
Whole genome shotgun sequencing of Brassica oleracea and its application to gene discovery and annotation in Arabidopsis
Abstract
Through comparative studies of the model organism Arabidopsis thaliana and its close relative Brassica oleracea, we have identified conserved regions that represent potentially functional sequences overlooked by previous Arabidopsis genome annotation methods. A total of 454,274 whole genome shotgun sequences covering 283 Mb (0.44×) of the estimated 650 Mb Brassica genome were searched against the Arabidopsis genome, and conserved Arabidopsis genome sequences (CAGSs) were identified. Of these 229,735 conserved regions, 167,357 fell within or intersected existing gene models, while 60,378 were located in previously unannotated regions. After removal of sequences matching known proteins, CAGSs that were close to one another were chained together as potentially comprising portions of the same functional unit. This resulted in 27,347 chains of which 15,686 were sufficiently distant from existing gene annotations to be considered a novel conserved unit. Of 192 conserved regions examined, 58 were found to be expressed in our cDNA populations. Rapid amplification of cDNA ends (RACE) was used to obtain potentially full-length transcripts from these 58 regions. The resulting sequences led to the creation of 21 gene models at 17 new Arabidopsis loci and the addition of splice variants or updates to another 19 gene structures. In addition, CAGSs overlapping already annotated genes in Arabidopsis can provide guidance for manual improvement of existing gene models. Published genome-wide expression data based on whole genome tiling arrays and massively parallel signature sequencing were overlaid on the Brassica–Arabidopsis conserved sequences, and 1399 regions of intersection were identified. Collectively our results and these data sets suggest that several thousand new Arabidopsis genes remain to be identified and annotated.
Arabidopsis thaliana is the first plant model species to have been sequenced on a genome-wide scale. Since the publication of its genome in the year 2000 (The Arabidopsis Genome Initiative 2000), there has been a continuous effort to complete sequence gaps and to improve gene annotation. Large-scale sequencing of expressed sequence tags (ESTs) and full-length cDNAs from Arabidopsis and other plants, coupled with better ab initio gene structure prediction programs, have contributed to improvements in the annotation of Arabidopsis (Wortman et al. 2003). Notable among them include the refinement of exon–intron boundaries and the addition of untranslated regions (UTRs) (Haas et al. 2002, 2003; Zhu et al. 2003). However, despite the availability of a great number of ab initio and database search–based gene prediction methods, identification and annotation of all the genes in any organism is still one of the major challenges in biology. This is partly because the existing EST/cDNA sequences in databases do not represent all transcription units and/or available gene-prediction programs are limited in their capacity.
Comparative genomics is rapidly emerging as a powerful tool for genome analysis and annotation. Over the course of evolution, functional regions such as exons and regulatory sequences tend to be more conserved than are nonfunctional regions, thus local sequence similarity has implications for biological functionality. Several recent studies have demonstrated that a comparative genomics approach is useful in refining gene predictions in human (Ansari-Lari et al. 1998; Bouck et al. 2000; Mallon et al. 2000; Flicek et al. 2003; Gugio et al. 2003), Drosophila (Bergman et al. 2002), yeast (Brachat et al. 2003), and Plasmodium (Carlton et al. 2002).
B. oleracea and A. thaliana diverged 15–20 million years ago (Yang et al. 1999). Earlier comparative mapping studies of Brassica and Arabidopsis using molecular markers revealed extensive synteny between these two species, suggesting that knowledge gained in one species can be productively applied to the other (Lan et al. 2000; Babula et al. 2003) although deviation from colinearity was also noted (Kowalski et al. 1994; Sadowski et al. 1996; Ryder et al. 2001). Nucleotide sequence conservation between these two species has been reported to be in the range of 75%–90% in exons, whereas in introns and intergenic regions, it is ≤70% (Quiros et al. 2001). Thus a genome scale comparison of Arabidopsis with Brassica at the sequence level provides an excellent opportunity to test the applicability of this phylogenetic footprinting approach in the annotation of plant genomes. To this end, we and the groups at Cold Spring Harbor Laboratories and Washington University carried out Brassica whole genome shotgun (WGS) sequencing, collectively generating 538,418 good reads. These data were aligned with and compared to Data Release 4.0 of The Institute for Genomic Research (TIGR) (ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/PREVIOUS_RELEASE_VERSIONS/release4.tar.gz) Arabidopsis genome sequence to discover new genes and refine the existing Arabidopsis annotation. Our in silico analysis was augmented with in vitro validation of conserved sequences for transcription by using PCR. This work, a similar effort by our colleagues at Cold Spring Harbor (Katari et al. 2005), and the use of the Brassica WGS sequence alignments, along with Arabidopsis cDNA alignments to the Arabidopsis genome to train TWINSCAN (Korf et al. 2001; P. Hu and M. Brent, in prep.), represent the first efforts to utilize comparative whole genome sequencing to improve gene annotation in plants. The Gramene database (www.gramene.org) also provides comparative alignments between maize and rice genomic sequence, but these have not yet been utilized in the improvement of gene models.
Results
Brassica genome sequencing
The WGS effort at TIGR resulted in 415,093 distinct high-quality reads of 667 bases average length. The distribution of these sequences across the various libraries constructed is shown in Supplement 1, Table S1. These sequences were submitted to the Genome Survey Sequence (GSS) division of GenBank. Accession numbers can be found in Supplement 2. Another set of 122,897 high-quality reads generated by Cold Spring Harbor Laboratory and Washington University in St. Louis (MO) were acquired from GenBank (Katari et al. 2005). Prior to the identification of conserved sequences, these sequences were searched against chloroplast, mitochondrial, ribosomal DNA, transposon-related sequences and repeats, and matching sequences were removed. The filtered combined sequence set consists of 454,274 reads with an average read length of 623 bases, representing a total of 283.0 Mb (0.44× coverage) of the B. oleracea genome, estimated at 650 Mb (Paterson et al. 2001).
Mapping Brassica WGS sequences to the Arabidopsis genome
The individual Brassica WGS sequences were mapped to the Arabidopsis chromosome sequences by using BLASTZ (Schwartz et al. 2000, 2003) at its default settings. Since many reads mapped to more than one genome location, the position of each WGS sequence was determined by the highest scoring alignment cluster. Of the 197,344 Brassica sequences that could be mapped to the Arabidopsis genome, 144,821 were found to intersect 84% (23,005 out of 27,384) of the annotated genes and 54% (84,481 out of 157,737) of the annotated exons. Incremental sampling of the Brassica sequences that matched the Arabidopsis genome provides an indication of the extent of gene and exon coverage as a function of the number of sequence reads (Fig. 1). As expected, gene coverage exceeds exon coverage since Arabidopsis annotation version 4.0 reports an average of 5.3 exons per gene.
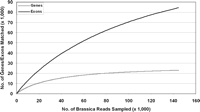
Reconstruction by incremental sampling of the relationship between the number of Brassica reads matching the Arabidopsis genome and the fraction of Arabidopsis genes and exons intersected by those reads.
The average coverage of the Arabidopsis genome by Brassica reads on a nucleotide basis (using only alignments from the topscoring read) was calculated within 5-kb windows across each chromosome, scoring only nucleotides in aligned positions. The distribution of these matches along the Arabidopsis chromosome 1 is shown in Figure 2. The chromosome has Brassica matches along its entire length, with an average coverage of 1.76 Brassica nucleotides per matching Arabidopsis nucleotide. The lower level of coverage between -14.8 and 15.2 Mb (the location of the centromere) presumably reflects the fact that the Brassica sequence set used had been filtered against known Arabidopsis repeats that include the well-characterized 180-bp centromeric repeat, sequences that are present in chromosome 1. A similar trend was observed for the other chromosomes (data not shown).
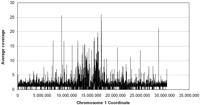
Coverage of the Arabidopsis chromosome 1 with Brassica WGS sequences. The Brassica sequence data set that had been filtered to remove known organellar, ribosomal, and repeated sequences, including transposons, was searched against the five Arabidopsis chromosomes by using BLASTZ. Each sequence is represented only once at the location determined by its highest scoring cluster of BLASTZ alignments. Coverage is expressed on a nucleotide basis and calculated in 5-kb windows.
Distribution of conserved regions across the Arabidopsis genome and the prediction of novel genes and exons
To evaluate the relationship between Brassica–Arabidopsis sequence conservation and Arabidopsis gene structure, the filtered Brassica sequences were aligned against a sample of 1000 Arabidopsis genes whose structures were supported by full-length cDNAs. An example of such alignments to a single Arabidopsis gene (At1g20090) is shown in Figure 3. It can be seen that conserved sequences invariably coincide with exons. Overall, the analysis showed that exons occupy 87% of the merged Brassica reads, indicating that sequence conservation does extend across the splice sites and a limited distance into the introns.
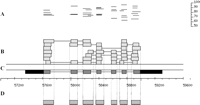
Alignment of Brassica WGS reads with Arabidopsis genomic DNA in the region of At1g20090, a gene whose structure is supported by full-length cDNA evidence. (A) The upper part of the figure shows the length and percentage identity for each BLASTZ alignment segment of Brassica sequence to the Arabidopsis genome after filtering by percentage identity and match length (see Methods). Note that occasionally two overlapping HSPs with the same percentage identity will appear merged into a single line. (B) Each box corresponds to a BLASTZ alignment shown in A. Boxes joined by black lines indicate multiple alignment segments within a single Brassica WGS sequence. (C) The dark and light boxes (expressed sequence) joined by the black line (introns) show the gapped alignment of the experimental cDNA to genomic DNA, with the coding sequence represented in gray and the UTRs in black. (D) The boxes below the BAC nucleotide scale show the extent of the CAGSs constructed from these sets of overlapping BLASTZ alignments.
BLASTZ alignments against this set of cDNA-validated gene models were evaluated with respect to sensitivity and specificity for detection of nucleotides in exons by using sets of alignments having a range of either minimum percentage identity or minimum match length. Specificity showed steady increase with both match length and percentage identity up to a certain point, whereas the opposite was true for sensitivity (Fig. 4). To identify suitable parameters for genome-wide identification of regions of sequence conservation between Arabidopsis and Brassica, we evaluated the product of sensitivity (Sn) and specificity (Sp) to achieve a reasonable compromise between these two parameters. These curves show broad optima at ∼40 bp match length and 60% identity. Thus these values were chosen for the global analysis, recognizing that the price of increased sensitivity would be an increased number of false positives. In cases where Brassica sequences generated overlapping alignments (high scoring pairs [HSPs]) to the same region of the Arabidopsis genome, the overlapping HSPs were mapped onto the Arabidopsis genome, and each region of contiguous or overlapping matches fulfilling the specified criteria was termed a CAGS (conserved Arabidopsis genomic sequence) (Fig. 5). Overall, the 454,274 Brassica sequences produced 10.27 × 106 hits of 51 bp mean length to the Arabidopsis genome, a number that was reduced to 2.7 × 106 alignments of 114 bp mean length after filtering for minimum length (40 bp) and identity (60%) (Table 1). These alignments were collapsed onto the Arabidopsis genome as illustrated in Figure 5B and produced 229,735 CAGSs of 170 bp average length. Among these, 74% (169,357) intersected existing gene models (i.e., overlapped by 25% of their length) and 26% (60,378) fell into intergenic regions (Table 1). CAGS overlapping existing gene models tend to be longer than those matching the intergenic regions. The average coverage on a nucleotide basis for these genic and intergenic CAGSs was 4.04 and 5.78 respectively. These values are higher than that given above for chromosome-wide coverage since a single read frequently generates clusters of HSPs at more than one chromosome location and will thus contribute to several CAGSs. The proportions of CAGSs with different levels of coverage is similar for both genic and intergenic CAGSs and is shown in Supplement 1, Figure S1.
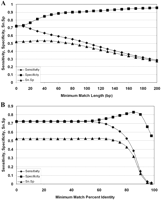
Sensitivity and specificity for exon detection by BLASTZ alignments: (A) as a function of match length, with minimum percentage identity set to zero; and (B) as a function of percentage identity with minimum match length set to zero. All BLASTZ alignments were filtered at different levels of match length and percentage identity, and specificity and sensitivity for identification of bases within true exons by these alignments were calculated.
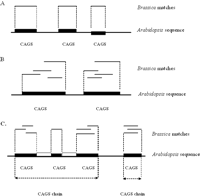
Illustration of conserved Arabidopsis genome sequence (CAGS). (A) CAGS with single Brassica matches. Thin lines represent Brassica–Arabidopsis HSPs; thick bars, the corresponding Arabidopsis genomic sequence. (B) Overlapping Brassica matches (HSPs) are merged to produce one CAGS for that region. (C) CAGS chains. Intergenic CAGSs that fall within 416 bp of one another were chained together to represent a single feature. Open boxes between CAGS represent regions <416 bp that may correspond to introns between CAGS.
Number, mean match length, and standard deviation of high scoring pairs (HSPs) and CAGS
For the purposes of enumerating intergenic regions that might represent novel genes or other conserved regions such as noncoding RNAs, regulatory elements, and conserved noncoding sequences (CNSs) (Kaplinsky et al. 2002), individual CAGSs were joined together into “chains” (Fig. 5C) if they were separated by ≤416 bp, a figure that represents the mean + 1 SD of the entire set of Arabidopsis introns and includes 93% of the introns. At the same time, only 12.1% of intergenic distances in the Arabidopsis genome fall below this value, so that the chance of merging two functionally distinct features into a single CAGS chain using this criterion is low. This process yielded 27,374 chains of intergenic CAGSs, approximately half of which are singletons (Table 2). The distribution of chains is proportional to the chromosome length, indicating that the CAGSs are uniformly distributed across the genome. The chains containing multiple CAGSs could represent novel multiexon genes conserved between Arabidopsis and Brassica. For later analysis we have divided these 27,374 chains into those that lie close to existing genes, using the intron + 1 SD criterion, that may represent extensions of existing gene annotations (11,688 perigenic CAGSs) and those that are separated by more than this distance and are thus likely to represent novel structures (15,686 truly intergenic CAGSs). The sequences of the complete set of intergenic CAGSs are provided in Supplements 3 and 4.
CAGS chains in intergenic regions of the Arabidopsis genome
Improving existing gene annotation using CAGSs
A set of 370 BACs, each containing intergenic chains comprised of three or more CAGSs, was identified and subjected to manual curation by using our ATH1 database and Annotation Station, resulting in the creation of 104 new gene models, identification of an additional 295 pseudogenes, and updates to 60 existing genes models. The Brassica alignments alone do not provide sufficient information for the creation of new gene models or the precise definition of splice junctions, even with careful manual curation, much less simply by computation. Thus we have not attempted to use the remaining set of CAGSs for automated gene refinement.
Investigation of expression from CAGSs
Transcriptional activity was assessed from 192 conserved regions selected approximately equally from the singleton and paired (chains of two) CAGS data sets, without regard to their location with respect to neighboring genes. PCR was performed on a population of cDNAs pooled from diverse Arabidopsis tissues that have been shown to contain transcripts for 70%–80% of all hypothetical genes assayed (Xiao et al. 2002). The sequences of the CAGSs examined and primers used are provided in Supplements 5 through 7. PCR products were sequenced to determine whether they indeed arose from the target locus. The results are summarized in Table 3. It is striking that although singleton and paired CAGSs yielded approximately the same the number of highquality sequences, the proportion of these sequences that were derived from the expected target was much higher for the paired (∼45%) than for the singleton CAGSs (∼18%). The reason(s) for this difference are not clear. All the paired CAGS sequences were unique in the genome in that there were no other regions with ≥80% identity to any part of the CAGS. Among the singleton CAGSs, 86% were unique while the remainder had one or more additional matches of >80% identity over ≥80% of their length (from two to nine repeats). However, among the singleton CAGSs that were repeated in the genome, 50% of those that sequenced successfully matched their target, while the other 50% did not. Thus the higher proportion of sequences from singleton CAGSs that did not match their targets cannot be ascribed to their repetitive nature but is more likely due to the idiosyncracies of PCR possibly coupled with the shorter target regions. The average length of the paired CAGS targets was 382 ± 9 bp (SEM), whereas for the singleton CAGSs it was 176 ± 9 (SEM). Table 3 also shows the breakdown of expression between perigenic (27%) and truly intergenic CAGSs (32%).
Success rate in validating expression from singleton and paired CAGS
Cloning of full-length cDNAs and its application to genome annotation
Those regions showing evidence of expression were subjected to 5′ and 3′ RACE analysis (Frohman et al. 1988). The resulting sequences from each location (33 paired and 18 singleton CAGSs, with seven loci producing no good sequences) were assembled by using TIGR assembler (Sutton et al. 1995) as described previously (Xiao et al. 2002) and a total of 70 transcript assemblies submitted to GenBank as FL-cDNAs or partial cDNAs (GenBank accession nos. AY299234–AY299303). Subsequently, the same sequences were aligned against the Arabidopsis genome and assembled into a minimal set of distinct transcripts by using the PASA pipeline (Haas et al. 2003). Of the 70 assemblies, 63 aligned properly to the genome sequence and were collapsed by PASA into 49 alignment assemblies. From these, 21 new genes were modeled at 19 loci (two having splice variants), new splice variants were added to two existing genes and the structures of another 17 existing genes were updated. The other nine alignment assemblies matched unannotated intergenic regions but provided insufficient information to instantiate new gene models. In almost every case, the regions in which the experimental data supported the annotation of a novel gene were already spanned by one or more gene predictions. However, there were always significant inconsistencies between the gene predictions in that region such that no new gene could have been modeled on the basis of predictions alone. Furthermore, the experimentally supported gene model always differed at one or more splice sites from any of the predictions. A list of new genes instantiated and existing genes matched and updated in this study is shown in Table 4. Some examples of newly created genes are shown in Figure 6. Examples of updates to gene models can be found in Supplement 1, Figure S2. We also compared the size and structure of the newly modeled genes with all genes that have FL-cDNA support. The two sets of cDNAs have comparable lengths, but the average CDS length and exon count is lower in the newly modeled genes (Table 5).
Gene models created or modified based on this study

Screen shots of new genes modeled based on RACE results. The images are taken from Annotation Station software. (From top to bottom) The tracks in the gene viewer show the spliced alignment of the assembled sequences from the RACE products (alignAssembly); the location of the paired CAGS and the primers used to test for its expression and subsequently for performing 5′ and 3′ RACE reactions (alignAssembly CAGS); the location of all CAGS in this region (blastZ:bog_seq); predictions of genemarkHMM, genescan+, glimmerA, and Twinscan; version 4 of the TIGR Arabidopsis annotation and the current curations of gene models in this region that incorporate the experimental evidence derived from this study; and the TIGR BAC identifier (the final + sign showing on some of the evidence lines indicates the strand of the alignment). (A) Screen shot of evidence relating to AGI locus At2g14635. The gene model on the left (At2g14635) did not exist prior to this work. Genemark.hmm made no prediction, and other predictions were discordant. The current working model was created based on the alignment of the assembly produced from the sequenced RACE products. (B) Screen shot of evidence relating to AGI locus At4g27415. There are no gene predictions in this region and no working model in version 4 of the TIGR Arabidopsis annotation. The sequenced RACE products generated two assemblies that resulted in the creation of two splice isoforms (At4g27415.1 and At4g27415.2).
Comparison of newly created genes with the complete set of cDNA-supported genes
Global expression profiling of CAGSs
In a recent publication, Yamada et al. (2003) described the development of an Arabidopsis whole genome tiling array and its deployment to characterize genome-wide transcription. Their “intergenic clusters” correspond to regions of the genome that were unannotated in TIGR release 3.0 but to which Arabidopsis ESTs could be mapped. Most of these clusters were incorporated into annotated gene models in later releases. Their “intergenic regions” contained neither an annotated gene nor an intergenic cluster. We have mapped both the intergenic clusters and the intergenic regions from the Yamada et al. (2003) data set onto our intergenic chains of CAGSs and find that 102 expressed clusters and 458 expressed intergenic regions intersect our 15,686 intergenic CAGS chains. Similarly, we have taken the massively parallel signature sequencing (MPSS) data provided by Blake Meyers (Meyers et al. 2004) and find that 1060 CAGSs of the 15,686 CAGS chains contain a significantly expressed MPSS signature. Together these two data sets provide evidence for expression of 1399 intergenic chains of CAGS.
Discussion
So far, the annotation of Arabidopsis and other plant genomes has relied on ab initio gene prediction and alignment of known features (ESTs, cDNAs, proteins, functional domains, etc.) against the target genome. In this work we have described a comparative genomic approach to annotation by using genomewide alignment of WGS sequences from a fairly close relative (B. oleracea) to a finished and annotated genome (Arabidopsis) to direct both in silico and experimentally based improvements to the genome annotation.
Mapping Brassica reads onto the Arabidopsis genome
At 0.44× coverage of the B. oleracea genome, the number of Arabidopsis genes having a Brassica match (84%) appears to be approaching saturation, while the number exons matched (54%) is far from saturated. Thus further sequencing of Brassica should lead to matches to more exons, a fraction of which would be part of novel genes. The relatively high level of coverage of the Arabidopsis genome by <0.5× coverage of the B. oleracea genome is most likely due to the fact that this genome is triplicated with respect to Arabidopsis (Cavell et al. 1998; Lagercrantz 1998). It is also interesting to note that in spite of the prior removal of sequences matching transposable element ORFs and known Arabidopsis repeats, there is still a higher density of Brassica alignments near the centromeres of each chromosome (e.g., between ∼12.5 and 14.8 Mb and between 15.2 and 16.5 Mb on chromosome 1). This suggests the presence in the Brassica genome of additional classes repetitive sequences not removed by our filtration process that are also found in (peri)-centromeric regions of the Arabidopsis genome.
Sequence conservation between Brassica and Arabidopsis and the development and chaining of CAGSs
BLASTZ produced a very large number of HSP alignments between the individual Brassica reads and the Arabidopsis genome. To maximize the number of conserved regions identified at the expense of specificity, values of minimum match length (40 bp) and minimum percentage identity (60%) toward the low side of the broad maxima for Sn.Sp were used to generate CAGSs for further analysis. Although the cut-off value of 60% identity for exon definition is lower than that reported by Quiros et al. (2001), our Sn.Sp analysis and the results exemplified in Figure 3 demonstrate that this lower cut-off for exon–intron discrimination is valid and effective. Furthermore, we subsequently analyzed nucleotide sequence conservation across 18 pairs of B. oleracea–A. thaliana orthologs by using recently acquired data (data not shown). In this data set, average sequence conservation between coding sequences was 71% (range = 52%–96%), while the identity across introns was 59% (range = 31%–75%). Compared with cut-offs of 60 bp match length and 70% identity, the criteria selected generate 30% more CAGSs for a 12% decrease in specificity, a compromise that seems reasonable.
One possible factor contributing to the effectiveness of the lower values for exon detection is the fact that in these Brassica–Arabidopsis alignments (CAGSs), no distinction is made between alignments between possible orthologs and paralogs. For alignments between a paralogous Brassica–Arabidopsis pair, it is likely that both exon and intron similarity are reduced while the exon–intron contrast is maintained. We should also note that, based on our experience with Arabidopsis genome annotation (Wortman et al. 2003), alignments between paralogous genes are often informative in defining gene structure.
Consistent with observation from many comparative genomics studies, the majority of the 229,735 CAGSs matched existing gene models, although the boundaries of conservation delineated by the BLASTZ alignments often do not coincide with experimentally determined gene boundaries. Approximately one-fourth (60,378) of the CAGSs fell between annotated genes and were classified as intergenic. The average coverage per nucleotide of 4.04 and 5.78 in the genic and intergenic CAGSs, respectively, likely reflects contributions both from the triplicated nature of the Brassica genome and the contribution to CAGSs of both orthologous and paralogous sequences that cannot be distinguished with the limited Brassica sequence available.
Expression of CAGS chains
Expression analysis showed that ∼30% (58 of 192) of CAGSs investigated were expressed. Cloning and sequencing of 5′ and 3′ RACE products from these regions allowed us to create 23 new gene models and to update the annotation of another 17 genes. Additional evidence for CAGS expression comes from our analysis of published data on whole genome tiling arrays (Yamada et al. 2003) and MPSS (Meyers et al. 2004), which revealed that ∼10% of all intergenic CAGSs may produce transcripts. The higher proportion of expressed CAGSs detected by our PCR-based approach (∼30%) likely reflects the greater sensitivity of this method. Taken together, these results suggest that between 10% and 30% of the ≥15,000 intergenic chains of CAGSs may correspond to novel and as yet to be annotated genes. Further analysis of the possible identity of CAGSs as genes will require targeted approaches such as high-throughput RACE/PCR, since current microarray technologies are limited in their ability to detect low level transcripts from such regions. Other clues as to the function of CAGSs may come from the study of T-DNA, transposon, or RNAi mutants that disrupt their function.
Use of CAGS in modeling gene structures
Because CAGSs do not precisely define exon boundaries, they cannot be used to automatically model or update gene structures. However, they do serve to flag regions where manual intervention can improve gene models as described above for 370 BACs examined. They also provide credibility for hypothetical gene predictions. Release 4.0 of the TIGR annotation contained 4253 hypothetical genes, of which 3433 overlapped one or more CAGSs.
The Brassica–Arabidopsis genome alignments generated in this study were used to train a novel gene prediction program, Twinscan (Korf et al. 2001). Of 2229 genes predicted by Twinscan in previously unannotated regions of the genome, 1674 coincide with our CAGSs. Based on experimental evidence, Twinscan is reported to perform somewhat better than other ab initio gene prediction programs on Arabidopsis (M. Brent, pers. comm.). Although, our limited investigations reported here support this observation, they also indicate that not all Twinscan predictions are correct and that expressed genes can be found in regions defined by CAGS chains that are devoid of a Twinscan prediction.
Summary
In this study, we have shown that WGS sequencing of a related species can be a valuable guide to identify genes that were not properly annotated and also to locate regions of the genome that have escaped the annotation process. Expression analysis of a sample of CAGS using PCR-based methods indicated that as many as 30% of the CAGS chains might be expressed, and showed that RACE can be effectively used to obtain full-length sequences and model gene structures. In future studies, we plan to use the global expression profiling data to target those regions most likely to harbor expressed genes for FL-cDNA cloning. In addition to its use to identify and improve the annotation of protein coding genes in Arabidopsis sequence conservation with Brassica, the conserved sequences can also be used to identify other genome features such as promoter elements (Colinas et al. 2002) and noncoding RNAs (Rivas and Eddy 2001), but the sequence information reported here has not yet been fully exploited for these purposes.
Methods
DNA sequencing
Several small (2–3 kb) and one large (10–15 kb) insert libraries were constructed in the high- or medium-copy vectors pHOS1 and pHOS2 by standard in-house methods (Fleischmann et al. 1995) using either total or “nuclear” DNA (generously provided by Dr. Pablo Rabinowicz) isolated from TO1000DH3, a doubled haploid derived from a rapid cycling Brassica oleracea kindly provided by Dr. Tom Osborn. End sequences were generated on ABI PRISM 3700 DNA sequencers with ABI “Big Dye” dye terminator cycle sequencing kits.
In silico identification of conserved segments
The filtered sequences were aligned against version 4.0 of the TIGR Arabidopsis Genome Annotation by using BLASTZ (Schwartz et al. 2003), the sequence alignment program underlying PIPMaker (Schwartz et al. 2000), at its default setting. To determine search parameters that would best discriminate between the alignments of the Brassica reads to Arabidopsis exons relative to introns, a sample of 1000 Arabidopsis genes that were supported by FL-cDNAs and had matches to Brassica sequences were selected, 200 from each of the five chromosomes, and concatenated. The Brassica WGS sequences were searched against this data set, and the resulting alignments were filtered by using a range of values
for minimum percentage identity and minimum match length. Sensitivity and specificity values for the identification of exons
from BLASTZ alignments were calculated at each nucleotide position from regions of the genomic sequence where exons and introns
intersected with the regions spanned by BLASTZ alignments. Sensitivity (probability that a base known to be in an exon is
found within a BLASTZ alignment) and specificity (probability that a base found within a BLASTZ alignment is actually in an
exon) were calculated as follows: 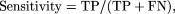
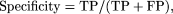 where TP is true positive (base known to be in an exon is found in BLASTZ alignment), FP is false positive (base known to
fall outside an exon is found within BLASTZ alignment), and FN is false negative (base known to be in an exon is found outside
BLASTZ alignment).
where TP is true positive (base known to be in an exon is found in BLASTZ alignment), FP is false positive (base known to
fall outside an exon is found within BLASTZ alignment), and FN is false negative (base known to be in an exon is found outside
BLASTZ alignment).
BLASTZ alignment segments were collapsed into CAGSs by using appropriate values for minimum match length and minimum percentage identify as described in Figure 5. Before further analysis, CAGSs were again searched against known features (annotated proteins, transposable elements, and repeat sequences, etc.) to eliminate any sequences that had passed through the earlier filtering steps. CAGSs that overlapped an existing gene annotation by 25% of their length were initially designated as genic and the remainder as intergenic. Intergenic CAGSs that were separated by less than a specified distance were chained together on the assumption that these CAGSs represent parts of the same feature. Subsequently, these intergenic CAGSs were broken down into “truly intergenic” and “perigenic” using similar criteria.
Laboratory validation of CAGS chains
A sample of 192 of the longer singleton and paired CAGS chains was selected for in vitro validation. Primers were designed within the CAGS, one in each CAGS in the case of paired CAGSs, avoiding regions close to the gap. PCR was performed by using a cDNA population derived from a variety of Arabidopsis tissues and treatments as template (Xiao et al. 2002). PCR products were treated with a mixture of alkaline phosphatase and exonuclease to remove residual primers and deoxynucleotide triphosphates and then sequenced from both ends using the same PCR primers. To obtain RACE products for the expressed regions, a second set of nested primers was designed and used for 5′ and 3′ RACE. Four to five independent clones from each of the 5′- and 3′-RACE products were sequenced from both ends by using generic sequencing primers, and the sequences from each locus tested were assembled by using TIGR assembler (Sutton et al. 1995). The assembled cDNA sequences for each gene were aligned against the corresponding CAGS sequences by using the AAT program (Huang et al. 1997). This was followed by manual inspection and submission of the assemblies to GenBank.
Acknowledgments
We thank Drs. Tom Osborn and Pablo Rabinowicz for providing the Brassica seed and DNA, respectively. We gratefully acknowledge the contributions and support of the TIGR sequencing, bioinformatics and information technology and Arabidopsis annotation groups. We also thank Dr. Michael Brent and Dr. Blake Meyers for discussions and access to data for Twinscan predictions and MPSS, respectively, as well as Dr. Steven Salzberg for advice on the specificity and sensitivity analysis. This project was supported by National Science Foundation (Cooperative Agreement DBI 9813586).
Footnotes
-
[Supplemental material is available online at www.genome.org. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: T. Osborn and P. Rabinowicz.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3176505.
-
↵1 Current address: Pioneer Hi-Bred International, Inc./A DuPont Company, Johnston, IA 50131-0552.
-
↵2 Corresponding author. E-mail cdtown{at}tigr.org; fax (301) 838-2808.
-
- Accepted January 5, 2005.
- Received August 21, 2004.
- Cold Spring Harbor Laboratory Press








