombe DSB hotspots showed one that was constitutively open (mbs1), while two others become open only as meiosis proceeds (tdh1+, ade6-M26) (Hirota et al. 2007). Mutations in S. pombe chromatin remodeling proteins such as Snf22, Gcn5, and Ada2 disrupt meiotic MNase cleavage patterns at ade6-M26 and also result in decreased recombination at this site (Hirota et al. 2008). snf22Δ mutants fail to exhibit the ∼2.4-fold meiotic induction of MNase sensitivity at the ade6-M26 break point seen in wild-type cells and have a concurrent ∼6.2-fold site-specific reduction in recombination at ade6-M26 (Hirota et al. 2008).
While studies at selected individual loci suggest that DSBs and crossovers occur preferentially at nucleosome-depleted regions (Shenkar et al. 1991; Ohta et al. 1994, 1999; Wu and Lichten 1994; Borde et al. 1999; Gerton et al. 2000), whether this relationship would hold in an unbiased survey of nucleosome-depleted regions across the genome was unknown. To address the relationship between nucleosome occupancy and meiotic recombination directly, we used formaldehyde assisted isolation of regulatory elements (FAIRE) to assess genome-wide associations between DSB hotspots and open chromatin in yeast. We also assessed whether the distinct regulation of recombination at subtelomeric borders and pericentromeric regions is accompanied by particular chromatin configurations in meiosis.
Results
Monitoring of meiotic entry and progression
To validate meiotic entry, synchrony, and progression in our time course, we analyzed the transcriptional profile of several key meiotic genes by expression microarray (Fig. 1). We analyzed RNA isolated in a meiotic time course from each FAIRE biological replicate. This included the time points used in the FAIRE assay (YPD, 0, 1.5, and 3 h), in addition to RNA isolated from the same cultures at other time points. These include a time point prior to our “time 0,” consisting of premeiotic cells that had been starved of a fermentable carbon source for 8 h (−8 h acetate) and RNA collections at 4.5, 6, 9, and 12 h, which were taken subsequent to those analyzed by FAIRE. To assess meiotic entry, we monitored the master regulators of the meiotic program IME1 and IME2, which were up-regulated at the 0-h sample relative to YPD and the −8-h acetate sample. Both IME1 and IME2 exhibited maximum expression 1.5 h into meiosis (Fig. 1A). We also monitored the early meiotic genes HOP1, SPO11, RAD51, and DMC1. All early genes exhibited strong up-regulation 1.5 h into meiosis and generally were maximally expressed by 3 h.
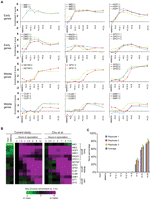
Monitoring meiotic entry and progression in the time course used for FAIRE. (A) To validate meiotic entry and synchrony in each biological replicate, we analyzed the transcription profiles of key meiotic progression genes by expression microarray. Time point is indicated on the x-axis, and expression relative to YPD is shown on the y. Biological replicates are plotted (1, blue; 2, red; 3, green). If only two samples are shown, the YPD probe was flagged for technical reasons, and thus no comparisons could be made. Early meiotic genes include IME1, IME2, HOP1, DMC1, RAD51, and SPO11. Middle meiotic genes include NDT80, SPS1, SPS2, CLB1, CLB6, and SMK1. (B) Comparison to meiotic transcription microarray data of Chu et al. (1998). Purple indicates positive meiotic enrichment, and green indicates negative meiotic enrichment. Transcription data is represented as a ratio between time point expression and 0 h expression. (C) Completion of meiosis as measured by spore counts. Spore counts were taken at each time point. Time point is indicated on the x-axis, and the percentage of sporulated cells is indicated on the y. Biological replicates are plotted (1, blue; 2, red; 3, green; average, purple).
Based on these profiles, we infer that DSB formation and strand invasion had begun in our 3-h sample. Although transcription analysis is an indirect means of determining the kinetics of DSB and joint molecule formation, our timing inferences match closely with those determined from two-dimensional gel analysis (Allers and Lichten 2001). Additionally, our averaged expression profiles are strikingly similar to previous meiotic expression array analysis (Chu et al. 1998) (Fig. 1B). Finally, by counting spores at each time point and 24 h past sporulation, we show that over three-quarters of cells in each replicate successfully completed meiosis (Fig. 1C).
Identification of meiotic open chromatin using FAIRE
We used FAIRE to study changes in chromatin during the transition from mitotic growth through the early stages of meiosis. FAIRE is a simple and inexpensive method for identifying sites of open chromatin that relies on phenol-chloroform extraction of sonicated, formaldehyde cross-linked chromatin to isolate nucleosome-depleted regions of DNA (Nagy et al. 2003; Hogan et al. 2006; Giresi et al. 2007). FAIRE has been used to positively select for nucleosome-depleted genomic sites throughout the mitotic cycle of S. cerevisiae (Hogan et al. 2006) and to isolate nucleosome-depleted active regulatory sites in human cells (Giresi et al. 2007; Eeckhoute et al. 2009). FAIRE has a strong anticorrelation with histone H3 and H4 ChIP-chip in mitotic yeast cells (Hogan et al. 2006).
We targeted the first 3 h of meiosis, which corresponds to the time DSBs are made and joint molecules are beginning to form (Allers and Lichten 2001). Four sample treatments were subjected to FAIRE: cells undergoing normal mitotic growth (YPD), premeiotic cells that had been starved of a fermentable carbon source for 16 h (YPA, 0 h), synchronized cells that had been incubated in sporulation medium for 90 min (1.5 h), and cells that had been incubated in sporulation medium for 180 min (3 h). Hereafter, we define a site of “open chromatin” as a FAIRE peak called at P <0.01 (see Methods). We identified 1204 sites that were open in at least one time point (Table 1; Fig. 2) and 510 sites that were open in all samples (e.g., CDC19) (Fig. 3). To further establish a firm relationship between FAIRE and nucleosome depletion in our samples, we compared our FAIRE enrichment profiles with H3 ChIP-chip enrichment profiles generated in an independent study from 0-, 1-, and 2-h meiotic samples (Borde et al. 2009). The H3 ChIP-chip data independently corroborated FAIRE enrichment profiles at individual loci (Fig. 3) and globally (overall Pearson correlation −0.381).
Sites of open chromatin are preferentially associated with DSB hotspots
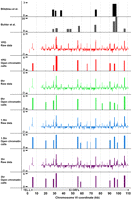
The distribution of open chromatin and DSB hotspots across a representative 110-kb genomic region containing 53 genes. Chromosome coordinates are plotted along the x-axis, with FAIRE enrichment (z score) plotted on the y. FAIRE data were loaded into the UCSC Genome Browser along with published DSB hotspots (black and dark gray) (Blitzblau et al. 2007; Buhler et al. 2007). FAIRE data from YPD (red), 0-h (green), 1.5-h (blue), and 3-h (purple) samples are plotted.
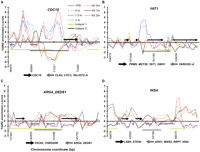
Chromatin at known hotspots is dynamic. Chromosome coordinates are plotted along the x-axis, with FAIRE enrichment (z score) plotted on the y. Positive z scores indicate relatively open chromatin, and negative numbers indicate relatively closed chromatin. FAIRE data from YPD (red), 0-h (dashed green), 1.5-h (blue fine dashes), and 3-h (purple and dotted) samples are plotted. Among the 1204 sites, 151 were open in all the meiosis samples (1.5 and 3 h), but not during mitotic growth (e.g., YAT1). Conversely, 156 sites were open during mitotic growth, but were not open in any of the meiotic samples. Meiotic histone H3 ChIP-chip data from 0 h (orange), 1 h (brown), and 2 h (dark brown and dotted) are plotted (Borde et al. 2009). Hotspot regions identified by Buhler et al. (2007) (light-green bars), and by Blitzblau et al. (2007) (dark-green bars) are shown. Arrows represent the coding regions of genes, with those above the x-axis coded from the Watson strand and those below coded from the Crick strand. (A) Chromatin dynamics at the CDC19 hotspot on chromosome I, an example of a hotspot with constitutively open chromatin. (B) Chromatin dynamics at the YAT1 hotspot on chromosome I, an example of a meiosis-specific site of open chromatin. (C) Chromatin dynamics at the ARG4_DED81 hotspot on chromosome VIII (Wu and Lichten 1994; Ohta et al. 1999), an example of a DSB hotspot with no observed open chromatin in any sample. (D) Chromatin dynamics at the HIS4 hotspot on chromosome III, an example of a hotspot at which the chromatin is open in YPD and closes as meiosis progresses.
Genomic regions that undergo high rates of meiotic recombination are generally associated with open chromatin
Evidence from locus-specific studies strongly suggests that meiotic recombination occurs preferentially in regions of open chromatin (Shenkar et al. 1991; Ohta et al. 1994, 1999; Wu and Lichten 1994). To examine this relationship genome wide, we compared our chromatin profiles with sites of meiotic ssDNA enrichment (DSB hotspots) from two published studies (Figs. 2, 3) (Blitzblau et al. 2007; Buhler et al. 2007). Of the 1157 (Buhler et al. 2007) and 258 (Blitzblau et al. 2007) defined DSB hotspots, 591 (51%) and 157 (61%), respectively, fall within a site of open chromatin from any of the samples (27% and 28% were expected by chance, respectively). To provide a more quantitative measure of overlap, we calculated the percentage of base-pair sites of open chromatin that intersected with DSB hotspots at each point in the time course (YPD, 0, 1.5, and 3 h). Strength of association is expressed as the ratio of observed to expected base-pair overlap, where the “expected” value is the average overlap measured in 10 permutations in which the genomic position of the open chromatin domains was randomized (Table 1; Methods). By this measure, open chromatin was strongly associated with both DSB hotspots (P << 0.0001) (Methods) and in no case were permuted open chromatin sites as strongly associated with hotspots as an experimental sample (Methods). This was true whether we examined sites that were open in at least one time point, or sites that were open in all time points.
In the 3-h sample, when we infer through up-regulation of SPO11, DMC1, and RAD51 and previous studies (Allers and Lichten 2001) that cells were processing DSBs into joint molecules, open chromatin had the strongest association with DSB hotspots: 2.5 (Buhler et al. 2007) and 3.3 (Blitzblau et al. 2007) times more highly associated than random. Sites that were open in all samples had an equally strong association with DSB hotspots, being 2.5 (Buhler et al. 2007) and 3.5 (Blitzblau et al. 2007) times more highly associated than random, implying that many hotspots exist in sites of constitutively open chromatin. Supporting this interpretation, meiosis-specific sites of open chromatin did not show a stronger association with DSB hotspots than sites that were constitutively open.
The association of open chromatin with DSB hotspots cannot be explained solely by the prevalence of open chromatin at gene promoters
Intergenic regions in yeast are typically depleted of nucleosomes (Polach and Widom 1995; Lee et al. 2004), and DSB hotspots are known to be associated with intergenic regions (Lichten and Goldman 1995; Petes 2001; Blitzblau et al. 2007; Buhler et al. 2007). To address the possibility that the association between open chromatin sites and DSB hotspots might be accounted for entirely by their co-occurrence in intergenic regions, we permuted the genomic location of sites of open chromatin such that the permuted location was restricted to intergenic regions. Even under these conditions, open chromatin identified by FAIRE was more highly associated with DSB hotspots than any of the permutations (Methods; Supplemental Table1). Thus, the open chromatin sites identified by FAIRE provide information regarding hotspot distribution that is independent of, and in addition to, the information provided by stratifying the genome into coding and noncoding regions.
Among open chromatin sites, the degree of chromatin openness is predictive of DSB hotspots
We asked whether FAIRE sites with higher signal, or “openness” were more likely to be DSB hotspots than FAIRE sites with lower signal. To this end, we conducted receiver operating characteristic (ROC) curve analysis (Zweig and Campbell 1993) (Methods) on each time point, using either regional P-values or probe z scores to quantify openness (Fig. 4). “True-positives” were defined as DSB hotspots identified in the literature (Blitzblau et al. 2007; Buhler et al. 2007). Five categories of open chromatin sites were analyzed: (I) open in any of our samples; (II) open in all of our samples; (III) open in at least one meiotic sample (1.5 or 3 h) but not YPD; (IV) open in all meiotic samples (1.5 and 3 h) but not YPD; (V) open in mitotic growth but closed in all meiotic samples.
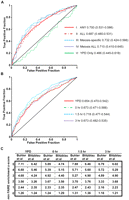
The degree of chromatin openness is predictive of DSB hotspots. (A) ROC curve analysis of the five categories of open chromatin. True-positives and false-positives are defined with respect to DSB hotspots (Buhler et al. 2007) (Methods). The FAIRE sites plotted are ordered such that those to the left are most open according to FAIRE (lowest P-value), and those to right, while still significantly open, have higher P-values. Data from category I (solid red), category II (maroon dashes), category III (light-blue dashes), category IV (dark-blue dots), and category V sites (solid green) are plotted. AUC values are listed next to the sample name, and the minimum and maximum AUC values of the random permutations follow in parentheses. (B) ROC curve analysis of sites of open chromatin in individual time points. Data from YPD (red), 0-h (green dashes), 1.5-h (light-blue dashes), and 3-h (purple dots) samples are plotted. (C) DSB predictive value of individual probes with FAIRE z scores above the indicated threshold, expressed as likelihood ratios. Values in the table are TPF/FPF at decreasing z score thresholds, listed vertically on the left. Time points are organized on the horizontal axis. Predictive value is expressed with respect to two DSB hotspot datasets (Blitzblau et al. 2007; Buhler et al. 2007). Because this analysis is probe based, multiple true-positives may be derived from a single DSB hotspot.
The degree of openness (as measured by P-value) in meiosis-specific open sites (categories III and IV) was strongly predictive of DSB hotspots, with ROC area under the curve (AUC) values of 0.712 (category III) and 0.734 (category IV) (Fig. 4A). Degree of openness was also predictive of DSB hotspots in sites that were open in any sample (category I; AUC = 0.700) and sites that were open in all samples (category II; AUC = 0.687). Of the individual time points, openness was most predictive of DSB hotspots in the 1.5-h time point (AUC = 0.718) (Fig. 4B). Conversely, the degree of openness in sites that are mitosis-specific (category V) was not predictive of DSB hotspots (AUC = 0.466).
We also quantified the ability of FAIRE to predict DSB hotspots independently of peak calling. Using the FAIRE enrichment z score of each probe as an ordering statistic, we plotted the true-positive fraction (TPF) versus the false-positive fraction (FPF), and determined the likelihood ratio (TPF/FPF) at decreasing z-score cutoffs at each time point (Fig. 4C) (Zweig and Campbell 1993). The degree of FAIRE enrichment as measured by probe values is indeed predictive of DSB hotspots, with the 1.5-h time point again being the most predictive. A given probe measuring FAIRE enrichment at the 1.5-h time point with a z score of over 3 has a 5:1 true-positive to false-positive ratio with respect to DSB hotspot prediction.
A class of DSB hotspots occurs within closed chromatin as defined by FAIRE
Over one-third of DSB hotspots, including some well-studied recombination hotspots such as ARG4 (Ohta et al. 1994; Wu and Lichten 1994), HIS2 (Malone et al. 1992), BUD23 (also known as YCR047C) (Goldway et al. 1993; Wu and Lichten 1994), and CYS3 (Ohta et al. 1994) occur within chromatin that is not open, as measured by FAIRE, in any sample (ARG4) (Fig. 3). This suggests that neither meiosis-specific opening nor a level of open chromatin beyond what is found intrinsically at promoter regions is strictly required for the establishment of a DSB hotspot. We tested the possibility that the subset of DSB hotspots that do not occur in open chromatin actually occur in relatively open chromatin that falls just below our classification threshold. We divided the 296 consensus DSB hotspots that were held in common between the two datasets into 167 that occurred in a site of open chromatin and 129 that did not. In each grouping, we aligned the hotspots at their start coordinates and calculated the moving average of FAIRE enrichment at each position (Supplemental Fig. S1; Methods). Hotspots associated with a site of open chromatin exhibited maximum openness at the center of the defined hotspot region. In contrast, hotspots not associated with open chromatin exhibit a chromatin profile indistinguishable from the genomic average. We interpret this finding to mean that the DSBs that occur in sites we classify as closed, are in most cases truly closed according to FAIRE.
FAIRE failed to detect open chromatin at ARG4, which has previously been shown to be nuclease hypersensitive by independent groups (Ohta et al. 1994; Wu and Lichten 1994, 1995). This raises the possibility that FAIRE may be unable to identify a subset of open chromatin sites. It is possible that large non-histone molecular complexes prevent otherwise open sites to be isolated by FAIRE. Recent reports have shown that Spo11-binding sites do not necessarily incur DSBs (Kugou et al. 2009), and even the strongest DSB hotspots in S. cerevisiae undergo DSBs in fewer than 10% of DNA molecules (Baudat and Nicolas 1997). Therefore, some hotspots may be highly occupied by recombination complexes regardless of whether they actually incur a DSB. Additionally, DSBs may be required for a transition to open chromatin at some loci. Last, DSBs that occur in a small percentage of cells may be detected by nuclease sensitivity assays, which are likely more sensitive in this context, but not FAIRE. However, the assertion that some hotspots occur in closed chromatin sites is independently supported by H3 ChIP-chip data, because hotspots not associated with FAIRE enrichment also exhibited positive enrichment by H3 ChIP-chip (Fig. 2B; Supplemental Fig. S1) (Borde et al. 2009).
Many DSBs occur in chromatin that had been recently open during mitotic growth
We next asked whether some of the DSB hotspots that occur in closed meiotic chromatin were open during mitotic growth. Indeed, 62 of the 156 sites open during YPD and closed in all of the meiotic samples corresponded to published DSB hotspots (Blitzblau et al. 2007; Buhler et al. 2007) (expected = 34.69; P < 0.0001). This is unlikely to be due to a threshold effect, whereby the mitosis-specific sites of open chromatin barely miss the meiotic cutoff. The mean FAIRE enrichment z score of these sites in YPD (2.13) is significantly higher than any of the meiotic time points (average of all meiotic samples = 0.96; two tailed t-test P < 0.0001) (Supplemental Fig. S2). While the fact that a locus was open during mitosis was predictive of a hotspot, the degree of mitotic openness was not, in contrast to meiotic openess (Fig. 4A).
The prevalence of DSB occurrence in recently open chromatin could be due to hotspots specified by pre-meiotic events, such as transcription-factor binding or histone modification patterns. For example, the presence of histone modification H3K4me3 is strongly elevated 0–2 kb from DSB sites and this pattern is established in vegetative cells (Borde et al. 2009). Additionally, some hotspots are known to require transcription-factor binding to stimulate meiotic recombination. A particularly well-studied example is the HIS4 locus, which requires the binding of transcription factors Rap1, Bas1, or Bas2 for hotspot activity, although it is unknown whether binding is required during meiosis or mitosis (White et al. 1991, 1993; Kirkpatrick et al. 1999a). We find the HIS4 hotspot to be open in mitosis (max z score = 5) and closed by the 1.5-h time point of meiosis. Independent H3 ChIP-chip experiments show the HIS4 region to be closed from 0 to 2 h in meiosis (Borde et al. 2009) (HIS4) (Fig. 3).
Chromatin adjacent to subtelomeric sequences is open specifically during meiosis
Telomeres play a critical role in the meiotic process. Telomere clustering and attachment to the nuclear membrane has been postulated to be involved in homologous chromosome pairing (Rockmill and Roeder 1998; Zickler and Kleckner 1998). However, it has been proposed that since telomeres and subtelomeres are very high in repetitive and nonunique DNA (Barton et al. 2003), a homology-based pairing search using these regions would be difficult. Concordantly, in S. cerevisiae, crossovers are suppressed at telomeres and heterochromatic subtelomeres, presumably to suppress nonhomologous exchanges between the repetitive and nonunique sequences that occur at chromosome ends (Su et al. 2000; Barton et al. 2003, 2008). In contrast, euchromatic sequences directly proximal to the subtelomeres, which could play a role in the homology search (see Discussion), have crossover rates that are greater than twice the genome average (Barton et al. 2008). Based on this and the proposed role of telomeres for homologous chromosome pairing, we hypothesized that the euchromatic DNA sequences adjacent to the subtelomeres would exhibit meiosis-specific changes in chromatin architecture.
We found significantly elevated levels of meiosis-specific open chromatin proximal to the previously defined (Barton et al. 2008) subtelomeric-euchromatic borders (defined in Fig. 5A and legend; Methods; Supplemental Tables S3, S4). Sites open in all meiotic samples, but not open in YPD (Category IV) were enriched most dramatically, occurring 2.8 times more frequently than expected 0–10 kb proximal to the subtelomeric border (P < 0.001), 2.1 times greater 10–20 kb from the border (P < 0.05), and 2.1 times greater 30–40 kb from the border (P < 0.05) (Fig. 5B; Supplemental Table S3). Expected values were derived from 10,000 permutations of the open chromatin chromosomal positions, and P-values were calculated using a χ2-test (Methods; Supplemental Table S3). In DNA located 20–30 kb proximal from the subtelomeric border, the distribution of open chromatin at any of the time points was indistinguishable from the average of permuted distributions. DSB hotspots are under-represented from the telomere to 10 kb proximal to the subtelomeric border, and are enriched 10–40 kb proximal to the subtelomeric border (Fig. 5C) (Blitzblau et al. 2007; Buhler et al. 2007). Interestingly, recombination rate peaks 0–10 kb proximal to the subtelomeric border, decline, and then climb again at 30–40 kb from the border (Fig. 5D). Since category IV FAIRE data displays a similar pattern of enrichment, we speculate that meiosis-specific open chromatin may play a role in the elevated rates of recombination in these regions (Discussion).
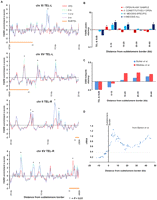
Regions proximally adjacent to the subtelomere borders exhibit meiosis-specific open chromatin. (A) Examples of meiosis-specific open chromatin at the subtelomeric borders. Distance from the subtelomeric border (coordinate 0) is plotted on the x-axis and FAIRE enrichment is plotted on the y-axis. Subtelomeres (orange bars) are shown. FAIRE data from YPD (red), 0-h (green), 1.5-h (blue dashes), and 3-h (purple dots) samples are plotted. Asterisks indicate sites significantly open (P < 0.01). (B) Analysis of telomeric regions. The x-axis reports the region analyzed and the y-axis reports the ratio of observed sites of open chromatin to the expected number from random sampling. (Red) Category I; (maroon) category II; (light-blue) category III; (dark-blue) category IV sites. The telomere and subtelomeres do not deviate from the genomic averages of expected/observed sites of open chromatin. Meiosis-specific open chromatin is most prominent in the 10 kb adjacent to the subtelomeres. In the 10–20-kb and 30–40-kb windows, only category IV sites are significantly above expected values. (C) The ratio of observed to expected number of DSB hotspots as plotted as a function from distance from the subtelomeric border. Data from Buhler et al. (2007) (blue) and Blitzblau et al. (2007) (red) are shown. (D) Recombination in the subtelomeric regions as measured by linkage mapping (Barton et al. 2008). Distance from the subtelomeric border is plotted on the x-axis and recombination rate (cM/kb) is shown on the y-axis. Recombination rates are presented as the average of the 15 reported rates closest to each position plotted. Subtelomeric regions are defined as having either >50% DNA sequence identity to another region of the genome or as having <60% nondubious ORFs using a 20-kb sliding window (Barton et al. 2008). The subtelomeric border is the point at which neither of these criteria is met. The gray line represents the genomic average recombination rate of 0.37 cM/kb (Barton et al. 2008).
Constitutive open chromatin is found at the centromeres and pericentromeric regions
Crossovers are repressed at S. cerevisiae centromeres and pericentromeric regions (Lambie and Roeder 1986; Chen et al. 2008), and exchanges at these regions are implicated in elevated frequencies of nondisjunction at meiosis II (Koehler et al. 1996; Lamb et al. 1996). However, in some studies, regions around centromeres have normal levels of meiotic DSBs (Blitzblau et al. 2007). This has led to the suggestion that DSBs near centromeres might be repaired by the sister chromatid (Chen et al. 2008), and that centromere-associated proteins such as cohesins and Sgo1 could create a chromatin environment that favors DSB repair from the sister. We speculated that repression of crossovers and noncrossovers near the centromeres is aided by a dense chromatin configuration present during meiosis, which predicts that these regions will not be enriched by FAIRE.
We analyzed the centromeres and associated regions 30 kb to the left and right of each centromere in 10-kb windows, using the same categorical groupings as above (I–IV) (Fig. 6; Supplemental Table S3). Contrary to our hypothesis, regions spanning centromeres were often open (12/16 centromeres, 1.29 expected P < 0.0001). Ten of the 16 centromeres were constitutively open (P < 0.0001), and none of the open sites that occurred within a centromere were meiosis specific. The pericentromeric sequences (defined as the DNA extending 10 kb on either side of the centromeres) were also constitutively open, containing more open chromatin than expected (P < 0.05 to the left; P < 0.01 to the right). Again, these sites of open chromatin were not specific to meiosis. This result does not support the hypothesis that dense chromatin near the centromeres plays a role in the suppression of crossovers and noncrossovers in the pericentromeric regions. Indeed, it raises the possibility that open chromatin somehow plays a role in this suppression.
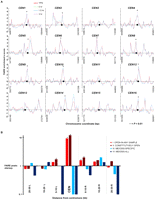
Centromeres and pericentromeric regions exhibit constitutively open chromatin. (A) FAIRE profiles of each of the 16 S. cerevisiae centromeres (CEN). Asterisks indicate sites significantly open at P < 0.01. Chromosomal coordinates are plotted on the x-axis and FAIRE enrichment is plotted on the y-axis. FAIRE data from YPD (red line), 0-h (green dashes), 1.5-h (blue dashes), and 3-h (purple dots) samples are plotted. (●) Centromeres. (B) Analysis of centromere-associated regions. The x-axis reports the region analyzed and the y-axis reports the ratio of observed sites of open chromatin to the expected number from random sampling. (Red) Category I; (maroon) category II; (light-blue) category III; (dark-blue) category IV sites.
A 15-bp DNA sequence motif is over-represented in meiosis-specific open chromatin
We examined whether sites of open chromatin might be specified by one or more DNA sequence motifs. When the open chromatin sites isolated either in any one of the conditions or all conditions were used as an input for the motif-finding algorithm BioProspector, no strong motif was identified. However, when sites of open chromatin specific for meiosis (category III or IV) were used as an input, we identified a 15-bp DNA sequence motif: SSGGTTCGANYCCSS (Fig. 7). This motif is similar to the B-box motif involved in regulation of tRNA expression (D'Ambrosio et al. 2008). Sites of open chromatin containing the B-box-related motif tended to be centered downstream rather than upstream of tRNA genes. Of the 29 meiotic open sites adjacent to a tRNA gene and containing a B-box related motif, 19 are downstream, eight straddle, and only two are upstream of the tRNA gene. Of the 55 sites that meet the same criteria, but are open in any one meiosis time point, the numbers are 34, 19, and two, respectively. Therefore, there is a strong bias for the B-box related motif to occur downstream of tRNA genes that are located in meiotic open chromatin.
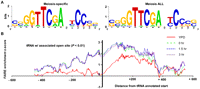
A 15-bp motif is over-represented in meiosis-specific open chromatin surrounding tRNA genes. (A) The 15-bp motif identified from FAIRE-enriched sites isolated from any meiotic sample, but not YPD, and FAIRE-enriched sites isolated from all meiotic samples, but not YPD, are represented (Crooks et al. 2004). (B) Open chromatin increases during meiosis at sites associated with tRNA genes. The position relative to tRNA annotated start sites (base pair) is plotted on the x-axis, and FAIRE enrichment is plotted on the y-axis. Cytoplasmic tRNA genes associated with a site of open chromatin were aligned at their annotated transcription start. Shown is a moving average of the average FAIRE-enrichment z score (window size = 15 probes). FAIRE data from YPD (red), 0-h (green dashes), 1.5-h (blue dashes), and 3-h (purple dots) samples are plotted. Regions corresponding to the functional tRNA have a median length of 72 bp (minimum of 70 bp and a maximum of 132 bp). The graph includes the 98 tRNA-associated sequences that overlap a site of open chromatin (P < 0.01).
Based on these results, we asked whether tRNA genes were associated with meiosis-specific open chromatin. We examined the chromatin profiles at all 275 cytoplasmic tRNA genes in the S. cerevisiae genome (Fig. 7). A total of 98 tRNA genes are associated with a site of open chromatin and 177 are not. Of these 98 tRNA genes, 65 were not open in YPD. Meiosis-specific open chromatin sites (Category III) were associated with tRNA genes far more than any other gene class, and over 25% of category III sites were associated with a tRNA gene, significantly greater than the expected proportion. However, meiosis-specific open chromatin at tRNA genes is unlikely to have a connection to recombination because DSB hotspots are not strongly associated with either tRNA coding sequences themselves or sequences 500-bp upstream and downstream of tRNAs (Supplemental Table S5). The enrichment of open chromatin downstream from tRNA genes suggests a mechanism other than transcriptional clearing of a tRNA gene promoter may be establishing these meiosis-specific chromatin changes.
Discussion
We used FAIRE on a synchronized meiotic time course in S. cerevisiae to investigate patterns of open chromatin during the first 3 h of meiosis. Up-regulation of SPO11, DMC1, and RAD51 in our samples and previous studies (Goyon and Lichten 1993; Allers and Lichten 2001) indicate that cells are processing DSBs into joint molecules during this time. An association between open chromatin and recombination hotspots had been established through many locus-specific studies, but whether and to what degree this association held on a genome-wide scale had yet to be determined.
Challenges
Several challenges were associated with our analysis of chromatin organization in meiosis. First, the biological function of open chromatin is likely dependent on chromosomal and physiological context. However, we used a strict P-value cutoff to define “open chromatin” uniformly across the genome. This cutoff likely sacrifices some biologically relevant true-positives for the sake of minimizing false negatives. ROC curve analysis indicates that sites identified at the low end of our P-value threshold are less likely to intersect with a DSB hotspot, and this trend intensifies as the cutoff is made more lenient. A second related challenge was defining a significant change in the openness of chromatin between samples or time points. Again, determining the degree of change likely to be linked to a biological outcome is difficult. Furthermore, we categorized sites as “open” or “not open” at individual time points without using quantitative information from adjacent time points to aid our calls.
Third, FAIRE may miss some sites such as the ARG4 locus that have been classified as open by other assays (Results). We speculate that sites that are highly occupied by recombination complexes could be identified as closed by FAIRE, but recognized as open by nuclease-sensitivity assays. Chromatin profiles generated by FAIRE may differ from those created by other assays such as DNase or MNase sensitivity or histone ChIP-chip. FAIRE probes chromatin with a reactive small molecule, while DNase and MNase probe chromatin by accessibility to a much larger enzyme. ChIP is dependent on the affinity of an antibody for its substrate. Each assay has its own advantages and disadvantages, and each is likely to be more sensitive than another in measuring some specific aspect of chromatin organization. Therefore, each assay reveals independent, nonredundant information about the chromatin profile at each locus, and carries a different definition as to what “open chromatin” means. DNase and MNase sensitivity assays may indeed be most sensitive in detecting open chromatin that occurs in sites that are highly occupied by recombination complexes, but not nucleosomal proteins. Conversely, openness as defined by FAIRE may in some cases fail to recognize sites that are highly occupied by non-histone proteins.
Fourth, we used tiling microarrays with a resolution of ∼270 bp, which is slightly less than the size of two nucleosomes. This resolution, together with the 250-bp minimum call size of our peak-finding algorithm, makes it possible that our analysis missed small sites of open chromatin that are relevant to our assertions. For example, DSB hotspot mapping on chromosome III by Southern blot analysis showed that some DSB hotspots can be resolved to ∼150 bp (de Massy et al. 1995; Liu et al. 1995; Baudat and Nicolas 1997). A fifth caveat stems from the low resolution of DSB hotspot mapping provided by the detection of meiosis-specific ssDNA. Although every DSB occurs at a single base, the ssDNA tails are often greater than 1 kb in length, and the corresponding hotspot calls can be 2 kb or greater in length (Blitzblau et al. 2007; Buhler et al. 2007).
Conclusions
Despite the challenges, our results provide clear evidence for the following:
-
DSB hotspots are generally associated with open chromatin.
-
A class of DSB hotspots occur in regions that are no more open than the genomic average, illustrating that open chromatin is not a requirement for DSB formation. The close spacing of our time points in relation to DSB formation, and the incomplete synchrony of meiotic samples makes it unlikely that we failed to capture opening of chromatin at these DSB hotspots. DSB hotspots that occur in closed chromatin could be specified by other properties such as histone modifications.
-
Among sites of meiotic open chromatin, the degree of chromatin openness as defined by P-value is predictive of DSB hotspots. However, among sites of mitotic open chromatin, the degree of openness is not predictive of meiotic DSB hotspots.
-
Regions 0–20 and 30–40 kb proximal to the subtelomeric borders are enriched for meiosis-specific sites of open chromatin (see pairing function section, below).
-
The chromatin surrounding centromeres is open in mitosis and the first 3 h of meiosis.
-
Many tRNA genes are open specifically in meiosis, with the open site usually occurring downstream of the annotated gene.
tRNAs and chromatin
The biological significance of meiosis-specific open chromatin around tRNA genes is not obvious to us. If this enrichment were connected in some way to recombination, we would expect an association between DSB hotspots and tRNA loci, which is not apparent. However, in S. pombe and humans, B-box motifs and tRNA-associated sequences do function in modulating chromatin modification (Kundu et al. 1999; Scott et al. 2006). They also serve as condensin and cohesin loading sites in mitotic cells in both S. cerevisiae and S. pombe (D'Ambrosio et al. 2008; Schmidt et al. 2009). In S. pombe, deletion of a ∼489-bp sequence containing two tRNA genes that form a cohesin-binding site resulted in a complete loss of cohesin loading at that site (Schmidt et al. 2009). Additionally, in S. pombe, the tRNA alanine (tRNAAla) gene specifies a heterochromatin boundary that is important for meiotic chromosome disjunction (Scott et al. 2006). Deletion of the centromere-proximal tRNAAla gene causes H3 histones dimethylated at lysine 9 (H3K9me2) to spread beyond the wild-type boundary at tRNAAla. This mutant also exhibits defective meiotic chromosome segregation (Scott et al. 2006), suggesting that in S. pombe chromatin changes directed by tRNAs are important for accurate meiotic disjunction. A recent study in S. pombe showed that long, polyadenylated, noncoding RNA genes often colocalize with meiotic DSB hotspots (Wahls et al. 2008), but did not address tRNA genes.
In S. cerevisiae, tRNA genes have been shown to play a role in the specification of heterochromatin boundaries. For example, the tRNA threonine (tRNAThr) gene is necessary and sufficient to stop the spread of heterochromatin from the HMR locus on chromosome III (Donze and Kamakaka 2001). Similarly, the tRNA glutamine (tRNAGln) is required for halting the spread of heterochromatin extending from rDNA arrays on chromosome XII (Biswas et al. 2009). The barrier activity of both tRNAThr and tRNAGln is abrogated in histone acetyltransferase mutants sas2 and gcn5, reinforcing an important function for tRNA in chromatin organization (Biswas et al. 2009).
A pairing function for meiosis-specific open chromatin at the subtelomeric borders?
We observed a strong enrichment of meiosis-specific open chromatin at the subtelomeric border regions. Approximately 8% of the sites we identified to be open during all meiotic time points, but closed in YPD exist 0–10 kb proximal to the subtelomeric border, which is significantly above the expected proportion of 3%. DSB mapping studies based on rad50S have reported an under-representation of meiotic DSBs 40–100 kb from the telomeres (Gerton et al. 2000; Borde et al. 2004; Robine et al. 2007). However, studies using ssDNA mapping in a dmc1 mutant strain report that the depletion of DSBs extends only 0–20 kb from telomeres, and that regions 20–120 kb from telomeres exhibit an over-representation of DSBs (Blitzblau et al. 2007; Buhler et al. 2007). It is attractive to propose that meiosis-specific opening of the chromatin in the regions proximal to the subtelomeric border (which are generally 0–40 kb from the telomere) provides access for recombination protein complexes and a unique substrate for homology searching to facilitate homologous pairing. It will be interesting to see whether mutants defective for pairing also show an absence of meiosis-specific open chromatin in these regions. Some factors that promote meiosis-specific open chromatin have been identified in S. pombe (Yamada et al. 2004; Hirota et al. 2007; Hirota et al. 2008), and the idea that meiosis-specific open chromatin aids chromosome pairing predicts that mutants for this process will be pairing defective.
Future applications in other systems
FAIRE is a low-cost, reproducible method that can be used to identify sites of open chromatin. Our results demonstrate that chromatin that is constitutively open in mitosis or meiosis is correlated with DSBs, and therefore FAIRE holds promise for the identification of putative hotspots in organisms in which meiotic synchrony cannot be established. It is also useful in organisms that lack convenient mutations that arrest meiosis after ssDNA has formed. FAIRE may be especially useful in organisms in which meiotic prophase staging is possible, such as Caenorhabditis elegans and Arabidopsis thaliana. In C. elegans, oocytes at all stages of meiotic prophase exist within each gonad in an unambiguous temporal/spatial arrangement (Zalevsky et al. 1999), although it may be challenging to obtain the estimated 500,000 germ cells required for FAIRE. In A. thaliana meiotic staging is more difficult, but cells undergoing meiosis can be quickly and cheaply isolated by capillary collection (C Chen, A Smith, H Ma, unpubl.).
Methods
Strains and growth conditions
Vegetative cells were grown in YPD (1% yeast extract, 2% peptone, 2% dextrose) to an OD600 of 0.6–0.8. Synchronous sporulation was carried out for each biological replicate (three total) by using YPD overnight cultures of wild-type cells (SK1 strain SHy02 [MATa/MATα ho∷LYS2/ho∷LYS2 leu2∷hisG/leu2∷hisG lys2/lys2 ura3/ura3]) to inoculate YPA (1% yeast extract, 2% peptone, 2% potassium acetate) cultures at an OD600 of 0.1. We grew the cultures at 30°C to an OD600 of 0.8–1.2 (about 16 h), collected cells by centrifugation, and washed with SM (2% potassium acetate). We then resuspended cells at an OD600 of 2.0 in SM and incubated 600-mL cultures in 2-L flasks at 30°C, shaking at 265 rpm.
Sample collection and FAIRE
Cells used for FAIRE were collected during vegetative growth (100 mL of culture per sample) and after 0, 1.5, and 3 h after placement in SM (50 mL of culture per sample). Three biological replicates of each time point were taken. To monitor meiotic entry, progression, and synchrony in our biological replicates, we collected RNA samples for expression microarray analysis at each FAIRE time point. Additionally, we collected RNA from premeiotic cells that had been starved of a fermentable carbon source for 8 h (acetate) and from synchronized cells that had been incubated in sporulation medium for 4.5, 6, 9, and 12 h to assay the transcriptional profile of key early, middle, and late meiotic genes. Total RNA was isolated from frozen samples using hot acid-phenol as previously described (Ausubel 1997). FAIRE samples were fixed with 1% formaldehyde for 20 min at room temperature and washed twice with 1X cold PBS. The samples were then snap frozen in a dry-ice/ethanol bath and stored at −80°C. FAIRE was carried out as described (Hogan et al. 2006). Briefly, cells were thawed and lysed with glass beads and the chromatin was sheared to an average size of ∼800 bp by sonication as measured by agarose gel electrophoresis. We conducted a phenol-chloroform extraction on the cross-linked samples to remove proteins and protein-associated DNA. The resulting DNA samples were further purified using Zymo Clean and Concentrator kit per the manufacturers' protocol (Zymo Research). Genomic reference DNA samples were generated using the YPD samples without formaldehyde cross-linking.
FAIRE amplification and labeling
FAIRE and reference DNA was amplified from ∼100 ng starting material using the Whole-Genome Amplification kit (Sigma) (O'Geen et al. 2006). Two micrograms of amplified DNA was labeled with either Cy3 or Cy5-conjugated dUTP using the BioPrime Array CGH kit (Invitrogen). For replicates 1 and 3 the FAIRE samples were labeled with Cy5 and the reference Cy3. A dye swap was performed on replicate 2. DNA concentration and dye incorporation were measured using a Nanodrop spectrometer. Four micrograms of each fluorescently labeled sample and reference were used in the hybridization.
Expression sample labeling
For expression experiments, total RNA was reverse-transcribed using SuperScript II reverse transcriptase (Invitrogen) incorporating amino-allyl dUTP (Sigma) at a ratio of 3:2 with dTTP. Genomic DNA (used as a reference) was amplified with Klenow (NEB) incorporating amino-allyl dUTP (Sigma) at a ratio of 3:2 with dTTP. Reactive Cy3 or Cy5 (Amersham) was coupled to the amino-allyl of the resulting DNA fragments in the presence of sodium bicarbonate.
Microarray hybridization and image acquisition
FAIRE samples were hybridized simultaneously with the reference at 52°C for 16 h in a Maui hybridization chamber to yeast whole-genome 4 × 44 k tiling microarrays (Agilent; ∼270-bp resolution). Expression samples were hybridized simultaneously with the reference sample to yeast whole-genome PCR product spotted microarrays containing coding and noncoding regions at ∼1-kb resolution. The arrays were scanned with an Axon 4000 scanner, and data was extracted using GenePix 6.0 software. Only spots of high quality by visual inspection, with fewer than 10% saturated pixels in either channel, with a background corrected sum of medians for both channels greater than 500, were used for the analysis.
Data processing and statistical analysis
The ratio of FAIRE intensity (sample) to genomic DNA intensity (reference) for each spot on the microarray was converted to a log2 ratio, which is referred to as a “FAIRE log ratio.” Mitochondrial probes were removed from the analysis prior to data processing. FAIRE log ratios were converted to z scores by centering at a mean of zero and scaling the standard deviation to one. The median z score was determined for each sample across all three biological replicates and the values were then recentered and scaled. These rescaled median z scores were used as input for the peak-finding algorithm ChIPOTle (V1.02) (Buck et al. 2005) with the following parameters: Gaussian background distribution, step-size 125 bp, and window size 500 bp (Buck et al. 2005). Peaks representing sites of open chromatin were collected from all samples using a Bonferroni corrected threshold of P <0.01. Sites of open chromatin were initially organized as an output from each time point (YPD, 0, 1.5, and 3 h). Among samples, sites were considered to be coincident if they shared >50% bp overlap or an identical high spot, defined as the probe with the highest z score. For analysis using these groupings, the site with the most significant P-value was used.
Overlaps with published hotspot coordinates as defined by ssDNA enrichment (Blitzblau et al. 2007; Buhler et al. 2007) or by genome tiling recombination maps (Mancera et al. 2008) were calculated using the Galaxy web application (Giardine et al. 2005). Overlaps with ORFs were calculated by determining the intersecting regions with samples and the Saccharomyces Genome Database (SGD) ORF annotations using Galaxy (Giardine et al. 2005). Regions were considered to overlap between two datasets if at least 1 bp overlapped. As a control, 10 permutations of each sample set of regions were generated using a PERL algorithm, which centers the start and end coordinates of each region on a random probe from the array (Giresi et al. 2007). Percent overlap was determined for each permutation as with the samples using Galaxy to generate a distribution of random overlaps for each sample. We tested the distributions of random overlaps for normality using a Shapiro-Wilk goodness of fit test (Shapiro and Wilk 1965). The normal distribution could not be excluded in any case, and we used a Z-test to determine the P-values of our experimental overlaps (Sprinthall 2003). The means of the distributions of random overlaps were used as the expected values.
We presented FAIRE enrichment over DSB hotspots and tRNA-associated sequences as moving averages of probe z scores. This was done by first determining all probes that fell within DSB hotspots or within 500 bp of a tRNA-annotated start or stop. We then aligned DSB hotspots (Buhler et al. 2007) (all are 2 kb) and tRNAs at their annotated starts. At each position within the aligned DSB hotspots or tRNA associated sequences, we calculated the moving average of the closest 15 probes.
We assessed the ability of FAIRE to predict DSB hotspots using ROC curve analysis (Zweig and Campbell 1993). Briefly, a ROC curve is a plot of the rates at which true-positives and false-positives are compiled with respect to an ordering statistic. In this case, the ordering statistic is the P-value of a site of open chromatin. A true-positive occurs when a FAIRE site intersects (250-bp overlap) with the coordinates of a DSB hotspot (Blitzblau et al. 2007; Buhler et al. 2007), and a false-positive occurs when a site does not intersect with a DSB hotspot. We ordered FAIRE sites by P-value and plotted the FPF, scaled to 1, on the x-axis vs. the TPF, scaled to 1, on the y-axis. We measured the AUC, which is a summary of the predictive value of the ordering statistic. An AUC value of 0.5 indicates a random predictor, while an AUC value of 1.0 indicates a perfect predictor. We compared each result with 10 control ROC curves generated by random placement of the analyzed FAIRE sites (as above). Additionally, we performed ROC curve analysis to quantify the ability of raw FAIRE data to predict DSB hotspots. We conducted the analysis as above ordering probes by FAIRE enrichment z score and defining a true-positive as when the probe intersected (44-bp overlap) a DSB hotspot (Blitzblau et al. 2007; Buhler et al. 2007). We calculated likelihood ratios (TPF/FPF) at various z score thresholds to express the predictability of FAIRE enrichment z scores in each time point.
Chromatin profiles at the regions associated with centromeres and telomeres were analyzed by calculating the total counts of sites of open chromatin that intersect the regions of interest (either centromeric or telomeric associated regions). Our experimental values were compared with the counts of intersecting regions expected from 10,000 random permutations of each sample. The mean counts of the random permutations were used as the expected values for experimental samples and the statistical significance of the expected overlaps to observed overlaps were assessed by a χ2-test. This method of analysis was also used to compare the overlap of sites of open chromatin with DSB hotspots where the centers of the permuted sites were restricted to intergenic space.
Motif determination
Over-represented sequence motifs were determined for each group of regions of open chromatin using BioProspector (Liu et al. 2001), set to find 10-, 13-, and 15-bp motifs. Yeast intergenic sequences were used as the background model.
Acknowledgments
We thank Arnold Barton and David Kaback for providing the chromosomal coordinates for the computationally defined boundaries between the subtelomeres and euchromatin. We also thank Paul Giresi for help with data analysis and three anonymous reviewers for very helpful comments. L.E.B. and G.P.C. thank NSF (MCB-0618691) and DOE (DE-FGO2-05ER15651) for financial support. This work was supported by NIH grant R01-GM072518 to J.D.L. We also thank The University of North Carolina at Chapel Hill's Office of the Vice Chancellor for Research and Economic Development for providing support for Open Access publication.
Footnotes
-
↵4 Corresponding authors.
E-mail gcopenhaver{at}bio.unc.edu; fax (919) 962-1625.
E-mail jlieb{at}bio.unc.edu; fax (919) 962-1625.
-
[Supplemental material is available online at http://www.genome.org. The microarray data from this study have been submitted to Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE18256.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.096297.109.
-
- Received May 20, 2009.
- Accepted September 24, 2009.
-
Freely available online through the Genome Research Open Access option.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








