
Large-Scale Comparison of Fungal Sequence Information: Mechanisms of Innovation in Neurospora crassa and Gene Loss in Saccharomyces cerevisiae
Abstract
We report a large-scale comparison of sequence data from the filamentous fungus Neurospora crassa with the complete genome sequence of Saccharomyces cerevisiae. N. crassa is considerably more morphologically and developmentally complex thanS. cerevisiae. We found that N. crassa has a much higher proportion of “orphan” genes than S. cerevisiae, suggesting that its morphological complexity reflects the acquisition or maintenance of novel genes, consistent with its larger genome. Our results also indicate the loss of specific genes from S. cerevisiae. Surprisingly, some of the genes lost from S. cerevisiae are involved in basic cellular processes, including translation and ion (especially calcium) homeostasis. Horizontal gene transfer from prokaryotes appears to have played a relatively modest role in the evolution of the N. crassa genome. Differences in the overall rate of molecular evolution between N. crassa andS. cerevisiae were not detected. Our results indicate that the current public sequence databases have fairly complete samples of gene families with ancient conserved regions, suggesting that further sequencing will not substantially change the proportion of genes with homologs among distantly related groups. Models of the evolution of fungal genomes compatible with these results, and their functional implications, are discussed.
Sequence comparisons are often used in comparative genomics to infer sequence/function relationships in one organism based on similarities to sequences in other organisms, but it is also instructive to ask about differences between organisms or their genomes and to ask how such differences arose. We have conducted a large-scale comparison of sequence information from the filamentous fungusNeurospora crassa, the unicellular fungus Saccharomyces cerevisiae, and sequences from nonfungal organisms, to investigate patterns of fungal genome evolution. A large number of N. crassa EST sequences are available (Nelson et al. 1997; this paper), as is the complete genome sequence of S. cerevisiae(Goffeau et al. 1996). N. crassa and S. cerevisiaeare ascomycete fungi and are estimated to have diverged from each other at least 310 mya (Berbee and Taylor 1993) and probably >400 mya (Taylor et al. 1999). This represents sufficient time for substantial differences to have arisen, but it is substantially more recent than the divergence of the fungi from other eukaryotes, >1 bya (Knoll 1992; Feng et al. 1997).
The N. crassa genome is approximately three times the size of the S. cerevisiae genome. N. crassa also exhibits much greater morphological and developmental complexity (Springer 1993), suggesting that N. crassa has a substantially greater number of genes. The number of genes in N. crassa has been estimated to be 1.5–2.2 times greater than that of S. cerevisiae (Kupfer et al. 1997; Nelson et al. 1997). A previous analysis of ESTs from N. crassa indicated that it has a much higher proportion of genes without identifiable homologs (commonly designated “orphan” genes) than S. cerevisiae (Nelson et al. 1997), a finding that we demonstrate more rigorously here.
These differences in genome size, gene number, phenotypic complexity, and proportion of orphan genes raise various possibilities regarding the evolution of fungal genomes. On the one hand, it is possible thatS. cerevisiae has been “streamlined” by the loss of genes, with a corresponding loss of phenotypic complexity (e.g., multicellularity). This hypothesis is consistent with phylogenetic analyses of the fungi that indicate that the unicellular fungi arose from multicellular ancestors (Bruns et al. 1992; Berbee and Taylor 1993; Liu et al. 1999). Some genes that are present in N. crassa but not in S. cerevisiae do reflect the loss fromS. cerevisiae of genes present in the common ancestor of these organisms (Braun et al. 1998). Gene loss might result in a concentration of widely conserved genes that are essential for life (e.g., Mushegian and Koonin 1997; Snel et al. 1999), providing an explanation for the lower proportion of orphan genes in S. cerevisiae. On the other hand, addition of a large number of genes to the N. crassa lineage subsequent to its divergence from the ancestor of S. cerevisiae could also explain the differences in genome size, developmental complexity, and—if the acquired genes were either truly novel or free to diverge radically from their sources—proportions of orphan genes.
We reasoned that comparison of N. crassa sequences to the complete S. cerevisiae genome and nonfungal sequence databases would provide us with insights bearing on these alternatives. For instance, genes present both in N. crassa and in other nonfungal eukaryotes but absent from S. cerevisiae are likely to reflect genes that have been lost from the S. cerevisiaelineage. Clearly, such gene losses could have substantial functional significance. Genes that are present in both N. crassa and prokaryotic organisms but not in S. cerevisiae or nonfungal eukaryotes are plausible candidates for horizontal transfer into theN. crassa lineage. If a large number of candidates for gene loss from S. cerevisiae or horizontal transfer into N. crassa were identified, these mechanisms could account for much of the difference in genome sizes and gene numbers between the two fungi. Although examples of both classes were identified by this study, a relatively modest number of candidate lost or transferred genes were identified, indicating that alternative explanations for the differences between N. crassa and S. cerevisiae must be sought.
RESULTS
In this study, we conducted large-scale homology searches using BLAST (Altschul et al. 1997) comparing N. crassa query sequences to three distinct databases: “SC,” the set of translated ORFs from the complete S. cerevisiae genome; “NF,” a set of translated ORFs from the nonfungal sequences in the public sequence databases; and “HMEST,” the human and mouse EST database. The NF and HMEST databases were largely independent, because NF contained annotated protein sequences from largely full-length cDNAs and genomic DNAs, whereas HMEST contained partial cDNA sequences from randomly sampled genes of humans and mice. For comparison, S. cerevisiae sequences (a set of ESTs and the translated ORFs from the complete S. cerevisiae genome) were also searched against NF and HMEST. These searches revealed several distinctive patterns of homolog distribution, summarized below. To facilitate interpretation of these patterns, additional ad hoc searches, described below, were performed against several additional data sets. Details regarding the custom sequence sets (databases) used for homology searches are provided in Table 1 and in Methods.
Sequence Sets Used in Analyses
A Relatively Low Proportion of Expressed Sequences in N. crassa Can Be Identified by Homology Searches
We reported previously that only 33.6% of N. crassa cDNAs were clearly homologous to proteins in the National Center for Biotechnology Information (NCBI) protein database, according to ungapped BLAST-X searches using 1865 N. crassa ESTs (Nelson et al. 1997). Here, we extend this observation by analyzing a larger number of sequences, refining our methodology, and analyzing sets of “control” sequences from S. cerevisiae. Before conducting searches, N. crassa ESTs were grouped into “discontigs” (sets of sequences that may not overlap but have a known spatial relationship, such as the sequences derived from both ends of a single cDNA clone; e.g., see Skupski et al. 1999). Thus, homology searches were conducted using 3578 N. crassa ESTs, grouped into 1197 discontigs. Because the discontigs are, for the most part, from distinct genetic loci, this constitutes some 10%–15% of the genes inN. crassa, based on the estimates of gene number by Kupfer et al. (1997) and Nelson et al. (1997).
These searches resulted in the identification of clear homologs (E ≤ 10−5) outside of the fungi for only ∼33% of loci (Table 2). In contrast, we found that >57% of predicted genes from S. cerevisiae have clear homologs in the same databases. This reflects more than the differences between the partial sequences obtained by EST projects and the full-length sequences obtained by genomic sequencing projects, because a higher proportion of S. cerevisiae ESTs also have identifiable homologs (Table 2). The differences are also not explained by the types of reads obtained by the Neurospora Genome Project, because a lower proportion of N. crassa sequences were identified for both 5′ and 3′ reads (data not shown). The fractions of columns containing mismatches or gaps in the contigs generated by TIGR Assembler (which reflect sequencing errors) are similar for the N. crassa and S. cerevisiae EST data sets (data not shown). Thus, compared with S. cerevisiae, it appears that a substantially greater proportion of expressed sequences from N. crassa represent orphan genes. This phenomenon has also been observed for complex multicellular eukaryotes such as plants and animals (Waterston and Sulston 1995; Delseny et al. 1997).
Percentages of Sequences with Detectable Homologs in Various Databases
The Low Proportion of Identified Genes in N. crassa Does Not Represent Accelerated Molecular Evolution
One possible explanation for the observed difference between N. crassa and S. cerevisiae would be accelerated sequence divergence in N. crassa, resulting in a larger proportion of sequences that cannot be identified by homology searches. Such a global acceleration of molecular evolution has been suggested forCaenorhabditis elegans (Mushegian et al. 1998) and also for the fungi as a group (Feng et al. 1997; Stassen et al. 1997). However, comparisons of divergence from nonfungal sequences for paired orthologous sequences from N. crassa and S. cerevisiae indicate that the rates of molecular evolution inN. crassa and S. cerevisiae are similar (Fig.1). Randomly chosen N. crassa sequences were paired with their closest homolog from S. cerevisiae, and both members of such a pair were used as queries against the NF database;N. crassa sequences with no clear homolog in S. cerevisiae were excluded from the analysis. Although different loci within an organism may evolve at substantially different rates, for a given pair of homologous N. crassa and S. cerevisiaesequences, the degrees of divergence of these sequences from their nonfungal homologs are approximately equal, as indicated by similar scores for the best match.
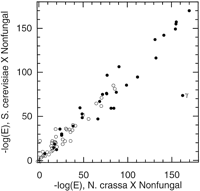
Rates of divergence are similar for N. crassa and S. cerevisiae. Pairs of homologous N. crassa and S. cerevisiae sequences were analyzed using BLAST against NF (a database of nonfungal protein sequences); each pair is represented by a point in the plot, with the x-axis showing the negative log of the E-value [−log (E)] of the best database match to the N. crassa query and the y-axis showing −log (E) of the best match to the S. cerevisiaequery. (○) Pairs for which the N. crassa sequence was (part of) an EST from our data set; in these cases, the N. crassa contig and the paired S. cerevisiae sequence were trimmed to the region of overlap, as described in Methods. (●) Pairs for which the N. crassa and S. cerevisiae sequences were complete protein sequences. The outlying point in this plot, labeled “γ,” is γ-tubulin (see text).
This analysis did identify one protein from S. cerevisiae that is substantially more divergent from nonfungal homologs than is the homologous N. crassa protein. The divergent protein (Fig. 1, point “γ”) corresponds to γ-tubulin, an S. cerevisiae protein that has been established on the basis of detailed analyses to have undergone an unusual degree of divergence from orthologous γ-tubulins present in other organisms (Keeling and Doolittle 1996). Thus, for a limited number of genes, S. cerevisiae may actually exhibit accelerated evolution relative toN. crassa (also see Stassen et al. 1997). However, the two organisms appear to have similar rates of evolution for most genes for which homologs may be identified, suggesting that the high proportion of orphan genes in N. crassa does not reflect a global acceleration of molecular evolution in that organism.
Comparisons of Different Databases Identify Patterns of Genome Evolution
Comparisons of homology searches conducted with N. crassaqueries against different databases reveal several distinct patterns of homolog distribution. Figure 2 compares the results of searches for homologs of N. crassa sequences in nonfungal organisms (x-axis) and in S. cerevisiae(y-axis). A majority of loci (discontigs) from N. crassa did not exhibit significant similarity to sequences in any of the databases, giving rise to points in the figure that lie near the origin. Many N. crassa loci have homologs in both S. cerevisiae and nonfungal organisms, corresponding to points away from both axes; most of these points lie near the line y = x,indicating—perhaps surprisingly—that they are not substantially more similar to homologous S. cerevisiae sequences than to nonfungal sequences. Loci with significant similarities to nonfungal organisms but with no detectable homologs in S. cerevisiaeappear as points near the x-axis (but away from the origin); they constitute potential cases of genes lost from S. cerevisiae or cases of horizontal transfer into N. crassa. Loci with homologs in S. cerevisiae but with no significant similarity to any known nonfungal proteins, constituting proteins that may be restricted to the fungi, appear as points near they-axis (away from the origin). These general patterns and the interpretation of specific cases are considered in more detail in the following sections.
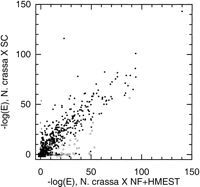
Comparison of homology searches against nonfungal sequences and againstS. cerevisiae sequences. Each point represents a singleN. crassa discontig, with the x-axis showing the negative logarithm of the E-value [−log (E)] of the best match in either NF or HMEST and the y-axis showing −log (E) of the best match in SC. Open circles represent possible cases of gene loss, horizontal transfer, or divergent orthologs (discontigs appearing in Tables 4 T5 T6). Gray circles represent possible cases of fungal specific genes (discontigs appearing in Table 3).
A Small Set of Fungal-Specific Proteins Can Be Identified
Although most N. crassa genes with identifiable homologs have both nonfungal and S. cerevisiae homologs, a small number of discontigs have homologs in S. cerevisiae but not in the non-fungal databases (Fig. 2). These may represent fungal-specific proteins, proteins that have diverged sufficiently that nonfungal homologs are not detected or proteins for which nonfungal homologs exist but have not yet been sequenced. Searches of the NF database using the full-length S. cerevisiae homologs of N. crassa discontigs revealed that some of these reflect artifacts of using partial sequences because the S. cerevisiae sequences had clear nonfungal homologs (E ≤ 10−5). However, nine cases remain candidates for fungal-specific genes (Table3). There appears to be some functional coherence to these cases. Three candidates appear to be cell wall components (such as Gas1p; see Popolo and Vai 1999), which may contribute to unique features of fungal cell walls, and two candidates correspond to classes of transcription factors that have not been reported outside of the fungi [the homologs of Ecm22p, a Gal4p-domain (C6 binuclear zinc cluster) protein (see Henikoff et al. 1997), and Sok2p, an APSES DNA-binding domain protein (see Aramayo et al. 1997)].
Fungal-Specific Genes Present in the NGP Data Set
Additional searches of these nine cases were conducted against sequence sets from other fungi. Homologs of all nine could also be identified in genomic sequence from Candida albicans (data not shown), and homologs of all but two could be identified in the available sequence data from Schizosaccharomyces pombe (Table 3). In sharp contrast, we were unable to identify homologs for any of the genes in the nonascomycete fungi (data not shown) and were only able to identifyAspergillis nidulans homologs in four cases (Table 3). This is likely to reflect limited sampling in these organisms, but some of these candidate fungal-specific proteins may actually be limited to the ascomycete fungi. These results suggest that most candidate fungal-specific genes can be identified in other fungal lineages. However, the identification of so few candidates suggests that the number of proteins that are present in both multicellular and unicellular fungi, but are not found in other groups of organisms, is quite small.
A Set of N. crassa Sequences with Nonfungal Homologs LackS. cerevisiae Homologs
Over 40 N. crassa genes were identified that have clear nonfungal homologs (E ≤ 10−5 against NF or HMEST) but no identifiable S. cerevisiae homologs (E > 0.1) (Fig. 2; Tables 4 and5). Nearly 20 other N. crassa genes have nonfungal homologs that are substantially better matches than are the most similar S. cerevisiae sequences (BLAST E-values for the best hit in the NF data set at least a factor of 1010 smaller than the best S. cerevisiaehit; Fig. 2; Tables 5 and 6). These two situations probably result from one of three evolutionary events: loss of a gene from the S. cerevisiae lineage, horizontal transfer of a gene into the N. crassa lineage, or exceptional divergence of a gene in S. cerevisiae.
Genes Lost from S. cerevisiae: N. crassa Discontigs with Nonfungal Homologs that Lack Detectable S. cerevisiae Orthologs
Candidates for Horizontal Gene Transfer into N. crassa:Discontigs with Apparent Orthologs Only in Prokaryotes
N. crassa Discontigs Whose Closest S. cerevisiaeHomolog Appears to Be a Divergent Ortholog
Examination of specific cases allows us to distinguish among these possibilities. In the majority of cases (36; Table 4), absence of a clear homolog in S. cerevisiae is most parsimoniously interpreted as the result of gene loss, because apparent orthologs of the N. crassa loci are present in other complex eukaryotes. In 13 cases (Table 5), the best match with the N. crassa sequence was a prokaryotic gene, and no closely related eukaryotic homolog was clearly identified. These sequences may reflect horizontally transferred genes, but this assignment should be viewed as tentative because additional sequencing of eukaryotes may reveal closer matches, in which case they would be reinterpreted as genes lost from S. cerevisiae. In the remaining 14 cases, an S. cerevisiaehomolog was identified but was not as close a match as a nonfungal eukaryote homolog, similar to the situation described above for γ-tubulin. These could, in principle, involve either the loss of the S. cerevisiae ortholog from an ancient family of duplicated genes or a case of accelerated divergence in S. cerevisiae. Ten of these sequences appeared to represent cases of gene loss in which a paralogous sequence was retained (also listed in Table 4), whereas four cases appeared to represent divergent orthologs (Table 6), based on our criteria for orthology (see Methods). The putative divergent orthologs involve homologs of calmodulin, ALG-2, calnexin, and UDP–glucose glycoprotein transferase. Strikingly, the first three of these genes encode Ca2+-binding proteins (see below), whereas the fourth (UDP–glucose glycoprotein transferase) shares a functional role with calnexin: They are both components of the endoplasmic reticulum quality control machinery (Parlati et al. 1995;Fernandez et al. 1996). Thus, there is functional coherence to this set of genes that appear to have undergone unexpected degrees of divergence.
Many of the genes that appear to have been lost in S. cerevisiae can be found in other fungi. Only 13 of the 46 (28%) candidates for gene loss have no apparent ortholog among the available fungal sequences, probably at least partly because of incomplete sampling. The nonascomycete fungi have the smallest number of orthologs in this category (4 sequences), whereas S. pombe has the largest number (18 sequences). These differences probably reflect both the potential for gene loss in these fungi and the availability of sequences. Only 14 of the 46 cases had orthologs in the availableC. albicans sequences, indicating that some gene loss occurred after the divergence between C. albicans and S. cerevisiae.
Genes that Are Lost or Excessively Divergent in S. cerevisiae Indicate Functional Differences
Some of the proteins that have been lost or show unexpected divergence in S. cerevisiae are involved in basic cellular processes, such as translation, the ubiquitin system, peroxisome function, and ion homeostasis (Tables 4 and 6). Consistent with such loss or divergence reflecting functional adaptations specific toS. cerevisiae, we found instances of functionally related proteins in the set of genes lost from S. cerevisiae, such as the p40 and Int-6 subunits of the translation initiation factor eIF3 (Asano et al. 1997). Perhaps most striking are the changes in genes that are involved in ion homeostasis, especially Ca2+homeostasis. The marked divergence of the Ca2+-binding proteins calmodulin, ALG-2, and calnexin was discussed above (Table 6). Cases of gene loss include annexin (Ca2+-and phospholipid-binding protein; Braun et al. 1998), DdCAD-1 (aDictyostelium discoideum Ca2+-dependent cell–cell adhesion protein; Wong et al. 1996), and a homolog of the mammalian voltage-activated shaker K+ channels (e.g., McCormack et al. 1995; see Table 4). The presence of homologs of annexin and of shaker K+channels in plants (Tang et al. 1995; Braun et al. 1998) further supports the view that such genes have been lost from S. cerevisiae, because the plants are likely to represent an outgroup to the animals and fungi (Baldauf and Palmer 1993).
Few Additional Homologs of N. crassa Sequences Could Be Identified in A. nidulans
Ozier-Kalogeropoulos et al. (1998) found that a high percentage of genes from the budding yeast Kluyveromyces lactis were homologs of S. cerevisiae genes previously considered orphans. Because K. lactis is closely related to S. cerevisiae(these yeasts diverged ∼80 mya; see Berbee and Taylor 1993), we reasoned that a similar survey of N. crassa sequences using a relatively closely related organism, such as the filamentous ascomyceteA. nidulans, might allow the identification of many orphanN. crassa sequences. In our data set, 342 N. crassadiscontigs (29%) had clear homologs in a database of 13404 A. nidulans ESTs, which extended the total number of discontigs with a clear homolog in any database (those listed in Table 2 and the A. nidulans database) to 555 discontigs (from 40% to 46%). Because the sequences available from A. nidulans probably represent somewhat more than half of the expressed genes (see Methods), this suggests that the availability of additional sequences from A. nidulans may allow the identification of clear homologs for slightly >50% of the N. crassa sequences examined in this study. However, these results suggest that the identification of homologs for many N. crassa orphan sequences will require the availability of sequences from fungi that are more closely related thanA. nidulans, which diverged from N. crassa ∼280 mya (Berbee and Taylor 1993).
Coverage of EST and Non-EST Databases Is Very Similar
Just as comparisons of homology search results against nonfungal andS. cerevisiae databases reveal patterns of genome evolution, comparisons of search results against two distinct databases of sequences from nonfungal organisms can provide information regarding the completeness of these databases. Our original reason for conducting searches using both NF (protein sequences from nonfungal organisms) and HMEST (human and murine ESTs) was to determine whether searching ESTs from humans and mice would substantially increase the number of N. crassa sequences for which a homolog was identified, relative to searching the NF database alone. However, our results showed this not to be the case; the results of homology searches against HMEST and NF using the N. crassa discontigs are compared in Figure3 and Table 2.
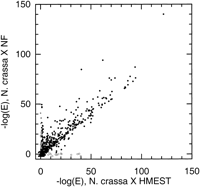
Comparison of homology searches against nonfungal protein sequences and against human and mouse ESTs. Each point represents a single N. crassa discontig, with the x-axis showing the negative logarithm of the E-value [−log (E)] of the best match in HMEST and the y-axis showing −log (E) of the best match in NF. Open circles represent possible cases of incomplete sampling in NF [discontigs with clear homologs (E ≤ 10−5) in HMEST but no detectable homolog in NF]. Gray circles show possible cases of incomplete sampling in HMEST or of genes not present in animals (discontigs with clear homologs in NF but none in HMEST; listed in Table 7).
A majority of N. crassa loci did not exhibit significant similarity to sequences in either database (points near the origin in Fig. 3). A small number of N. crassa loci with significant matches to human or mouse EST sequences but no detectable homologs in the database of nonfungal protein sequences (points near thex-axis and away from the origin) constitute cases of gene families that have not been sequenced outside the fungi except in EST projects. A modest number of N. crassa loci have detectable homologs in the nonfungal database but not in the EST data set (points near the y-axis and away from the origin in Fig. 3). These could reflect incomplete sampling in HMEST or genes with restricted distribution outside the fungi (see below). Most N. crassaloci with significant identity to proteins in NF also have significant identity to proteins in HMEST (points near or above the liney = x in Fig. 3; the tendency for points to lie above y = x generally reflects matches to complete sequences in NF and partial sequences in HMEST, giving better BLAST scores against the NF database).
We found that only 33 (2.8%) of N. crassa discontigs had clear homologs (E ≤ 10−5) in HMEST but not NF; of these, 15 (1.3% of the total number of discontigs) have clear homologs in SC, whereas 18 (1.5%) are found clearly only in HMEST. However, the number of discontigs for which there are clear homologs in NF but not HMEST is larger (98, or 8.2%). A priori, this could reflect less complete sampling in the EST database or the limitations of the partial sequences present in EST databases. However, closer inspection reveals that most of the N. crassa genes with homologs in NF but not HMEST also lack known homologs in both placental mammals andC. elegans (Table 7). Therefore, the absence of homologs in HMEST may reflect the true distribution of these genes. The majority (>65%) of N. crassa sequences with homologs in NF but not HMEST have biological functions related to metabolism (Table 7), including functions like the biosynthesis of vitamins and amino acids, suggesting that these sequences may correspond to proteins that have been lost in the animals.
N. crassa Genes with Homologs in NF but Not HMEST
DISCUSSION
Background
Most comparative genomics to date has focused on prokaryotes, reflecting the availability of multiple complete genome sequences from prokaryotes and the relatively high proportion (usually ∼70%) of prokaryotic genes for which homologs may be identified in other organisms (Koonin et al. 1997). Genomic analyses of the ascomycete yeast S. cerevisiae have been nearly as successful in finding homologs in other organisms, with standard homology searches resulting in the identification of homologs for >60% of the genes (Koonin et al. 1994; Goffeau et al. 1996). However, genomic analysis of other eukaryotes may be substantially more difficult. The proportion of genes in Arabidopsis thaliana and C. elegans that can be identified by homology searches is much lower than for prokaryotes orS. cerevisiae (Waterston and Sulston 1995; Delseny et al. 1997; The C. elegans Sequencing Consortium 1998). A detailed comparison of the S. cerevisiae and C. elegansgenomes indicates that 51% of S. cerevisiae sequences have readily identified homologs in C. elegans, whereas only 26% of C. elegans proteins have readily identified homologs inS. cerevisiae (The C. elegans Sequencing Consortium 1998). This suggests that the relatively high proportion of proteins with “cross-phylum” homologs in S. cerevisiae may be exceptional for eukaryotes.
Patterns of Genome Evolution in the Fungi
Based on evaluation of ESTs representing ∼10%–15% of the genes in N. crassa, we have extended a previous report (Nelson et al. 1997) that a smaller proportion of N. crassa genes have identifiable homologs than is observed for S. cerevisiae(Table 2) and various prokaryotes. This difference may be related to differences in the sizes of the S. cerevisiae and N. crassa genomes, ∼13.5 Mb and 43 Mb, respectively. Estimates of the total number of genes in N. crassa vary considerably (Kupfer et al. 1997; Nelson et al. 1997), but most estimates indicate that N. crassa has at least 50% more genes than S. cerevisiae. Our results bear on several of the possible mechanisms by which such differences might have arisen.
Gene loss in S. cerevisiae appears to have had an important functional impact, but the proportion of N. crassa discontigs corresponding to genes lost from S. cerevisiae that were identified by our analyses (46 out of 396 for which clear homologs were detected in the nonfungal or EST databases; Fig. 2; Table 4) cannot account for the magnitude of differences in gene number between N. crassa and S. cerevisiae. Furthermore, loss of genes fromS. cerevisiae does not inherently explain the relatively high proportion of orphan genes in N. crassa.
The results of various evolutionary and genomic analyses have led to contrasting views regarding the impact of horizontal gene transfer during evolution (Gogarten et al. 1996; Doolittle 1998; Woese 1998;Snel et al. 1999). At least some groups have proposed that it has played an important role in the evolution of eukaryotic genomes in general (Doolittle 1998) and fungal genomes in particular (Prade et al. 1997). Our analyses did reveal several possible cases of horizontal gene transfer from prokaryotes (Table 5), and many of the candidates for horizontal gene transfer do correspond to “operational” genes encoding enzymes involved in modular metabolic functions, as suggested by previous analyses (Rivera et al. 1998; Jain et al. 1999). However, even if all of the candidates for horizontal transfer identified by this study reflect authentic cases (13 out of 1197 discontigs analyzed), <2% of N. crassa genes are plausibly derived from the incorporation of prokaryotic genes subsequent to divergence of the N. crassa and S. cerevisiae lineages.
It has been suggested that many fungal proteins exhibit a higher rate of molecular evolution than do homologous vertebrate proteins (Feng et al. 1997; Stassen et al. 1997). A similar difference in rate of evolution between N. crassa and S. cerevisiae could potentially explain the higher proportion of orphan genes in the former relative to the latter. However, our results (Fig. 1) show that there is not a global difference in rate between the two fungi.
Implications of Genetic Innovation in N. crassa
If there has been substantial genetic innovation in the N. crassa lineage, it is reasonable to speculate that many of the complex developmental pathways exhibited by N. crassa are mediated by novel protein-coding genes. One class of functionally characterized orphan genes identified in our earlier analysis ofN. crassa ESTs corresponds to clock controlled genes regulated in response to light and circadian rhythms (Nelson et al. 1997). This is a well-characterized developmental pathway in N. crassa(Loros 1998) that is absent from S. cerevisiae. The current study identified an additional N. crassa gene (the NPH1 homolog; see Table 4) possibly involved in responses to light, as did additional analyses of N. crassa ESTs (nop-1; seeBieszke et al. 1999). However, some of the pathways that distinguishN. crassa from S. cerevisiae are found not only in filamentous ascomycetes related to N. crassa but also in other (nonascomycete) filamentous fungal lineages. Because these latter fungi are less closely related to N. crassa than is S. cerevisiae, a hypothesis of genetic innovation in N. crassa for these genes would require either convergent evolution or horizontal transfer between N. crassa and the nonascomycete filamentous fungi.
Furthermore, the mechanism by which N. crassa could have gained large numbers of genes is unclear. If the impact of horizontal transfer on the N. crassa genome has been relatively modest as our results suggest (see above), then more extensive genetic innovation would reflect either the duplication and divergence of genes (e.g.,Tatusov et al. 1997) or overprinting [the generation of novel genes from noncoding sequences, as proposed by Keese and Gibbs (1992) andOhno (1984)]. Gene duplication, long thought to be the primary mechanism responsible for the generation of novel genes (Ohno 1970;Kimura and Ohta 1974), does not explain our inability to identify homologs of any kind for most of the N. crassa transcripts analyzed. Furthermore, there are few large gene families in N. crassa (Nelson et al. 1997). This may be due to the fact that closely related sequences in the N. crassa genome are actively mutated by the RIP (Repeat InducedPoint mutation) process (Selker 1990). Finally, although the high proportion of orphan genes could be explained by extensive overprinting, because genes derived in this way would truly lack homologs, the source of the requisite unexpressed ORFs remains obscure (but for potential sources, see Ohno 1984; Keese and Gibbs 1992).
An alternative possibility is that many cases of gene loss in S. cerevisiae could not be detected by our methods. Such cases might be drawn from two sources. Some could reflect novel genes introduced into the early fungi and subsequently lost from S. cerevisiae. We would have been unable to detect loss of such genes by our methods because they lack nonfungal orthologs and the number of fungal sequences is still limited. Such a pattern would also explain the high proportion of orphan genes in N. crassa. The greater developmental complexity of N. crassa would reflect retention of phenotypes ancestral to the fungi and the genes necessary for the expression of those phenotypes. This would be consistent with phylogenetic analyses indicating that the unicellular yeasts evolved from multicellular ancestors (Bruns et al. 1992; Berbee and Taylor 1993; Liu et al. 1999), and it would explain the relative paucity of fungal-specific genes identified by this study. If this hypothesis is correct, it should be revealed in future genome projects with diverse fungi, with the result that genes currently unique to N. crassa and its close relatives will be found in more distantly related fungal lineages. However, the relatively low proportion ofN. crassa sequences with clear A. nidulans homologs suggests that few homologs for orphan sequences in N. crassa will be identified in distantly related fungi, unless it is possible to substantially increase the sensitivity of the methods used for database searches.
A second possible source of genes whose loss from S. cerevisiae could not be detected by the methods applied here would be genes that were inherited from the common ancestor of the eukaryotes but had limited functional importance and thus were under weak selective pressure. Such genes might both be disproportionately lost from S. cerevisiae and have a rate of divergence in N. crassa high enough to preclude detection of nonfungal homologs. It has been suggested previously that orphan genes reflect a class of rapidly evolving genes, based on the identification of a large number of such genes in Drosophila (Schmid and Tautz 1997) and the budding yeasts (Ozier-Kalogeropoulos et al. 1998). Significantly fewer phenotypically identified genes are found among the rapidly evolvingDrosophila genes, suggesting that the latter are more likely to have relatively modest and difficult to detect phenotypes and that the rapid evolution of these proteins reflects weak purifying selection (see Kimura and Ohta 1974). Disproportionate loss of such genes is plausible, as suggested by Braun et al. (1998). We found support for the notion that genes that have been lost (or underwent excessive divergence) in S. cerevisiae are under weaker selection, because the N. crassa discontigs with a clear homolog in the nonfungal data sets (E ≤ 10−5) that also have clear homologs in SC are generally more highly conserved (median nonfungal E = 8 × 10−22,n = 315) than those that lack clear homologs in SC (median nonfungal E = 2 × 10−10, n = 81).
Implications of Gene Loss in S. cerevisiae
Patterns of gene evolution may provide functional information about the genes identified using genome sequence data (Rivera et al. 1998;Pellegrini et al. 1999). Examination of the genes that appear to have been lost or are highly divergent in S. cerevisiae reveals a surprising number of genes involved in basic cellular processes. Presumably, these changes have had an impact on the biology of S. cerevisiae. This may be true even in cases in which a paralog of a lost gene remains in the S. cerevisiae genome, such as the shaker K+ channel identified by this study (Table 4). The shaker K+ channel homolog present in S. cerevisiae (YPL088w) shows greater similarity to a proteobacterial oxidoreductase than to eukaryotic K+ channels (data not shown), suggesting that YPL088w encodes an oxidoreductase unlikely to provide a biological activity that compensates for the absence of a shaker K+ channel.
Global changes in the ion homeostasis systems in S. cerevisiaeare strongly suggested by our analyses. One gene previously demonstrated to have been lost in S. cerevisiae encodes the Ca2+-binding protein annexin (Braun et al. 1998). Three of the four putative divergent orthologs in S. cerevisiae that were identified by this study are most closely related to the Ca2+-binding proteins calmodulin, calnexin, and ALG-2. Strikingly, there is evidence for functional divergence for two of the divergent S. cerevisiae genes (Geiser et al. 1991; Moser et al. 1995; Parlati et al. 1995). These data suggest that multipleS. cerevisiae Ca2+-binding proteins that localize to different subcellular compartments have undergone functional divergence from homologous proteins in other organisms and that this divergence occurred after the divergence of S. cerevisiae from other well-studied fungi, such as N. crassa, A. nidulans, and S. pombe.
It is believed that S. cerevisiae underwent a complete genome duplication after its divergence from K. lactis (Wolfe and Shields 1997) and that most duplicated sequences were subsequently lost (Keogh et al. 1998). One might suppose that the instances of gene loss revealed here occurred during this same period. However, the identification of so few C. albicans homologs (30% of the genes in Table 4) given that the C. albicans genomic sequence is >90% complete (see Methods) strongly suggests that some gene loss also occurred prior to the divergence between C. albicansand S. cerevisiae. Furthermore, inspection of searches involving K. lactis sequences (Ozier-Kalogeropoulos et al. 1998) and comparison with the results presented in this paper suggests that loss of genes from the S. cerevisiae lineage occurred both before and after its divergence from K. lactis (data not shown). Thus, it is likely that some level of gene loss has occurred at many stages during the evolution of S. cerevisiae and, presumably, other fungal lineages as well.
Coverage of the Nonfungal Database and the Mammalian EST Database
To understand the significance of the high proportion of N. crassa genes that are currently orphans, we must consider the completeness of the nonfungal databases. We found that nearly allN. crassa discontigs that had eukaryotic homologs in the NF database also had homologs among the mammalian ESTs (Fig. 3; Tables 2and 7). Likewise, few N. crassa discontigs have homologs in the human and mouse EST data set but not in NF. These results imply that incompleteness of the public sequence databases is not a major factor in the high proportion of N. crassa discontigs that lack nonfungal homologs and also that the sampling of conserved gene families is fairly complete in both the EST and non-EST sequence databases. That is, additional sequencing will reveal few additional broadly distributed, conserved gene families. Green et al. (1993)proposed that there is a limited number of “Ancient Conserved Regions”; our results suggest that we are rapidly approaching a complete set.
Summary
Our analyses suggest that the differences in genome size and proportions of orphan genes between N. crassa and S. cerevisiae reflect some combination of genetic innovation in theN. crassa lineage and loss of genes from the S. cerevisiae lineage. There remain mysteries associated with either of these possible avenues of genome evolution: The mechanism of genetic innovation in the N. crassa lineage is presently unclear, whereas extensive loss from the S. cerevisiae lineage would require the disproportionate loss of genes that do not have recognizable nonfungal homologs. It may be that relative to S. cerevisiae, N. crassa retains many more uniquely fungal processes. The loss of specific, functionally important proteins during the evolution of S. cerevisiae that we have documented shows that surprising biological inferences can be made by the types of large-scale comparisons performed here (also see Pellegrini et al. 1999). Our ability to identify various patterns of genome evolution using single-pass sequence data demonstrates the utility of EST projects for evolutionary and comparative genomic investigations (Braun et al. 1998). However, the absence of complete genomic sequence forN. crassa does mean that some questions may only be asked in one direction; for instance, we could identify cases of probable gene loss from S. cerevisiae but not cases of loss from N. crassa. The growing availability of sequence data from the fungi should allow further exploration of the patterns of genome evolution identified by this study.
METHODS
Generation of N. crassa cDNA Sequences
Partial cDNA sequences (ESTs) were generated as part of theNeurospora Genome Project (NGP). Current information on the NGP is available from the project's Web page (http://www.unm.edu/∼ngp) or by contacting M.A.N. or D.O.N. The sequences analyzed in this paper were generated either as described (Nelson et al. 1997) or using the Thermosequenase dye terminator premix kit (Amersham) according to the manufacturer's recommendations. The directionally cloned cDNA libraries have been described previously (Nelson et al. 1997); some additional sequences reported here were obtained after highly expressed messages reported in that paper were identified by hybridization as described by Ausubel et al. (1994) and removed from the arrays of clones that were sequenced. A total of 3578N. crassa ESTs from 2202 clones were analyzed in this paper; 1313 ESTs were derived using the T3 sequencing primer (5′ reads), and 2265 ESTs were derived using the T7 primer (3′ reads). Quality control procedures have been presented previously (Nelson et al. 1997), and the error rates for this data set are comparable with those seen in other EST projects (including the S. cerevisiae ESTs described below).
Assembly and Clustering of N. crassa ESTs
ESTs were assembled with The Institute for Genomic Research (TIGR) assembler using defaults for EST assembly (Sutton et al. 1995), resulting in 2093 contigs. To further group contigs that reflect transcripts of the same locus, the contigs were assembled into 1197 discontigs (discontiguous-sequence clusters) using both single-linkage clustering of sequences with gapped BLAST-NE-values ≤ 10−25 and grouping of T3 and T7 reads based on shared clone names. Because of problems associated with EST sequencing projects, such as lane-tracking errors, record keeping errors, and the presence of chimeric clones, some discontigs will contain sequences representing the transcripts of more than one locus. Based on analysis of apparent chimeric patterns in search results (data not shown), we estimate between 60 and 100 improperly clustered discontigs, indicating that the EST data set represents the transcripts of 1250–1300 loci.
Public Data Sets
Computational analyses were performed on several sets of sequences obtained from public databases. Details of these data sets are given in Table 1. The C. albicans data set is probably fairly complete, because the CAL data set contains 14.9 Mb of genomic sequence, which is 93% of the 16-Mb C. albicans genome (Keogh et al. 1998). This is supported by the fact that 233 out of 240 (97%) of N. crassa discontigs with identified homologs in each of SC, NF, and HMEST also had homologs in the CAL data set. The A. nidulansdata set is composed primarily of ESTs, making estimation of coverage more difficult, but 168 (68%) of these same 240 discontigs have homologs in ENI, suggesting that ENI may be 60%–70% complete.
Homology Searches
Homology searches were carried out with the gapped BLAST programs (Altschul et al. 1997), using executable copies obtained from the NCBI (v.2.0.5). Searches were performed as comparisons of protein sequences, with translation of nucleotide query or database sequences as necessary (Blast-P, Blast-X, TBlast-X). Nucleotide queries were preprocessed with NSEG to mask low-complexity regions, and protein query sequences (including six-frame translations of ESTs) were filtered with SEG (Wootton and Federhen 1996). Unix scripts and C programs were used to automate searches on large sets of query sequences and to extract summary information (e.g., identity and E-value of best hit).
Queries were considered to have a clear homolog forE-values ≤ 10−5. A discontig was considered to have a clear homolog if any of the constituent contigs had a clear homolog. This cutoff gives a probability of including a single false hit (type I error) for the entire set of N. crassa queries of <5%, based on Bonferroni correction for multiple comparisons. Queries were considered to have a possible homolog in a database for E-values ≤ 0.01; this weaker cutoff will result in a moderate number of false database matches but should increase sensitivity. Queries were considered to have no potential homologs in a database for E-values > 0.1, because any homologous sequences this divergent are beyond the commonly recognized “twilight zone” of evolutionary similarity (e.g., seeMushegian and Koonin 1996; Koonin et al. 1997).
We used homology searches to differentiate between orthology and paralogy (Fitch 1970) whenever possible. Homologous proteins were considered to be probable orthologs if comparisons between the N. crassa sequence, the best hit in the S. cerevisiae data set, and the best hit in the nonfungal data set form a symmetrical set, as described by Tatusov et al. (1997). We considered N. crassagenes to be candidates for genes resulting from horizontal transfer after divergence from S. cerevisiae if their best nonfungal hit was prokaryotic and they had no hit in the S. cerevisiaedata set or in other fungi that would suggest that the gene was present in the common ancestor of N. crassa and S. cerevisiae. For this analysis, we assumed the fungal phylogeny ofBruns et al. (1992), whose relevant features were confirmed by Liu et al. (1999).
Comparison of Divergence (Molecular Clock Analyses)
The N. crassa contigs described in this paper and a set of full-length N. crassa protein sequences obtained from the NCBI were searched against the SC and NF databases. Sequences with BLAST hits of E ≤ 10−5 against both SC and NF were identified and subjected to further analysis. Random subsets of full-length N. crassa protein sequences passing these criteria were chosen and paired with their best matches from SC. For pairs composed of an N. crassa contig, which was generally not full length, and a S. cerevisiae cDNA sequence, portions of both sequences that were not part of the region of overlap indicated by BLAST were removed, to ensure that the paired queries were comparable. The two members of each of the resulting pairs were searched against NF. Pairs for which the closest homologs in NF for either the N. crassa or S. cerevisiae sequence were clearly paralogs rather than orthologs (see above) were eliminated.
Acknowledgments
We are grateful to M.P. Skupski (National Center for Genome Resources) for providing special purpose data sets, to S. Kang and the students associated with the Neurospora Genome Project for expert technical assistance, and to audiences at the University of New Mexico, Los Alamos National Laboratories, the Ohio State University, the University of Washington, EMBL Heidelberg, and the Laboratory of Molecular Systematics at the Smithsonian Institution for insightful comments. We are grateful to the Albuquerque High Performance Computing Center (AHPCC) for computers and computational support and S. Blea for programming assistance. NGP sequencing was supported by UNM and NSF grant HRD-9550649 to D.O.N., M.A.N., M. Werner-Washburne, and R. Miller. A.L.H. was supported by NIH grant 5P20-RR11830-02 and the AHPCC.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 These authors contributed equally to this paper and should be considered cofirst authors.
-
↵5 Present address: Department of Plant Biology, The Ohio State University, Columbus, Ohio 43210 USA.
-
↵6 Corresponding author. Present address: Celera Genomics, Rockville Maryland 20850 USA.
-
E-MAIL aaron.halpern{at}celera.com; FAX (240) 453-3324.
-
- Received September 22, 1999.
- Accepted February 10, 2000.
- Cold Spring Harbor Laboratory Press








