
Poised for Contagion: Evolutionary Origins of the Infectious Abilities of Invertebrate Retroviruses
Abstract
Phylogenetic analyses suggest that long-terminal repeat (LTR) bearing retrotransposable elements can acquire additional open-reading frames that can enable them to mediate infection. Whereas this process is best documented in the origin of the vertebrate retroviruses and their acquisition of an envelope (env) gene, similar independent events may have occurred in insects, nematodes, and plants. The origins of env-like genes are unclear, and are often masked by the antiquity of the original acquisitions and by their rapid rate of evolution. In this report, we present evidence that in three other possible transitions of LTR retrotransposons to retroviruses, an envelope-like gene was acquired from a viral source. First, the gypsy and related LTR retrotransposable elements (the insect errantiviruses) have acquired their envelope-like gene from a class of insect baculoviruses (double-stranded DNA viruses with no RNA stage). Second, the Cer retroviruses in the Caenorhabditis elegans genome acquired their envelope gene from a Phleboviral (single ambisense-stranded RNA viruses) source. Third, the Tas retroviral envelope (Ascaris lumricoides) may have been obtained fromHerpesviridae (double-stranded DNA viruses, no RNA stage). These represent the only cases in which the env gene of a retrovirus has been traced back to its original source. This has implications for the evolutionary history of retroviruses as well as for the potential ability of all LTR-retrotransposable elements to become infectious agents.
What is the origin of vertebrate retroviruses? Phylogenetic analyses of their reverse transcriptase sequences strongly suggest that retroviruses are derivatives of retrotransposons that bear long terminal repeats (LTRs) (Xiong and Eickbush 1990; Feng and Doolittle 1992). The principal difference between LTR-retrotransposons and retroviruses is the acquisition by the latter of a third open reading frame (ORF): the envelope (env) gene. The envgene typically encodes a transmembrane protein and a host receptor-binding protein, which together can mediate infection and transmission of the viruses (Coffin et al. 1997). Because envgenes represent antigenic sites that elicit a host immune response, segments of this gene are under strong selective pressure to diverge. Both the antiquity of the original acquisition and the rapid sequence divergence have made it difficult to ascertain the origins of theenv gene in retroviruses. Indeed, it is unclear whether vertebrate env genes represent a single acquisition event or multiple events.
Vertebrate retroviruses do not represent the only lineage with anenv gene. Other instances of env-like gene acquisitions have taken place in the evolutionary history of LTR-retrotransposons. LTR-bearing retrotransposable elements and their related viruses can be divided into six clades, with the vertebrate retroviruses representing one of these (Fig. 1A). Of the other five clades, only the DIRS1 clade, with just three known representatives, lacks a third ORF. The Ty1-copia clade has one instance of an env-like gene acquisition in the SIRE-1 element from the soybean, Glycine max (Laten et al. 1998). The BEL clade contains two possible examples of an env-like acquisition: in the Cer7 element from C. elegans and the Tas element from Ascaris lumbricoides (Bowen and McDonald 1999;Felder et al. 1994). Finally, the Ty3-gypsy clade contains at least three putative instances of env acquisition: the insect gypsy-like elements (Song et al. 1994; Desset et al. 1999), the plant Athila-like elements (Wright and Voytas 1998), and the Osvaldo element from Drosophila buzzatii (Pantazidis et al. 1999). In most of the above cases, structural features reminiscent of retroviral envelope genes (leader peptide, N-glycosylation sites, and transmembrane regions) can be readily identified. However, of these various examples of the addition of a third ORF, only in the case of the gypsy group, termed the insect errantiviruses (Boeke et al. 1999) have virus-like particles generated by the elements been shown to be infective (Song et al. 1994).
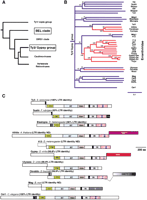
The Ty3/ gypsy family of LTR retrotransposons. (A) Schematic of the LTR-containing retrotransposable elements and related viruses, with the Ty3/ gypsy group highlighted. The LTR retrotransposons are divided into six groups (clades) based on a phylogeny of their RT domains (Xiong and Eickbush 1990). (B) A neighbor-joining phylogenetic analysis of representative sequences from the Ty3/ gypsy group. Lineages highlighted in red have been shown to contain a third ORF, putatively an env-like gene. Bootstrap values and divergence scales are indicated. Nodes with < 50% bootstrap support have been collapsed. (C) ORFs from representatives from the Ty3/ gypsy group are schematized to the scale indicated, with the various enzymatic and structural modules highlighted. Three instances of an env-like gene are represented. In some instances, the carboxyl-terminal extension to the core integrase domain contains a GPY/F domain (degenerate in Gypsy and Osvaldo). The Mag lineage bears a different carboxyl-terminal extension (X). The percent LTR identity is a good indicator of the age of a particular element insertion (LTRs are identical in sequence at the time of insertion).
Three possibilities can account for the origins of env-like ORFs within these retrotransposable element lineages. Many vertebrate retroviruses have incorporated host genes during their evolution: for example, src in the avian Rous sarcoma virus (Takeya et al. 1981). Thus, one possibility is that the env gene could have been acquired from a host genome, usurping the normal receptor-binding/ membrane fusion abilities of a host gene. A second possibility is that a serendipitous fusion of two protein domains leads to the de novo formation of env genes, which are now evolving under different selective constraints than either of the original proteins. A third possibility is that the element acquired its env-like gene from another infectious agent, utilizing the ready-made machinery of the latter for its own purposes. Plant caulimoviruses, which represent one clade within the LTR-retrotransposable element, may represent just such a fusion of an LTR-retrotransposable element with a plant virus. The cell-to-cell movement proteins of the caulimoviruses have been shown to be both functionally and phylogenetically related to those from a number of other plant viruses (Koonin et al. 1991). We show here that a similar acquisition event (i.e., from a viral source) has occurred multiple times in the evolutionary history of LTR-retrotransposons, leading to the founding of at least two and possibly three separate lineages of invertebrate retroviruses.
RESULTS
The Insect Errantiviridae Acquired a Baculoviralenv Gene
A phylogenetic analysis of representative members of the Ty3/ gypsy group based on the conserved reverse transcriptase (RT), ribonuclease H (RNH) domains is presented in Figure 1B (Malik and Eickbush 1999; Marin and Llorens 2000). The lineages in red represent those that have acquired an env gene downstream from their pol gene. The open reading frames of these putative retroviruses are compared with other representative members of the Ty3/ gypsy group in Figure 1C. The gag genes upstream of the pol genes are often characterized by two or three CCHC RNA-binding motifs in retroviruses (thick black lines), although these are apparently absent in many lineages of the Ty3/ gypsy group. The pol genes of these retroelements include the enzymatic protease (PR), reverse transcriptase (RT), ribonuclease H (RNH), and the integrase (IN) domains. Downstream from the IN domain, a carboxyl-terminal extension is often found. This extension usually includes a GPY/F domain (named after the most highly conserved residues), which may bear DNA-binding specificity. In one lineage, a chromodomain module (CD) is found downstream from the GPY/F domain (Malik and Eickbush 1999).
The three instances of an env gene-like acquisition, in the Athila (Arabidopsis thaliana), Gypsy (Drosophila melanogaster), and Osvaldo (Drosophila buzzatii) elements, have no detectable similarity to each other. Among these three lineages, only the gypsy-like elements have been extensively characterized. Several intact members have been found in insect genomes (see Desset et al. 1999 for a current listing), and the biological function of the env gene has also been elucidated (Song et al. 1994). Members of this lineage are referred to asErrantiviridae (Boeke et al. 1999). Phylogenetic analysis of the errantivirus env genes is largely congruent to that based on the RT/RNH domains (data not shown), supporting a monophyletic introduction of the env genes into Errantiviridae.
Because the sequences of many errantiviral elements have been determined, we investigated the origins of the env gene in this widespread lineage. Pairwise comparisons among the errantiviruses indicate that the env genes diverge more rapidly than thepol genes, but at about the same rate as the structuralgag genes (not shown). This high divergence is evident from a multiple alignment of the errantivirus env genes (Fig.2). The alignment presented does not include the (predicted) amino-terminal leader peptide and carboxyl-terminal transmembrane regions that are found in all errantiviruses, as these have poor sequence conservation. Apart from one block of conserved amino acids shown in the boxed region (Lerat and Capy 1999; Desset et al. 1999), there are only very short segments of similarity conserved among all the errantiviruses (shown overlined).

Multiple alignment of the errantivirus env genes and 'related' baculovirus ORFs. The alignment is shaded using MacBoxshade to a 50% consensus with gray and black shading indicating similar and identical residues, respectively. The boxed region corresponds to the Logo in Fig. 3. The different errantivirus sequences (accession numbers) used are ZAM (AJ000387), Tirant (Z93507), 17.6 (P04283), 297 (C24872), Idefix (AJ009736), and Gypsy (M38438) from Drosophila melanogaster; Tom (Z24451) from D. ananassae; TED (C36329) from Trichoplusia ni. Also shown are the baculoviruses,Spodoptera exigua nucleopolyhedrovirus SENV (AAF33539.1),Lymantria dispar nucleopolyhedrovirus, LDNV (AAC70316); andXestia c-nigrum granulovirus, XNGV (AF162221_27). Note that the homology extends to beyond the block (regions overlined) including cysteine residues that may be important for mediating interactions between the two proteolytic products of the env gene. Other baculovirus ORFs that show homology (not shown) are Autographa californica nucleopolyherdrovirus, ACNV (P41428); Orygia pseudotsugata nuclear polyhedrosis virus, OPNV (O10282);Bombyx mori nucleopolyhedrosis virus, BMNV (L33180).
We also used the complete env genes from the available insect errantiviruses to identify blocks of conservation using the BlockMaker program. As indicated by the multiple alignment, only one extended block of conserved amino acids was identified (Fig.3A). Using this segment of conserved amino acids, we then used the position-specific scoring matrix (PSSM) to search the non-redundant database using MAST. The MAST search successfully identified open reading frames (ORFs) from three insect baculoviruses: the Spodoptera exigua nucleopolyhedrosis virus or SENV (Ijkel et al. 1999), Lymantria dispar nucleopolyhydrosis virus or LDNV (Kuzio et al. 1999), and the Xestia c-nigrum granulovirusor XNGV (Hayakawa et al. 1999). These matches were at highly significant levels, indicated by the low probabilities of finding such a match based on chance alone (E-values). Thus, we conclude that theenv genes from errantiviruses and the baculoviral ORFs share common ancestry. (This conclusion is also borne out by a PSI-BLAST search using errantivirus env genes as query). In a previous study of the env genes (Lerat and Capy 1999), the same block of conserved amino acids was suggested to be in common between errantiviruses and vertebrate lentiviruses. We could not confirm this finding, as these lentiviral matches had E-values up to 1000, which are considered non-significant in our analysis.
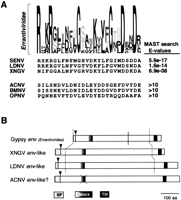
(A) Logos of the conserved block in the envelope genes of insect errantiviruses. In the Logos format, the height of each residue is proportional to its frequency, and the total height of all the residues in the position are proportional to the conservation (information content) at any particular position. Thus, the tallest residues represent invariant residues. This information is used to construct weighted queries to search the protein database. Highlighted below the Logo are the significant MAST matches and the E-values reported. Because blocks are ungapped, a gap was manually introduced in the gypsy sequence to correct obvious misalignments (Fig. 2). (B) Schematic ORFs of the env-related genes in errantiviruses and baculoviruses. Highlighted are the predicted leader peptide (SP, cleavage site shown by the arrow) and transmembrane regions (see Methods), as well as the conserved block of conservation (Fig. 2) common to all errantiviruses. The solid vertical line indicates the site of proteolytic cleavage for the gypsy env, whereas the dotted lines refer to the region aligned in Fig. 2.
To support the significance of the blocks-MAST approach, the complete baculoviral ORFs (referred to as env-like) were used to perform a complementary iterative database search (PSI-BLAST, Altschul et al. 1997). We detected the insect errantiviruses at highly significant levels (starting at E-values < 10−5) at the first iteration in the case of LDNV and SENV. Further iterations improved the identification of all the env genes of the gypsy family. BLAST results also revealed the presence of homologous ORFs in three other baculovirus genomes: Autographa californica nucleopolyhedrosis virus (ACNV), Bombyx mori nucleopolyhedrosis virus (BMNV), and Orgyia pseudotsugata nucleopolyhedrosis virus (OPNV). However, neither ACNV, BMNV, or OPNV possess the highly conserved block shown in Figure 3A (E-values > 10).
As shown in Figure 2, the limited sequence similarity between the errantivirus sequences can be extended to include the LDNV, SENV, and XNGV baculovirus ORFs. Indeed, there are few regions of the alignment where the errantivirus env genes are similar to each other, but not to the baculovirus ORFs. Comparison of these ORFs with the ACNV, BMNV, and OPNV ORFs revealed significant levels of similarity throughout their lengths except at, and upstream of, the block of conservation boxed in Figure 2; these ORFs (ACNV, OPNV, and BMNV) are not included in the alignment. Like the errantiviruses, all the baculovirus ORFs are predicted to have amino-terminal signal peptides and carboxyl-terminal transmembrane regions (shown in Fig. 3B).
What is the role of these baculoviral ORFs? In baculoviruses, an envelope analogous gene (gp64) has been documented as being crucial for infection from cell to cell, and from the gut to the hemocel (Oomens and Blissard 1999). However, gp64 homologs are missing from LDNV, SENV, and XNGV, the three baculoviruses represented in Figure 2. In these baculoviruses, the ORFs shown in Figure 2 have been suggested to represent the viral envelope genes based on predicted structural features (Fig. 3B; Kuzio et al. 1999). When we scan the LDNV, SENV, and XNGV genomes for additional envelope genes (i.e., ORFs containing amino-terminal signal, transmembrane domains, and glycosylation signals indicative of receptor-like genes as discussed in Methods) no other candidate ORFs can be identified. Our findings that these baculoviral ORFs are homologous with the env genes of errantiviruses strengthens the identification of these ORFs as the source of infectious ability for LDNV, SENV, and XNGV. Thus, we propose that extant Baculoviridae use two different genes for the purposes of mediating infection: the gp64 homologs and errantivirusenv-like homologs. In the case of the ACNV, BMNV, and OPNV baculoviruses that use gp64, the encoded env-like genes (Fig.3A) may no longer function as envelope genes and probably perform an unknown secondary role that accounts for their preservation. Using the analogy of the vertebrate retroviruses, the block of conservation observed in Figure 3A may correspond to a host receptor binding determinant, a possibility that can be experimentally tested. An inherent prediction of this finding is that site-directed mutagenesis of the conserved block in the env genes should have similar effects on the infectious abilities of both the errantiviruses as well as the (gp64-lacking) baculoviruses.
What was the direction of the lateral transfer of the envgene? Phylogenetic analyses confirm that the introduction ofenv genes into Errantiviridae was a monophyletic event (Fig. 1B). Because the baculoviruses were presumably always infectious agents (no other forms have been reported), and errantiviruses originated from a non-viral retrotransposon lineage (Fig. 1B) we can propose a baculovirus origin of the envgenes. It is logical that we have traced the origin of envgenes in errantiviruses to baculoviruses in two respects. First, whereas the Ty3 clade (Malik and Eickbush 1999) is found in fungi, plants, vertebrates, and even slime molds, errantiviruses are restricted to insects, the same host range as the baculoviruses. Second, LTR-retrotransposons have been found inserted into baculovirus genomes. For example, the TED retrotransposon found in the lepidopteran host Trichoplusia ni, is also found in the genome of the associated ACNV baculovirus (Friesen and Nissen 1990). Thus, LTR retrotransposons can insert into the viral genome, from which we have suggested they obtained their env gene.
The Nematode Cer Elements Acquired Their env Gene from Phleboviruses
Among the LTR-retrotransposons (Fig. 1A), relatively little is known about the BEL clade. The BEL clade has recently been the subject of phylogenetic scrutiny (Bowen and McDonald 1999). We present an updated phylogenetic analysis of the members of the BEL clade based on their reverse transcriptase (RT) and ribonuclease H (RNH) domains. The presented phylogenetic tree (Fig. 4A) points out distinct lineages, found in insects, nematodes, and vertebrates. The insect lineage consists of members from Drosophila melanogaster (BEL1–3), Anopheles gambiae (Moose),Drosophila simulans (Ninja) and Bombyx mori (Pao). Additional members have been identified in other mosquito genomes (Cook et al. 2000). Thus, this is a widespread lineage at least in dipterans. The nematode lineage presently consists of members fromCaenorhabditis elegans (Cer7–14) and Ascaris lumbricoides (Tas). However, screening of nucleotide databases reveals segments of BEL clade members in other nematode genomes (data not shown). The vertebrate lineage currently includes members from the pufferfish, Fugu rubripes. However, the identification of a BEL-like segment in the genome of the ascidian urochordate, Ciona intestinalis (accession no. AJ226777), strongly suggests that this clade is widespread in chordates. Indeed, members of the BEL clade have been identified in basally branching metazoans, like the blood fluke,Schistosoma mansoni (Tiao element accession no. AF073334), suggesting that the BEL clade is widespread in metazoans.
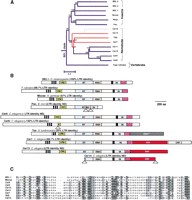
(A) Phylogenetic analysis of BEL clade members from insect, nematode, and vertebrate genomes. Bootstrap values and a divergence scale are indicated. (B) Schematic ORFs from representative members of the BEL clade with various enzymatic and structural features highlighted. Vertical gray lines indicate a termination codon or frameshift encountered. Lower triangles indicate larger deletions. Two different env genes are found in Tas and the Cer7, Cer13, and Cer14 retroviruses (Bowen and McDonald 1999; Felder et al. 1994). In Cer7, an additional accessory protein is found downstream from theenv gene (Bowen and McDonald 1999). A carboxyl-terminal extension to the core integrase domain, with a presumed DNA-binding role is also highlighted, and a multiple alignment presented in (C). There has been some confusion over the enzymatic domains encoded by the BEL clade based on the apparent absence of an intact ribonuclease H/integrase domain from one of the earliest members identified, Pao (B. mori). Closer inspection from pairwise comparisons reveals that this is the result of at least two large internal deletions in the open reading frame of the Pao element that was originally sequenced (Xiong et al. 1993). This is confirmed from comparisons to intact Pao elements from the B. mori genome whose sequence is present in the est (expressed sequence tags) databases.
Schematic ORFs from representative members of the BEL clade are presented in Figure 4B. Like the Ty3/ gypsy group, all members of the BEL clade appear to carry a carboxyl-terminal module in addition to the core integrase domain (IN) that includes the HHCC zinc finger-like motif and the catalytic D, D (35) E motifs. An alignment of this module is presented in Figure 4C. This extension is analogous to the GPY/F module in the Ty3/ gypsy group (Fig. 1C; Malik and Eickbush 1999). Whereas the function of these modules is unknown, they presumably bear (by analogy to mammalian retroviruses) DNA-binding determinants.
Also apparent from this schematic representation (Fig. 4B) is the presence of additional coding regions downstream from the integrase (and extension) domains in several nematode representatives: Tas, Cer7, and Cer13. For both Tas and Cer7, this downstream ORF has been referred to as the envelope gene by analogy to the vertebrate retroviruses and the insect errantiviruses (Felder et al. 1994; Bowen and McDonald 1999), although neither has been biochemically tested. We have identified the Cer13 and truncated Cer14 elements as new additions to the Bowen and McDonald (1999) study (accession nos. AC024209 andAL110479, respectively). Interestingly, whereas the envelope genes from Cer7, Cer13, and Cer14 are very similar to each other, they bear no detectable similarity to the envelope gene from Tas. Thus, if these additional ORFs do encode envelope genes, they must represent distinct acquisition events.
We next investigated the evolutionary origins of the Cerenv-like genes. We were surprised to find a strong similarity to a group of glycoproteins (G2) from Phleboviruses, a class of single-stranded RNA viruses. Indeed, these similarities had been noted previously and formed the basis of the Cer env genes being classified as part of the same family as the Phlebovirus glycoproteins in the Pfam database (Bateman et al. 1999). To confirm that these were true matches, and not just an artifact because of compositional bias, we used PSI-BLAST searches with the Cer13 env as query. This resulted in matches to the Cer7 and Cer14 envelope genes as well as Phleboviral glycoproteins at significant levels (E-values < 10− 21 ). In addition, when the second iteration was performed using a (surrogate multiple alignment) consensus of the Cer env genes alone (see Methods), it again found matches to these glycoproteins at significant levels (E-values < 10− 31 ), supporting the hypothesis that the Phleboviral G2 glycoproteins are homologous to the Cerenv genes.
Multiple alignment of these genes (env-like from Cer elements and G2 glycoproteins from Phleboviruses) and their schematic representation are presented in Figure 5. The domain enclosed by vertical lines in Figure 5A is shown in the alignment in Figure 5B. All these genes possess a predicted transmembrane domain at their carboxyl-terminal ends, which as expected, contains only weak sequence similarity. There are 19 cysteines that are conserved across all sequences and it is likely that disulfide bonds may play a role in the correct folding of this family of proteins, or alternatively, in mediating the interaction between the proteolytic products of theenv gene. In the case of the Phleboviruses, the G2 glycoprotein is processed from a single polypeptide translated from the M (medium) RNA that also encodes the non-structural protein N-Sm, and the G1 glycoprotein. The Cer13 element, which represents the most intact member of the nematode elements, has its env gene in the same frame as the rest of the domains, as does the truncated Cer14. Although the Cer7 env is apparently in a different frame, the frameshift occurs in the integrase extension domain, probably reflecting a defect of the Cer7 element rather than a true frameshift (Fig. 4B). Thus, it is likely that the env protein in the Cer retroviruses is also processed out of a single large polyprotein in a similar manner to the Phleboviridae G2 glycoproteins, i.e., by a proteolytic cleavage. In support of this model, the predicted proteolytic cleavage sites coincide exactly between the Cer env and the Phleboviral G2 sequences.

(A) Schematic representation of the env-like genes from Cer7 and Cer13, and the G2 glycoproteins from Phleboviruses. In the case of Cer7, a leader signal peptide has been proposed whose cleavage site is indicated. For the Phleboviruses, a polyprotein of three proteins, N-Sm, G1, and G2 is proteolytically processed into the individual proteins. It is likely that the Cer7 and Cer13env-like genes are processed in a fashion similar to G2. The transmembrane (anchors) regions of each protein are indicated and the thin gray lines indicate the area shown in the multiple alignment in (B). Although not indicated, several potential N-glycosylation sites are predicted in both env and glycoprotein genes. The multiple alignment is shaded to an 80% consensus using MacBoxshade with gray and black shading representing similar and identical resides respectively. In particular, note the 19 conserved cysteines believed important for the correct folding of the glycoproteins.
The PSI-BLAST searches we conducted also identified regions of similarity between the Cer env genes and glycoproteins of another class of plant single-stranded RNA viruses, the Tenuiviruses. Phylogenetic analysis based on alignments of the Cer env-like genes and the G2 glycoproteins from Phleboviruses and Tenuiviruses (Fig. 6) supports the model that the nematode Cer elements acquired their envelope gene from a Phleboviral-like ancestor. A homology among the Phleboviral and Tenuiviral glycoproteins has been noted in an earlier report (Estabrook et al. 1996). This, as well as similarity of other features, has suggested the phylogenetic classification of Tenuiviridae as sister-families to the Phleboviruses (Ramirez and Haenni 1994). Phleboviruses belong to the Bunyaviral genus of single-stranded RNA viruses. However, no other families of Bunyaviridae bear proteins that are homologous with the Phleboviral glycoproteins. This situation is analogous to the one in Baculoviridae, in which two different glycoproteins are found, one of which has been acquired by a retrotransposon lineage.
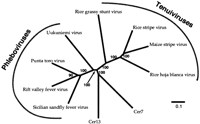
An unrooted neighbor-joining tree of the Cer env genes and the Phleboviral and Tenuiviral G2 glycoproteins. Bootstrap values and a divergence scale are indicated. The Phleboviral G2 glycoproteins presented are from the Punta Toro Virus (accession no. P03517 ), the Rift Valley Fever Virus (AAA47449), the Sicilian Sandfly fever Virus (AAA75043), and the Uukuniemi virus (P09613).
A Tas Element May Have Acquired Its env Gene from a Herpesvirus-Like Ancestor
Despite its phylogenetic proximity to the Cer retroviruses (Fig.4B), the Tas element does not encode a Cer envelope gene. Instead, Tas contains a different ORF that may represent yet another recentenv-like gene acquisition. Unfortunately, the Tas element sequence contains several frameshifts and termination codons, and represents a “dead” element. Its LTRs are more than 5% divergent, also suggesting that many mutations may have accumulated. A BLAST search using the Tas env gene did not reveal any significant similarities. However, a search of the BLOCKS + database (Henikoff et al. 2000) using the IMPALA search program (Schaffer et al. 1999) revealed a marginally significant best match (E-value 0.085) to gB glycoproteins from Herpesviridae, a class of double-stranded DNA viruses with no RNA stage (Fig. 7). The strength of the match could be underestimated for two reasons. First, comparisons involving only a single (dead) Tas env gene are expected to be weaker than those involving a group of closely related genes (like the errantiviral env genes). Second, the herpesviral gB glycoprotein blocks are biased because of an exclusive sampling from mammalian lineages. The actual viral source of the Tasenv gene is probably a phylogenetically distinct subclass.
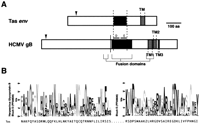
Possible similarity of the Tas env-like gene to herpesviral glycoprotein gB. (A) The Tas envelope gene consists of a predicted leader signal peptide and a carboxyl-terminal transmembrane domain. In the central portion, a region of homology is found between the Tas gene and a segment of the herpesviral glycoprotein gB. A schematic of the gB glycoprotein of the human cytomegalovirus is also presented. HCMV gB has three transmembrane domains at its carboxyl-terminal end (TM1–TM3). TM1 and two other segments implicated in the fusogenic (cell attachment and membrane fusion) properties of gB are indicated with brackets. The central segment that is believed to be responsible for fusion (indicated by a black box) corresponds to the blocks D and E shown in (B). (B) The gB glycoproteins are represented as a series of conserved blocks (Block PF00606- BLOCKS + database), of which only blocks D and E (shown in Logos format) show homology with Tas (corresponding sequence shown below each Logo).
The glycoprotein gB constitutes greater than 50% of the protein mass of the envelope and has been implicated in the viral attachment and fusion of herpesviruses (Britt and Mach 1996). Of the many glycoproteins encoded by Herpesviridae, the gB glycoproteins are primarily implicated in infection. Interestingly, the segment of gB glycoproteins believed to be largely responsible for the viral attachment to the cell surface is precisely the segment that has similarity to Tas (Fig. 7; Britt and Mach 1996). Thus, whereas the sequence similarity is not by itself conclusive, the biological function performed by glycoprotein B in herpesviral infections adds considerable support to a relationship between the Tas envelope andHerpesviridae gB glycoproteins.
DISCUSSION
One of the characteristic features of transposable elements has been their spectacular ability to undergo cross-species horizontal transfers. This has been best documented in the case of the DNA-mediated elements, P and mariner (Clark et al. 1994; Robertson 1997), but is also true for LTR-retrotransposons (Jordan et al. 1999). Whereas the mechanism for horizontal transfer still remains to be established for any transposon, one likely scenario is that they rely on other vectors for their horizontal spread. For example, a transposable element could insert from the host genome into an associated DNA-based viral genome, which can subsequently infect another host species. (In the case of RNA viruses, the retrotransposable element could simply be co-packaged within the viral capsid). Thus, the transposon can piggyback its way into a new genome. There are obvious limitations to tracking down a potentially short-lived, insert-bearing viral strain during the short period of the actual transfer. However, the TED retrotransposon-bearing ACNV baculovirus may represent just such an example (Frisen and Nissen 1990). Acquisition of an env gene, on the other hand, releases the retrotransposon from relying on another vector for jumping into different hosts, increasing the probability (frequency) of cross-species transfer.
We have uncovered multiple instances of such env acquisitions in the phylogenetic history of LTR retrotransposons (summarized in Fig.8). In three instances (the insect errantiviruses, and the nematode Cer and Tas retroviruses) we have traced the origins of this env gene. In all three cases, this origin is a virus. Thus, the env genes of both the insect errantiviruses and Tas retroviruses are derived from different lineages of double-stranded DNA viruses, whereas the Cer retroviruses are derived from single-stranded RNA viruses. Interestingly, a viral origin also explains the origin of the env-like cell-to-cell movement proteins of the plant caulimoviruses. However, caulimoviruses are thought to have arisen by the fusion of an LTR-retrotransposon with a single-stranded RNA virus from plants (Koonin et al. 1991). They have lost their ability to integrate into host genomes, and thus can no longer be considered strictly analogous to vertebrate retroviruses.
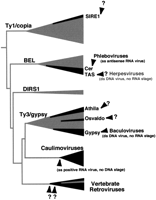
Schematic of env-like gene acquisitions in the evolutionary history of LTR-retrotransposons. The LTR retrotransposable elements are divided into six clades, each represented by a triangle. The height and width of the triangles represent the age (presumed without accounting for horizontal transfers) and current known diversity of each clade, respectively. Thus, although the DIRS1 clade has representatives in slime mold, fungi, and nematodes, indicating an ancient history, it is not as abundant as the other clades. Eight possible instances of anenv-like gene acquisition can be found and are indicated by the black regions. In four of these cases, the evolutionary origins of this env gene have been traced back to the viral source indicated. The strongest evidence was found in the Gypsy and Cer cases. The origins of the other four env-like genes remain unknown. In the case of the vertebrate retroviruses and the plant caulimoviruses, most members have an env gene, which has subsequently been lost in some endogenous vertebrate retroviruses. The exact number of env gene acquisitions in vertebrate retroviruses is unclear.
The ability to successfully trace the evolutionary origins ofenv genes depends on the age of the acquisition and the constraints under which the env genes have been evolving. Along these lines, it may no longer be possible to delineate the (potentially ancient) origin(s) of the env gene in vertebrate retroviruses, even as our knowledge of different viral glycoproteins increases. Better success may be predicted for other, more recent acquisitions of env genes. The vast numbers of mobile elements obtained from genome sequencing efforts have belied the notion that LTR retrotransposons are exclusively found in invertebrates, whereas retroviruses are exclusively found in vertebrate genomes. The transition from a non-viral retrotransposon to a retrovirus could have occurred as many as eight times (Fig. 8). In four of these instances, it is now possible to implicate other viral sources for the acquisition of infectious ability.
The mechanism of this acquisition could be very simple. During their conversion from an RNA genome to a double-stranded DNA (subsequently integrated), LTR retrotransposons and retroviruses undergo intermolecular strand-transfer events at their LTRs (for review, see inVarmus and Brown 1989). Recombination independent of sequence similarity has been proposed as the mechanism of retroviral transduction of cellular oncogenes (Swain and Coffin 1992). Similarly, a viral infection of a host cell that occurs simultaneously with the retrotransposition of an LTR-bearing element could lead to an illegitimate recombination intermediate in which the env gene is successfully acquired by the daughter element. The opportunism, shown by LTR retrotransposons in acquiring an env gene from another infectious agent, presents a general paradigm in which, potentially, any LTR retrotransposon can become a virus.
METHODS
Blocks (see www.blocks.fhcrc.org) for the gypsy class of insect errantiviruses were constructed using the Gibbs heuristics in the BlockMaker program (Henikoff et al. 1995). This program uses a sampling technique to identify conserved segments that are at least eight amino acid residues long. Using the BLOCKS identified, Motif Alignment and Search Tool (MAST) (Bailey and Gribskov 1998) was used to search the non-redundant Genbank database (seewww.sdsc.edu/MEME/meme/website/mast.html) as well as the identified ORFs. PSI-BLAST (Altschul et al. 1997) iterative database searches were also used for confirmation. In most cases, a single iteration, or simply a BLASTP search, sufficed. In the case of Cer retroviruses, PSI-BLAST searches in the second iteration were performed after unchecking all other matches. Thus, a surrogate multiple-alignment consensus of only the Cer retroviruses is used as a query in the second iteration. Multiple alignments were performed using CLUSTAL_X (Thompson et al. 1997) with minor manual modifications of the gaps. Blocks and alignments are presented using the Logos format and MacBoxShade. Phylogenetic analysis was performed using Neighbor-Joining (Saitou and Nei 1987) and maximum parsimony-heuristic (tree-bisection-reconnection branch swapping with the number of trees saved at each step limited to five) and branch-and-bound methods using the PAUP* package (Swofford 1999). Signal peptide predictions were made using SignalP (Nielsen et al. 1997) and transmembrane domains were predicted using PHD (Rost and Sander 1993).
WWW Resources
www.blocks.fhcrc.org A database of the most highly conserved segments of protein families.
www.sdsc.edu/MEME/meme/website/mast.html A tool to search sequence databases using protein motifs.
Acknowledgments
We thank Jorja Henikoff for her advice on using the BLOCKS + database and different searching tools. We also thank Kami Ahmad, Rahm Gummuluru, Pauline Ng, Jim Smothers, Danielle Vermaak, and Bas van Steensel for their comments on the manuscript. This work was supported in part by grants NSF MCB-9974606 to T.H.E. and NIH GM-29009 to S.H. H.S.M. is a postdoctoral fellow at the Helen Hay Whitney Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author. Present address: 1100 Fairview Avenue, A1-162, Seattle, WA 98109 USA.
-
E-MAIL hsmalik{at}fred.fhcrc.org; FAX (206) 667-5889.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.145000.
-
- Received April 20, 2000.
- Accepted June 29, 2000.
- Cold Spring Harbor Laboratory Press








