
Efficient Male and Female Germline Transmission of a Human Chromosomal Vector in Mice
Abstract
A small accessory chromosome that was mitotically stable in human fibroblasts was transferred into the hprt −hamster cell line CH and developed as a human chromosomal vector (HCV) by the introduction of a selectable marker and the 3′ end of anHPRT minigene preceded by a loxP sequence. This HCV is stably maintained in the hamster cell line. It consists mainly of alphoid sequences of human chromosome 20 and a fragment of human chromosome region 1p22, containing the tissue factor gene F3. The vector has an active centromere, and telomere sequences are lacking. By transfecting a plasmid containing the 5′ end ofHPRT and a Cre-encoding plasmid into the HCV+hamster cell line, the HPRT minigene was reconstituted by Cre-mediated recombination and expressed by the cells. The HCV was then transferred to male mouse R1-ES cells and it did segregate properly. Chimeras were generated containing the HCV as an independent chromosome in a proportion of the cells. Part of the male and female offspring of the chimeras did contain the HCV. The HCV+ F1 animals harbored the extra chromosome in >80% of the cells. The HCV was present as an independent chromosome with an active centromere and the human F3 gene was expressed from the HCV in a human-tissue-specific manner. Both male and female F1 mice did transmit the HCV to F2 offspring as an independent chromosome with properties similar to the original vector. This modified small accessory chromosome, thus, shows the properties of a useful chromosomal vector: It segregates stably as an independent chromosome, sequences can be inserted in a controlled way and are expressed from the vector, and the HCV is transmitted through the male and female germline in mice.
Stable transgenic eukaryotic cells (and mammalian animals) are currently essentially generated by the random integration of foreign DNA into the host genome. This introduction of foreign DNA mutates the host genome: The transgene can modify the properties of neighboring host genes, whereas the host genome itself can influence transgene expression (Robertson et al. 1995; Rivella and Sadelain 1998). In addition, often more than one copy of the transgene is introduced in the host genome (Hashido et al. 1995; Garrick et al. 1998) and the insertion of foreign DNA can lead to rearrangements and deletions (Choi et al. 1993; Strauss et al. 1993). Episomal vectors such as those containing the latent origin of replication oriP from the herpes Epstein–Barr-virus replicate once every cell cycle in cells expressing its transactivator EBNA-1 and allow the introduction and long-term expression of foreign DNA in human cell lines. However, these vectors are present at greater than one copy per cell and rely on the presence of the transacting viral protein EBNA-1 for replication and the origin of replication of oriP is not functioning in rodent cells (Sun et al. 1994; Wade-Martins et al. 1999).
The generation of an eukaryotic chromosomal vector would alleviate these drawbacks. Ideally, such a vector should (1) be mitotically stable without selection, (2) allow the integration of foreign DNA without size restriction at a well-defined locus, (3) allow the regulated expression of genes present on the vector, (4) be transferrable among different cell lines, and (5) show stable male and female germline transmission as an independent chromosome in transgenic animals. The construction of artificial mammalian chromosomes is one way to generate such a chromosomal vector.
Different strategies have been followed to generate mammalian artificial chromosomes (MACs). In the bottom-up approach, artificial chromosomes are generated de novo. In vivo self-assembled MACs were obtained after the introduction of human alphoid repeats in the human HT 1080 cell line, together with total human genomic DNA and telomeric repeats (Harrington et al. 1997). Two other groups generated de novo chromosomes by the introduction of yeast artificial chromosomes (YACs) carrying centromeric alphoid repeats capped with chimerical yeast–human telomeric repeats in human HT 1080 cells (Ikeno et al. 1998; Henning et al. 1999). MACs were also created by the lipofection of circular or linear P1 artificial chromosomes containing human alphoid repeats in the presence or absence of human telomeric arrays into the human HT 1080 cell line (Ebersole et al. 2000). If the linear constructs were used, human telomeric repeats had to be present for effective de novo chromosome formation. In all cases, the resulting de novo minichromosomes are estimated to be 2 Mb–10 Mb in size, which is likely to be the result of a multimerization of the input sequences.
In the top-down approach, nonessential chromosomes present in somatic cell hybrids are reduced in size either by telomere-associated chromosome fragmentation (TACF; Brown et al. 1994; Farr et al. 1995;Heller et al. 1996; Mills et al. 1999) or by irradiation microcell-mediated chromosome transfer (MMCT; Au et al. 1999; Handel et al. 1999; Hernandez et al. 1999). Minichromosomes (all containing α satellite repeats) of less than 2.5 Mb, thus, have been created. Finally, several authors explored the possibility of using naturally occurring minichromosomes (Raimondi et al. 1996; Guiducci et al. 1999).
Recently, we detected and characterized five mitotically stable small accessory chromosomes (SACs) in human fibroblasts derived from a patient (Vermeesch et al. 1999). Here, we describe the isolation of one of these SACs in a hamster cell line, its modification by integration of a selectable marker gene, and a loxP sequence for site-specific insertion of foreign sequences using the Cre recombinase and its characterization as an HCV in cell lines and in mice.
RESULTS
Isolation, Modification, and Characterization of a Human SAC
We previously characterized five mitotically stable SACs carrying a functional centromere, as indicated by the presence of CENP-C proteins and without telomere sequences indicating that they were circular chromosomes (Vermeesch et al. 1999). Permanent cell lines containing the SACs were obtained by fusing these fibroblasts to hamster CH cells. Forty-seven hybrid cell lines were screened for the presence of SACs by FISH using alphoid probes. To confer some properties of a vector to the SACs, the plasmid pBS-neo/loxP/3′HPRTwas introduced into an SAC (Fig. 1A). This plasmid contains the neomycin resistance gene under control of a thymidine kinase promoter, followed by a loxP sequence and the 3′ end of a human HPRT minigene. The neomycin resistance gene allows the positive selection of somatic cell hybrids containing the SAC, whereas the loxP/3′HPRTsequence provides an in vivo cloning site. To select for SACs with an integrated pBS-neo/loxP/3′HPRT, microcells were generated from the primary transfectants and were then size selected (Zhang et al. 1989). The size fraction with the smallest microcells was subsequently fused with hprt− CH cells. The resulting 88 G418-resistant hamster/human somatic cell hybrids were screened for the presence of an SAC by PCR and FISH (not shown). A hybrid cell line, E10B1, containing one human SAC was selected for further analysis.
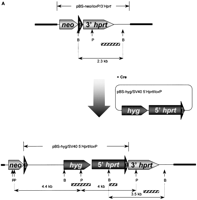
Modification and characterization of the small accessory chromosome (SAC). (A) Structure of the different vectors and strategy for introduction of new sequences into the SAC by Cre-mediated recombination. SAC sequences are indicated with a thick line, vector sequences with a thin line, and loxP sequences with an arrowhead. Neo, neomycine resistance gene driven by a thymidine kinase promoter; hyg , hygromycin resistance cassette driven by the PGK promoter; 5′- and 3′HPRT, human HPRT minigene driven by the SV40 early promoter; P,PstI cleavage site; B, BamHI cleavage site. Fragments used as a probe for Southern blot hybridizations are indicated with a shaded bar (not drawn to scale). (B) FISH with lissamine-labeled human CotI DNA (red signal) and biotine-labeled pBS-Neo/loxP/3′HPRT plasmid (green signal) on a methaphase of hybrid E10B1. The metaphase was counterstained with DAPI. (BI) Pseudocolored image, the circle shows the SAC; (BII) G-like banding derived from the DAPI channel. (C) In the last lane, inter-alu PCR products using E10B1 genomic DNA as a template are shown. Hamster and human genomic DNA were used as a negative and positive control, respectively. (D) FISH with biotin-labeled inter-alu PCR products on a metaphase of the human HT1080 cell line in which the SAC was introduced by MMCT. The circle indicates the SAC; arrowheads show the signals present on chromosome region 1p. (E) FISH with a FITC-(C3TA2)3 peptide nucleic acid probe (green signals) and a lissamine-labeled α-satellite 2 probe (red signals) on metaphase spreads of the SAC+ HT 1080 cell line did not show a green signal on the SAC (circle). (F) Magnification of the SAC shown in E without the red signal of the α-satellite 2 probe.
We hybridized metaphases of E10B1 with hamster CotI DNA. No hybridization signals were present on the SAC; however, all hamster chromosomes were brightly stained (not shown). Lissamine-labeled human CotI and biotine-labeled pBS-neo/loxP/3′HPRT cohybridized exclusively to the SAC in metaphase spreads of E10B1 (Fig.1B). The SAC, thus, is the only human chromosome present in this somatic cell hybrid and it is the only chromosome with an integrated pBS-neo/loxP/3′HPRT. Metaphases of the hybrid cell line were then hybridized with α satellite probes for chromosomes 2, 6, 12, and 14/22, respectively ,which were previously shown to hybridize to the SACs in the original fibroblasts. A signal was obtained only with the α-2 satellite probe (not shown). Because the α-2 satellite probe also hybridizes with the α-satellite repeats of human chromosomes 18 and 20, a Southern blot was performed to identify the origin of the alphoid array present in the SAC. E10B1 genomic DNA was digested with XbaI, size-separated with agarose gelelectrophoresis, blotted, and subsequently probed with a radio-labeled α-2 satellite probe (see below). The resulting band pattern corresponded with the higher order repeats of α-satellite DNA of human chromosome 20 (Rocchi et al. 1990; Baldini et al. 1992).
The isolation of the SAC in a hamster cell line allowed us to investigate the human sequences present on the SAC in more detail. Inter-alu PCR detected a discrete number of human sequences on the SAC (Fig. 1C). FISH with the inter-alu products of E10B1 on metaphase spreads of an SAC+ human cell line HT1080 (T. Voet and P. Marynen, unpubl.) showed signals on the SAC and unexpectedly on chromosome region 1p (Fig. 1D). Sequencing of a number of the inter-alu PCR products allowed us to generate primers for three STSs. YAC containing one or more of the three generated STSs were identified by screening the megaYAC library. All YACs contained fragments of human chromosome 1p22. Twelve STSs mapping to human chromosome 1p22 were then tested on E10B1 genomic DNA by PCR. A region of 1cM–2 cM spanning from STS WI-9122 to D1S2719 was found to be present in the SAC. In this region, at least three genes are present: down-regulator of transcription 1 (DR1), glutamate-cysteine ligase regulatory subunit (GLCLR), and human tissue factor (F3). FISH with an FITC-(C3TA2)3 peptide nucleic acid probe on metaphase spreads of the SAC+ HT 1080 cell line could not detect a signal on the SAC, suggesting that it was still circular (see Fig. 1E,F).
The mitotic stability of the minichromosome in the hamster cell line was measured after 109 population doublings in the presence or absence of G418 (Table 1). FISH was performed on metaphase spreads to detect an eventual integration of SAC sequences into the hamster chromosomes. After 109 population doublings, 95% of the cells grown in G418 medium contained one SAC, 4% contained two SACs, and in the remaining 1% no signal was seen. The latter might be cells that have lost the SAC or represent a failure of the FISH (e.g., due to loss of the SAC from the metaphase on the slide). In the absence of selection, 78% of the cells contained one independent SAC and 8% of the cells carried two SACs after 109 population doublings. Integration of the SAC into a hamster chromosome was never observed. This indicated that the loss rate of the SAC amounted to 0.23% per mitosis in the absence of any selective pressure. Immunofluorescence using an anti-CENP-C antibody resulted in two bright spots on the SAC, showing that its stability is due to the presence of an active centromere (not shown).
Mitotic Stability of the HCV
Recombination-Mediated Introduction of New Sequences in the SAC and Expression
To investigate the site-directed introduction of new sequences into the SAC, we constructed a plasmid pBS-Hyg/SV405′HPRT/loxP containing a hygromycin resistance expression cassette followed by the 5′ end of the humanhprt minigene controlled by the SV40 early promoter and aloxP sequence (Fig. 1A). The hprt − E10B1 hybrid was cotransfected with pOG231, a Cre-expression plasmid, and pBS-Hyg/SV40 5′HPRT/loxP. Homologous recombination at the loxP sequence would then reconstitute the human HPRT minigene (Fig. 1A) and these clones, thus, can be selected in HAT medium. As negative controls, E10B1 was mock-transfected or transfected with pBS-Hyg/SV405′HPRT/loxP or pOG231 only. No cells survived HAT selection in any of the negative controls. Over 200 resistant clones grew out of the cells cotransfected with pOG231 and pBS-Hyg/SV40 5′HPRT/loxP. This accounts for a recombination efficiency of 1.6 × 10−5/cell.
The correct reconstitution of the human HPRT minigene was demonstrated by PCR analysis on genomic DNA isolated from 10 clones, with HPRT primers spanning the loxP site. The predicted 2,1-kb PCR product was obtained with genomic DNA from all clones but not with control genomic DNA derived from untransfected E10B1 cells (result not shown). In addition, the DNA from the clones was digested with either PstI or BamHI, size fractionated by agarose gel electrophoresis, blotted, and then probed with a fragment of either the hygromycine resistance gene, the 5′HPRT gene or the 3′ HPRT gene. Upon hybridization of the PstI-digested DNA with the hygromycine probe, two signals of 4 kb and 4.4 kb, respectively, are expected (Fig. 1A). In five out of nine clones (Fig. 2, upper panel, lanes 1, 2, 6, 7, and10) this was indeed the case, whereas in the other clones some rearrangements and/or amplifications did occur. In two clones a signal was visible at 7.2 kb. When the same Southern blot was hybridized with the 5′ end of the HPRT gene, a signal at the same position was obtained (Fig. 2, middle panel). This suggests that the 7.2-kb band results from the integration of multiple copies of pBS-Hyg/SV405′HPRT/loxP at the loxP site. Upon hybridization the BamH1 digested DNA with the 3′HPRT probe, a hybridization signal of 2.3 kb is expected before integration and a 3.5-kb hybridization signal upon correct integration. This 3.5-kb fragment is present in all clones. However, in four clones (Fig. 2, lower panel, lanes 4, 6,8, and 9), the original 2.3-kb fragment was also present. We suggest that this might be due to a duplication of the circular SAC as a result of Cre-mediated sister chromatid exchange. Indeed a Cre-mediated homologous recombination occurring between the two loxP sites present on each sister chromatid of the circular SAC after DNA-replication could result in a duplication of the SAC. Subsequently, if in one of the two sites an integration of plasmid pBS-Hyg/SV40 5′HPRT/loxP took place, both the original 2.3 kb and the rearranged 3.5 kb would be present. Taken together we conclude that four clones out of 10 (clones 1, 2, 7, and 10) had a correctly integrated plasmid pBS-Hyg/SV40 5′HPRT/loxP without obvious rearrangements.
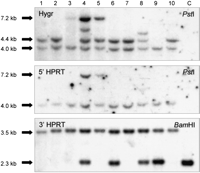
Southern analysis of the Cre-mediated introduction of a plasmid into the HCV. The pBS-Hyg/SV405′HPRT/loxP plasmid was integrated into the HCV by cotransfection with the Cre-encoding plasmid pOG231. Correct integration reconstitutes an HPRT minigene. Southern blots with DNA isolated from 10 independent HAT-resistant clones digested with either PstI or BamHI (lane1–10) or untransfected E10B1 cells (lane C) were hybridized with probes for the hygromycin gene (upper panel), the 5′ end of the HPRT minigene (middle panel) and the 3′ end of the HPRT minigene (lower panel), respectively. A scheme of the probes and of the expected signals is shown in Fig. 1A.
To confirm that the HAT resistance of the clones was the result of the correct reconstitution and expression of the human HPRTminigene, RT-PCR was performed on RNA isolated from three clones. The amplified cDNA was of the correct size, and subsequent sequencing of the RT-PCR product confirmed expression of the human HPRTminigene. The same amount of clones grew out when the cells of four tested clones were grown in medium containing hygromycin or HAT, suggesting that all HPRT+ cells also express the hygromycin resistance gene in pBS-Hyg/SV405′HPRT/loxP from the SAC. The SAC, thus, shows a number of salient features of a chromosomal vector and will be called an HCV from now on.
Transfer of the HCV to Mouse ES Cells and Generation of Chimeras
Using MMCT, the HCV was transferred into a male mouse ES cell line (R1). Nine G418-resistant hybrids containing the HCV were obtained; five of them were expanded and characterized. The five hybrids were maintained with and without G418 selection for 40 population doublings and the presence of the HCV was investigated by FISH with labeled human Cot1 DNA (Table 1). Chromosome loss rates of the different ES clones in the absence of selection were low and varied between 2.66% and 0.26% per mitosis. FISH analysis using human Cot1 DNA as a probe confirmed the presence of the HCV as an independent chromosome in the ES cells (not shown). No FISH signal was visible on the HCV with either a mouse or hamster Cot1 probe, indicating that little or no mouse or hamster DNA was integrated into the HCV (data not shown). This experiment also showed that no hamster chromosomes were cotransferred to the ES cells. To determine whether an active centromere was present on the HCV, immunofluorescence was performed with anti-CENP-C and FISH with human Cot1 DNA on metaphase spreads of the ES cell hybrids. A CENP-C signal was visible on all mouse centromeres, as well as on the HCV, although fainter (not shown). Hence, the stable segregation of the HCV is due to the presence of an active centromere.
We injected male ES clone G or I cells, characterized with a HCV loss rate of 0.26% and 0.82%, respectively, into C57BL/6 blastocysts to create chimerical mice carrying the HCV. Two male chimeras (derived from clone G) and one female chimera (derived from clone I) were obtained from three independent experiments. PCR experiments on tail DNA of the chimeras using primers spanning the loxP site and primers derived for human 1p22 sequences present in the HCV were positive for the male chimeras and negative for the female chimera. FISH analysis performed on cultured tail fibroblasts from the male chimeras with a human Cot1 probe showed that the HCV was present as an extra human minichromosome in 16% and 22% of the cells, respectively.
Germline Transmission of HCV in Mice
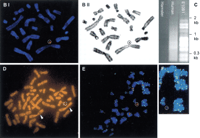
The two male chimeras were mated with female C57BL/6 and dominant-agouti offspring was obtained from both (Table2). PCR detected the HCV in 22% and 44% of the agouti offspring, respectively. FISH analysis on primary tail fibroblasts of five of the F1 transchromosomal mice indicated that the HCV was still present as an independent human chromosome among the 40 normal mouse chromosomes. No signal could be detected on the HCV by FISH using mouse Cot1, mouse major, and minor satellite DNA as a probe, indicating that little or no mouse DNA was integrated into the HCV (Fig. 3 A–D). In each of the five transchromosomal mice tested, ∼85% of the nuclei from the tail fibroblasts contained a single HCV, the remaining nuclei showing no signal (not shown). No nuclei were observed with two or more signals. Immunofluorescence staining with anti-CENP-C, followed by FISH with a centromere 2 alphoid probe, detected a CENP-C signal on both kinetochores of the HCV (Fig. 3 E,F). FISH with a peptide nucleic acid telomere probe showed that the HCV contained no telomeric sequences (Fig. 3 G,H). Taken together this indicates that germline transmission did not change major functional properties of the HCV.
Germline Transmission of the HCV by the Two Male Chimeras
Characterization of the HCV in F1 transchromosomal mice. Metaphase spreads of tail fibroblasts of HCV+ F1 mice were used for FISH. A cohybridization was performed with a biotinylated mouse Cot1 DNA probe (detected in green) and a lissamin-labeled human CotI DNA probe (red). (A) The green channel detecting the mouse sequences; (B) The red (human Cot1) and blue (DAPI counterstain) channels. (C,D) The detection of major and minor mouse satellite sequences (biotinylated, detected in green) and of the HCV (lissamin-labeled human Cot1 DNA, red), respectively. The left panels show the green channel; on the right the composite images are shown. Next, a codetection was performed of the HCV using a biotine-labeled alphoid 2 probe (green) and of CENP-C proteins by immunostaining (red signal). (E) The composite image (DAPI counterstain). The red CENP-C signal on the HCV is hidden by the strong green alphoid 2 signal. (F) The red channel only, of the image shown in E. Finally, a slide was sequentially hybridized with a FITC-labeled peptide nucleic acid telomere probe (G, green signals) and a lissamin-labeled human alphoid 2 probe (H, red signal). The poor quality of the metaphase inH results from the combination of the different protocols used for FISH with a peptide nucleic acid probe and a DNA probe. A circle shows the position of the HCV.
To investigate the tissue distribution of the HCV in the F1 mice, an HCV+ mouse was sacrificed and DNA was isolated from different tissues. A Southern blot with XbaI-digested DNA was then hybridized with a human alphoid 2 probe (Fig.4). DNA of the E10B1 HCV+ cell line was included as a control. Identical signals from the higher order repeats of the alphoid DNA of the HCV were obtained for all tissues tested, the hamster hybrid showing that the HCV was present in all mouse tissues with a similar copy number. Interphase FISH using a human alphoid 2 probe on liver, lung, and white blood cells of two HCV+ F1 mice was in agreement with the Southern blot results. Inter-alu PCR with tail DNA of five HCV+F1 mice generated a discrete number of products indistinguishable from those obtained with DNA of E10B1 (not shown). Taken together with the Southern blot data, this shows that male germline transmission of the HCV did not result in gross rearrangements of the HCV.
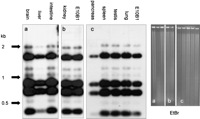
Tissue distribution of the HCV. Southern blot analysis of the HCV. DNA prepared from different tissues of an HCV+ F1 mouse was digested with XbaI, size-separated, and blotted. The left panel shows hybridization with a human alphoid-2 probe. The signal obtained for the different tissues is identical to the signal obtained for the E10B1 clone. The right panel shows the ethidium bromide–stained agarose gels.
Next male and female F1 HCV+ mice were mated with C57BL/6 mice. F2 HCV+ mice were identified by PCR with 1p22 primers on genomic tail DNA (Table 3). Both male and female transchromosomal mice showed efficient germline transmission of the HCV. Thirty-one percent and 35% of the newborn mice, respectively, were found to contain the HCV. FISH analysis with a centromere 2 alphoid probe and a mouse Cot1 probe confirmed these results (not shown).
Germline Transmission of the HCV by HCV+F1 Mice
Expression of HCV Genes in Transchromosomal Mice
Tail fibroblasts of the F1 HCV+ mice did proliferate in medium containing 800 μg/mL G418, whereas fibroblasts of HCV− F1 agouti offspring died rapidly in this medium, showing expression of the neomycin resistance gene from the HCV. When an equal amount of tail fibroblasts of two transchromosomal mice was seeded in medium with or without G418, 91% (100 G418-resistant colonies against 110 in the control) and 83% (96/115) of the cells, respectively, were G418 resistant. This is consistent with the number of HCV+ cells as detected by FISH, suggesting that all HCV+ cells do express the neomycin gene.
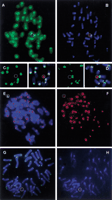
The presence of the human TF gene on the HCV provided an opportunity to evaluate expression of a human gene driven by its own promoter in the HCV+ mice. cDNA was synthesised from different tissues of a male and a female HCV+ F1 and F2 mouse and used as a template for PCR reactions, with primer sets selectively amplifying either the human or the mouse F3 gene. The identity of the PCR products was confirmed by sequencing. Figure5A shows that the expression of humanF3 mRNA is variable in different mouse tissues but that the expression levels are very similar in different transchromosomal animals of two generations. The highest expression was observed in the brain, kidney, and intestine; low expression was seen in muscle, whereas very little human F3 mRNA could be detected in liver. A Western blot stained with rabbit antihuman F3 detects similar amounts of F3 in kidney samples of four transchromosomal mice with an Mr identical to the one observed for human kidney (Fig. 5B).Taken together, these results also suggest that no variegation occurs. When the expression of F3 in kidney was analyzed by immunostaining of tissue sections, the epithelia of the glomeruli, and some tubuli of HCV+ animals were clearly positive, whereas in HCV− kidneys the glomeruli were negative (Fig. 5C). As shown by Luther et al. (1996), this is the typical human expression pattern of F3 in the kidney.

Expression of human F3 in HCV+F1 and HCV+F2 mouse tissues. RNA was isolated from different tissues of a male (I) and a female (II) HCV+ F1 or HCV+ F2 mouse and brain of a normal control mouse. RT-PCR assays were developed detecting specifically human or mouse F3 mRNA. Equal amounts of cDNA were used for 30 cycles of PCR with the human F3primers (hTF panels) or with the mouse F3 primers (mTF panels). A human fetal brain control is shown in laneC1 , lane C2 shows a normal mouse brain control. (A) RT-PCR experiments with cDNA derived from tissues of transchromosomal F1 and F2 mice. B, brain; k, kidney; l, liver; I, intestine; m, muscle; n, negative control. (B) Western blot with 25 μg of total kidney proteins extracted from human kidney (hu), kidneys of four transchromosomal mice (lanes F1 I, F1 II, F2 I, and F2 II, respectively) and a normal mouse (m), stained with rabbit antihuman F3. (C) Immunostaining with rabbit anti human F3 of kidney from an HCV+ F1 mouse and an HCV− littermate as a control shows positivity of the epithelia of the glomerulus, a typical human expression pattern only in the HCV+ kidney. Thelower panel shows a human control kidney.
DISCUSSION
Mitotic Stability of the HCV
The mitotic stability of an HCV in different genetic backgrounds is an important aspect determining its experimental usefulness. Our choice of the original fibroblasts from which the HCV was isolated, in part, was driven by the observation that the different SACs in the cells carried a functional centromere and were mitotically stable. After transfer into hamster cells and introduction of the loxP site and a selectable marker, the HCV maintained its mitotic stability, showing a loss of <0.23% per mitosis in the absence of selection. This can be explained by the presence of an active centromere. Several studies using linear human minichromosomes already showed that these segregated properly in human or hamster cells in the absence of selection (Farr et al. 1995; Raimondi et al. 1996; Harrington et al. 1997; Ikeno et al. 1998). There is some evidence, however, that the copy number of smaller minichromosomes (2.4 Mb) is more variable in human and hamster cell lines (Mills et al. 1999).
Interestingly, the HCV showed also stable segregation in mouse male R1 embryonic stem cells, showing <1% loss per mitosis in four out of five ES clones tested. Shen et al. (1997) introduced human minichromosomes derived by TACF of the human Y chromosome into the CGR8 ES cell line. These minichromosomes were rapidly lost from the ES cells in the absence of selection, suggesting that human centromeres function poorly in ES cells. One of the minichromosomes did segregate in a stable way in the ES cells and it was shown that this particular minichromosome acquired mouse centromere sequences. These findings were extended by showing that one of the Y-derived minichromosomes was mitotically stable in mouse L cells but not in ES cells (Loupart et al. 1998), indicating that the instability of the minichromosome is defined by particular properties of the ES cell line and not by species differences.
These data suggest that the stability in ES cells of the HCV described in present work could be due to the acquisition of mouse centromeric sequences. This could then explain the different loss rates observed for the HCV in the different ES clones (2.6%– 0.26%). Although we cannot exclude this, we suggest that this is unlikely and that these differences–which are small, as the highest loss rate observed is similar to that of the stable human–mouse chimerical minichromosome reported by Shen et al. (1997)–are of epigenetic origin. Indeed, no FISH signals were seen with labeled mouse Cot1 DNA on the HCV in any of the five cell lines tested, although this does not exclude the insertion of small amounts of mouse satellite DNA or single-copy sequences. However, if the stability of the HCV would be dependent on the insertion of mouse DNA, this should have happened independently in each of the ES clones generated, whereas in the experiments of Shen et al. (1997), the capture of mouse DNA sequences was the exception, even in the presence of selective pressure.
Another group (Tomizuka et al. 1997; Tomizuka et al. 2000) showed mitotic stability of a human chromosome–14 fragment in female mouse TT2F ES cells, showing a loss rate of <0.1%. However, this data was based on only one clone. By FISH it was shown that this human chromosomal fragment 14 did not incorporate mouse centromeric sequences. Another fragment of human chromosome 2 was found to be much less stable (3.2% loss/doubling) in these ES cells. Hernandez et al. (1999) reported the introduction of human Y–derived minichromosome into ES cells, but it is difficult to compare the mitotic stability to this of the HCV because the number of population doublings analyzed was much lower.
The mitotic stability of the HCV is not limited to mouse ES cells as F1 HCV+mouse liver, lung, and white blood cells were shown to carry the HCV in >85% of the cells by interphase FISH. Furthermore, analysis of tail fibroblast metaphases showed that the HCV was present as an independent chromosome. These data are corroborated by the Southern blot data that show the presence of equal amounts of HCV-derived human alphoid sequences in all tissues tested. When approximately 62 population doublings are needed to produce mature sperm in mice (Drost and Lee 1995) and the chromosome loss rate of the HCV is 0.26% in the used ES cells, then 85% of the cells should carry the extra human chromosome. This is in perfect agreement with the number of HCV+ cells absorbed by Southern blot and FISH. Additionally, it suggests that human genes located on the HCV do not confer a selective disadvantage to the cells retaining the HCV in mice. The HCV appears also to be structurally stable as it did not acquire sufficient mouse sequences to be visualized by FISH and the inter-alu PCR pattern obtained with DNA from the F1 HCV+ generation was identical to the one obtained from the E10B1 hybrid. Taken together with the Southern blot experiment discussed above, these data argue against gross rearrangements of the HCV.
Meiotic Stability of the HCV and Germline Transmission
The high stability of the HCV in mouse male R1-ES cells suggested the possibility to use this chromosome to generate transchromosomal mice. Two normal male chimeric HCV+ mice were obtained and mated with female C57Bl/6 mice to test the germline transmission of the extra human minichromosome. The HCV was efficiently transmitted by both chimeras (to 22% and 44% of the agouti pups, respectively). In addition both male and female F1 HCV+ mice efficiently transmitted the HCV to 31% and 35% of their offspring, respectively. As discussed above, the HCV in the mice seems very similar to the original HCV, as was characterized in the hamster hybrid cell line E10B1. No particular phenotype was associated with the presence of the HCV in any of the HCV+ mice born.
Hernandez et al. (1999) reported that 41 male chimeras were generated using ES cells carrying a human chromosome 21–derived minichromosome, but none of them could transmit the additional human chromosome through the germline. Shen et al. (2000) reported the germline transmission of a human/mouse minichromosome only by the female chimeras and detected only sporadic, unpredictable transmission by male animals. Of the nine generated male chimeras, none transmitted the human minichromosome.Tomizuka et al. (1997) generated chimeras carrying minichromosomes derived from human chromosomes 2, 14, and 22, respectively. The chimeras of either sex containing the chromosome 14–- and 22–derived minichromosomes never yielded dominant-agouti offspring. Male germline transmission was only reported for one out of four chimeras carrying a chromosome 2–derived artificial chromosome (2 out of 17 dominant-agouti pups). In contrast, all four female chimeras carrying the same minichromosome showed efficient germline transmission. Recently, efficient germline transmission of a different human chromosome–14 fragment by male and female F1 and F2 mice was shown (Tomizuka et al. 2000). However, the efficiency of germline transmission by F1 and F2 male mice containing the human chromosome–2 fragment was much lower when compared with the efficiency of germline transmission of the same minichromosome by female mice. Co et al. (2000) created transchromosomal mice with a satellite-DNA-based artificial chromosome, but male germline transmission was not reported.
From these and other data, it can be concluded that germline transmission of an extra (mini)chromosome by male mice might be very inefficient. Earlier reports argued that the presence of one or more unpaired chromosome(s) disrupts the meiotic process in male mice by complete meiotic breakdown during the first meiotic division (Burgoyne and Mahadevaiah 1993). In female meiosis an unpaired X was shown to segregate as an intact chromosome only in part of the first meiotic divisions. Also the unpaired X affected alignment and segregation of other chromosomes in the complement. Nevertheless, the majority of oocytes from XO females were able to complete the first meiotic division (Hunt et al. 1995). This could explain the sex difference observed for the germline transmission of human minichromosomes in the literature.
In contrast to these results, the HCV was efficiently transmitted through both the male and the female germline. This suggests that the HCV is not recognized as an unpaired chromosome during gametogenesis in the mouse. It is unlikely that this would be the result of the small size of the HCV. We have not been able to determine, in an unambiguous way, the size of the HCV, as it does not migrate into PFGE gels, nor could it be detected on Southern blots of PFGE experiments performed after irradiation of the plugs. The intensity of the DAPI staining, however, indicates that the HCV has about 20% of the size of the smallest human chromosome and it, thus, can be estimated at 5 Mb–10 Mb. This is well within the range of the other minichromosomes that have been generated. The major structural difference between the HCV and the artificial chromosomes reported by others (Tomizuka et al. 1997; Ikeno et al. 1998; Hernandez et al. 1999; Shen et al. 2000) is the absence of detectable telomere repeats, suggesting that the HCV is a circular chromosome. Telomeres have been shown to be involved in the spatial organization of chromosomes during meiosis, mediating the clustering at the centrosome that is located at the nuclear envelope in a particular phase of the prophase of meiosis I (the bouquet phase; De Lange 1998). It is thought that this arrangement would facilitate the pairing of homologous chromosomes. In fission yeast, in which similar clustering (to the spindle pole body) is defective as a result of mutations of the telomere proteins taz1 or lot2, recombination between homologous chromosomes is affected and there is a marked inhibition of the meiotic process (Cooper et al. 1998; Nimmo et al. 1998). A chromosome without telomeres would escape from the nuclear reorganization induced by the telomere relocation and one could hypothesize that as a consequence of its aberrant nuclear localization, it could escape detection as an asynapted chromosome at that stage. Here, it should be noted that the human chromosome–14 fragment (Tomizuka et al. 2000) showing efficient germline transmission was not analyzed for the presence of human or mouse telomeric repeats.
In mitosis a stringent checkpoint mechanism operates at the metaphase/anaphase transition to ensure that a bipolar spindle is formed and that all the chromosomes are aligned at the spindle equator before anaphase is initiated. The signal for delay is thought to come from the kinetochores, which are not under tension (Li and Nicklas 1995). The HCV carries an active centromere, as indicated by its mitotic stability and the detection of CENP-C proteins on the chromosome. LeMaire-Adkins et al. (1997) showed that in the female mouse meiosis the metaphase/anaphase checkpoint was absent. The available evidence, however, indicates that in the male meiosis, this checkpoint is stringent, raising the question how the HCV escapes from this control in the male meiosis. In XO mouse oocytes, equational division of the unpaired X is a frequent event, suggesting that individual chromosomes are able to form two fully functional kinetochores at meiosis I (Hunt et al. 1995). The questions whether this is also the case in the male meiosis and how the presence of a single chromatid would affect meiosis II in the male mouse remain unanswered at present. The HCV+ male and female transmitting mice provide a unique instrument to investigate these issues.
It should be noted that a higher incidence of small litters are generated by HCV+ F1 males than by HCV+ F1 females. Whether this is significant or strain dependent has to be analyzed. Comparison of litter sizes generated by transchromosomal mice reported by other groups is not possible because either male germline transmission could not be obtained, could be obtained only sporadically (Tomizuka et al. 1997; Hernandez et al. 1999; Co et al. 2000; Shen et al. 2000), or independent litter sizes are not mentioned (Tomizuka et al. 2000).
Gene Expression from the HCV
The generation of HPRT+ and HYG+ E10B1 cells by the simultaneous reconstitution of a human HPRT minigene and site-specific insertion of a hygromycin marker gene in the HCV shows that expression of genes present on the HCV occurs in this hamster cell line.
The proportion of G418 fibroblasts derived from HCV+ F1 mice is similar to the proportion of HCV+ fibroblasts detected by FISH. This suggests that no extensive, strong-position-effect variegation occurs. In this context, it is interesting to note that although CENP-C is present on the HCV, a comparison of DAPI-stained images of the HCV with the centromeres of the mouse chromosomes shows that heterochromatin formation is very limited on the HCV (not shown).
We showed that a fragment of human chromosome 1p22 containing the humanF3 gene is present on the HCV. An RT-PCR analysis shows that there is variable expression of the human F3 gene in the different tissues tested. Parry et al. (1998) analyzed the expression of a human F3 minigene containing a 2100-bp F3promoter fragment, the F3 cDNA sequence and intron 1 of theF3 gene. The transgenic mice expressed low levels (∼1%) of human F3 with a murine expression pattern and the investigators suggested that this was due to the lack of yet-unidentified transcription factors in the mice (Parry et al. 1998). Our RT-PCR results suggest that higher expression levels can be obtained by the introduction of the complete human F3 gene on an HCV. Interestingly, the epithelia of the glomeruli of the HCV+mice are clearly positive for F3, which is the expression pattern observed in humans (Flossel et al. 1994). This shows that the murine expression pattern observed from the human minigene results from the absence of human regulatory sequences, rather than from the absence of the appropriate transcription factors (unless encoded by a syntenic gene). These expression data illustrate the potential of the HCV in the analysis of gene regulation, the rescue of knockouts of regulated genes, the generation of mouse models for human disease, and the production of relevant proteins in transchromosomal animals. Finally, the HCV has potential in human ex vivo gene therapy because human embryonic stem cell lines are becoming available (Reubinoff et al. 2000).
In conclusion, we show here that a modified naturally occurring accessory chromosome has the properties of a useful chromosomal vector: New sequences can be inserted into the vector in a specific site and be expressed, the vector can be transferred between different cell lines, and the vector has a functional centromere and is mitotically stable in hamster and mouse cells. Additionally, it shows efficient transmission through both the female and the male germline in mice.
METHODS
The isolation of DNA and RNA from cells and tissues, Southern blot analysis, and DNA sequencing were performed according to standard procedures.
Plasmids
Plasmid pBS-neo/loxP/3′HPRT was obtained from Smith et al. (1995). Plasmid pBS-Hyg/SV40 5′Hprt/loxP was derived from plasmid pBS-Hyg/TKHprt −Δ3′/loxP (Smith et al. 1995). The latter was digested with EcoNI and blunted, followed by a BssHII digestion. In this way, the thymidine kinase promoter of the hprt gene was removed with 1.1 kb of intron 1 HPRT sequences. The SV40 promoter was derived from plasmid pUT SV1 (Drocourt et al. 1990) by a BamHI digestion, followed by blunting and a MluI digestion. The 402-bp SV40 early-promoter fragment was ligated into theEcoNI/BssHII site of pBS-Hyg/TKHprt −Δ3/loxP.
Cell Cultures
HT1080 cells and CH cells were grown in DMEM/F12 (Life Technologies) supplemented with 10% FCS, TES and HEPES, and high-glucose concentration. ES cells (R1, 129/SV × 129/SV-CP) were grown as described by Nagy et al. (1993). E10B1 cells were grown in DMEM /10% FCS with 15 μg/mL 8-azaguanine to select forhprt − cells.
Transfections
pOG231 (18.75 μg) and pBS-Hyg/SV40 5′Hprt/loxP (25 μg) were co-transfected into 12.5.106 E10B1 cells resuspended in 0.5 mL ice-cold DMEM /10% FCS by electroporation at 230 V and 960 μF. Twenty-four hours after transfection, selective HAT medium was added. Colonies were visible after ∼8 d and were then picked. Plasmid pOG231 was obtained from O'Gorman et al. (1997).
Microcell Mediated Chromosome Transfer
Colcemid was added to a final concentration of 0.5 μg/mL to 2 × 107 donor E10B1 cells growing in the log phase. After 48 h, the cells were trypsinized and pelleted by centrifugation at 800 rpm for 10 min. The pellet was resuspended in 2 mL serum-free medium at room temperature and added to a 40-mL 1:1 v/v Percoll/DMEM solution containing 20 μg/mL cytochalasin B. The mixture was centrifuged in a prewarmed centrifuge at 43,000g (19,000 rpm) for 70 min at 34°C–37°C. The microcells, visible as two bands in the Percoll gradient, were diluted in 50 mL serum-free medium, pelleted by centrifugation at 800 rpm for 10 min and washed with 5 mL phosphate buffered saline. To eliminate contaminating donor cells, the microcells were incubated in a tissue culture flask for 2 h at 37°C in 5 mL DMEM/10% FCS. Viable donor cells adhered to the bottom of the flask, whereas the microcells remained in the medium. Microcells were collected from the suspension by centrifugation at 800 rpm for 10 min.
The microcells, resuspended in 1 mL DMEM with 100 μg phytohema-agglutinin, were added to 107 log phase ES cells in suspension and allowed to adhere to the recipient cells by incubation for 10 min at 37°C. The medium was removed and 1 mL prewarmed (37°C) 50% w/v polyethylene glycol 1500 was added for 1 min. The cell suspension was washed three times with 5 mL DMEM and incubated in 5 mL DMEM/10% FCS with penicillin/streptomycin. After 24 h the cells were transferred to a 175 cm2 Falcon in selective medium (DMEM/10% FCS + 400 μg/mL G418). After ±8 d single colonies became visible.
Immunofluorescence
CH or ES cells were trypsinized and centrifuged and the cell pellet was resuspended in hypotonic solution (0.075 M KCl) at 1 × 105 cells/mL. Slides were prepared as described byJeppesen et al. (1992). Briefly, 0.5-mL portions of the cell suspensions were cytocentrifuged onto glass microscope slides using a Cytospin 2 (Shandon) at 2000 rpm with high acceleration for 10 min. The slides were immersed in KCM (120 mM KCl, 20 mM NaCl, 10 mM Tris-HCl, 0.1% [v/v] Triton X-100) for 10–20 min at room temperature. Immunofluorescence was performed without fixation as described by Page et al. (1995). Anti-CENP-C (serum from rabbit 554) was a gift from W.C. Earnshaw. Anti-CENP-C was diluted 1:500 in 1 × TEEN (1 mM triethanolamine-HCl at pH 8.5, 0.2 mM NaEDTA, 25 mM NaCl) with 0.1% Triton X-100 and 0.1% BSA. The slides were covered with a parafilm coverslip and incubated for 30 min at 37 °C and then washed 3 times for 5 min at room temperature in 1 × KB (10 mM Tris-HCl at pH 7.7, 0.15 M NaCl, 0.1% BSA). The primary antibody was detected with Cy3-donkey-anti-rabbit (Jackson Laboratories) diluted 1:200 in KB. The slides were incubated for 30 min at 37°C, washed 1 time in 1 × KB, fixed in 10% formalin in KCM for 10 min at room temperature, rinsed for 10 min with distilled water, and fixed in 3:1 methanol/acetic acid for 15 min at room temperature and air dried. The slides were analyzed by fluorescence microscopy and subsequently used for FISH.
Fluorescent In Situ Hybridization
The centromere 2 alphoid DNA probe was the 1.4-kb insert of pBS4D (from M. Rocchi). Human, mouse, and hamster CotI DNA were obtained from Life Technologies. Probes were labeled with either biotine-16-dUTP, fluoresceine-12-dUTP (Life Technologies), or lissamine-5-dUTP (DuPont NEN) using the Nick Translation System (Life Technologies); denaturation of the metaphase slides and probes, hybridization, and subsequent detection of the fluorescent signals were performed as described by Kermouni et al. (1995). FISH with a FITC-(C3TA2)3 peptide nucleic acid probe to detect telomere sequences was performed as described by Vermeesch et al. (1998). Chromosomes were counterstained with DAPI and the slides were mounted in Vectashield mounting medium (Vector Laboratories). The signal was visualized by digital-imaging microscopy using a cooled charge-coupled device camera (Photometrics).
PCR
The PCR reactions to amplify the reconstituted human hprtminigene were performed under standard conditions with the 10 pmole of each primer, 5′-TGGCGTCGTGATTAGTGATG and 5′-CCTGACCAAGGAAAGCAAAG, using 300 ng DNA and 2.5 units TAQ DNA polymerase (Roche) in 50 μL PCR reactions.
Inter-ALU PCR was performed on genomic DNA of the E10B1 cell line with the primers 5′-GGATTACAGGYRT GAGCCA and 5-RCCAYTGCACTCCAGCCTG in 50 μL reactions with 300 ng DNA template.
Forward and reverse primers of the used STSs of human chromosome 1p22 are indicated respectively. D1S2849: 5′-AGCTGAGATCGTGCCA and 5′-TCCCTAACCCTCCAGACT; WI-5213: 5′-TGCAATATCCATTAATAGAACACAA and 5′- TAATTTGGTAGAACTTTCCTCTTTG; WI-1423: 5′-CAAACAGAGATATCATTTGTCAGGT and 5′-TCTATAC GGGCATAAGCAATCT; D1S2868: 5′-AGGTATAATCTG CAATAAAAAACTT and 5′-AAAGTAAAACAATATGAAGC CAC; WI-9122: 5′-CAACAAAAACAGTGAAAGAAATGC and 5′-GCACACAACAGATGAAACTTATAGC; D1S236: 5′-AAACCACCTACCAATGTCTGTC and 5′-GAAGCTGTCGT TATGGGGT; D1S2664: 5′-CAGCCCACAGAATAACACTG and 5′-TTCATGCTATGATTTTCCGC; D1S2719: 5′- CCCTTAAC CACTTCTGGATT and 5′-CATGCTTTTGTTGGCTTATTAC; WI-7967: 5′- GTGAAAAATTAACTTTTTGTGTGGC and 5′-CACCAAAAATAAGCAATTTCAGC; D1S497: 5′- AGCTCTTG GAGAGACTGGC and 5′-AGCTCTGGAAGATTGCTGATT; D1S2753: 5′- ACAGTACGCTCCATCACG and 5′-TGCACCCTATGACTCAACTC; D1S2808: 5′- AGTTCCTGC CCACAAAG and 5′-GCCATCTCCAGTAAAATGC; three generated STSs: 5′-CCACCTCCTCACAAAATGCT and 5′-GCCTCCTGGTACAATCTTGC, 5′-CAGCCTGGCAT GTAGAAGGT and 5′-TCCTACTCCACACACCTCCC, and 5′-TGAGCCACTCTATCTGGCCT and 5′-CAGGAGCCCATT GTTCTAGC.
For the selective amplification of human or mouse TF , the following primers were designed. HuTFf 5′-GTGTATGG GCCAGGAGAAAG and huTFr 5′-TGTTATGTGCAAAAGGT GCC amplified a 428-bp humanTF fragment, whereas mTFf 5′-CCGAGCAATGGAAGAGTTTC and mTFr 5′-AGATTTTC CAAAAGCCCAC amplified a 496-bp mouse TFcDNA fragment. The forward and reverse primer for each reaction were derived from different exons to avoid amplification of genomic DNA contaminating the RNA preparations. cDNA was synthesised from 1 μg of total RNA according to standard procedures in a volume of 20 μL. One μL aliquots of the synthesis were used for PCR. The annealing temperature was 57°C and 30 cycles of amplification were performed. When the human primers were used, a PCR product was obtained using cDNA synthesized from human fetal brain RNA (Stratagene) and not with brain cDNA prepared from a control mouse, whereas the reverse was true when the mouse TF primer set was used.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 These authors contributed equally to this work.
-
↵5 Present address: Center for Human Genetics, Flanders Interuniversity Institute for Biotechnology, University of Leuven, Campus Gasthuisberg, Herestraat 49, B-3000 Leuven, Belgium.
-
↵6 Corresponding author.
-
E-MAIL Peter.Marynen{at}Med.KULeuven.ac.be; FAX 32-16-347166.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.159901.
-
- Received August 10, 2000.
- Accepted November 3, 2000.
- Cold Spring Harbor Laboratory Press








