
A Genetic Linkage Map for Zebrafish: Comparative Analysis and Localization of Genes and Expressed Sequences
Abstract
Genetic screens in zebrafish (Danio rerio) have isolated mutations in hundreds of genes with essential functions. To facilitate the identification of candidate genes for these mutations, we have genetically mapped 104 genes and expressed sequence tags by scoring single-strand conformational polymorphisms in a panel of haploid siblings. To integrate this map with existing genetic maps, we also scored 275 previously mapped genes, microsatellites, and sequence-tagged sites in the same haploid panel. Systematic phylogenetic analysis defined likely mammalian orthologs of mapped zebrafish genes, and comparison of map positions in zebrafish and mammals identified significant conservation of synteny. This comparative analysis also identified pairs of zebrafish genes that appear to be orthologous to single mammalian genes, suggesting that these genes arose in a genome duplication that occurred in the teleost lineage after the divergence of fish and mammal ancestors. This comparative map analysis will be useful in predicting the locations of zebrafish genes from mammalian gene maps and in understanding the evolution of the vertebrate genome.
A powerful combination of genetics and embryology has established the zebrafish (Danio rerio) as an important model organism for the analysis of vertebrate development, physiology, and behavior. Genetic screens have identified mutations in >600 genes with essential functions in the embryo (Driever et al. 1996; Haffter et al. 1996). The transparency and external development of the embryo allow exquisite manipulations, such as dye-labeling, transplantation, and in vivo time-lapse imaging, that illuminate the function of mutated genes at the cellular level (Kimmel 1989; Schier and Talbot 1998). Moreover, molecular analysis of zebrafish mutations has revealed new genes and gene functions (Postlethwait and Talbot 1997; Schier and Talbot 1998). This emphasizes both the potential of the system and the need to develop strategies and infrastructures that will facilitate the cloning of genes defined by zebrafish mutations.
Three approaches have been used to clone mutant loci in zebrafish: insertional mutagenesis, positional cloning, and the candidate approach (Postlethwait and Talbot 1997; Beier 1998). The candidate approach has been used most widely, and >10 loci have been cloned as candidates identified by criteria such as expression pattern and map position (e.g., Schulte-Merker et al. 1994; Talbot et al. 1995; Brand et al. 1996). Zebrafish cDNA sequencing projects will enhance the candidate approach by generating expressed sequence tags (ESTs) that correspond to many new genes. Expression analysis and mapping can then assess these genes as candidates for mutations. As more genes are localized, mapping a mutation becomes an effective method to test many candidates in parallel. Thus construction of gene maps for zebrafish will accelerate the molecular analysis of mutations by providing a large pool of candidates that can be efficiently evaluated with straightforward mapping experiments.
A recent study reporting the map locations of 144 zebrafish genes provided a basis for comparing the genomes of zebrafish and mammals (Postlethwait et al. 1998). Analysis of zebrafish and mammalian gene maps revealed extensive conservation of synteny—genes that are on the same chromosome (syntenic) in zebrafish tend to have syntenic orthologs in mouse and human. This finding raised the possibility that comparative analysis may predict the positions of zebrafish genes from mammalian gene maps. Mapping of additional genes will enhance comparative mapping by (1) identifying the locations and borders of segments of conserved synteny, and (2) determining the extent to which gene order is maintained within a conserved syntenic segment.
We have determined 104 new map positions of zebrafish genes by scoring single-strand conformational polymorphisms (SSCPs; Brady et al. 1997;Förnzler et al. 1998) in a haploid mapping panel. To allow straightforward comparison between our map and those produced with other crosses (e.g., mutant mapping crosses), we also scored 53 previously mapped genes (Postlethwait et al. 1998) and 217 simple sequence length polymorphism (SSLP; Knapik et al. 1998) markers in our mapping panel. These map positions define new candidates for mutations, and phylogenetic analysis of mapped genes defines new regions of conservation between zebrafish and human.
RESULTS
Map Construction
To construct a zebrafish linkage map, we scored a total of 390 PCR-based genetic polymorphisms in a mapping panel comprised of 48 individual haploid progeny of a Tü × TL female. These polymorphisms included 105 SSCPs from fully sequenced cDNAs and ESTs (of which 104 were mapped, see below) that defined previously unmapped genes (Fig. 1; Table1). We also developed SSCPs to map 10 sequence-tagged sites (STSs) derived from genomic clones in BAC, PAC, and YAC vectors and other sequences (Table2). To allow comparison between our map and others constructed with different crosses, we analyzed 275 previously mapped markers in our mapping panel. Of these markers, 217 were SSLPs (Knapik et al. 1998), 53 were SSCPs linked to previously mapped genes (Table1), and 5 were STSs associated with cloned random amplified polymorphic DNAs (RAPDs) (Table 2).

Examples of polymorphisms scored in the 48 individual haploid siblings (lanes 1–48) that comprise the mapping panel. (A) SSCP derived from EST AA494741. (B) SSLP marker z1273 (Knapik et al. 1998).
Genes and ESTs Mapped by SSCP
Mapped STSs
By linkage analysis and reference to previous maps (Knapik et al. 1998;Postlethwait et al. 1998), we ordered 389 of the 390 polymorphisms into a map with 25 linkage groups, each representing one zebrafish chromosome (Fig. 2). The map contains 25 gaps whose supporting lod score is <3, which corresponds to a gap of 23 cM or greater for a mapping panel of 48 haploid individuals. All of these large gaps were flanked by previously mapped markers, allowing the positions and approximate sizes of the gaps to be determined from previous maps. The 389 markers occupied a total of 273 unique map positions, or bins (average of 1.42 marker/bin), where markers in each bin are separated from markers in other bins by at least one crossover. The linkage groups in the map encompassed 2894 cM, using the Kosambi mapping function to estimate the number of double-crossovers (2640 cM with no mapping function). This corresponds to 99% coverage of the genome, using 2900 cM as the total length of the female meiotic linkage map (Postlethwait et al. 1994; Johnson et al. 1996). Accordingly, we were able to assign map positions to 114 of the 115 new polymorphisms we developed, as these showed significant linkage (lod > 3.0) to a previously mapped gene or SSLP marker.
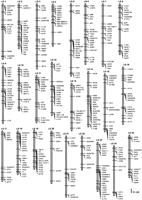
Genetic linkage map of the zebrafish genome. Shown are the positions of 389 markers genotyped on a single mapping panel consisting of 48 haploid zebrafish embryos. The map includes 157 gene and EST markers, 15 STS markers, and 217 SSLP markers. The SSLPs, which are designated as “z” and “gof” markers, are described in Knapik et al. (1998)and Goff et al. (1992), respectively. ESTs with mammalian orthologs are named by their human counterparts. ESTs with no clear orthology are named by their GenBank accession nos. Primer sequences and GenBank accession nos. for mapped genes, ESTs, and STSs are shown in Tables 1and 2. Linkage group nomenclature and orientation follows Postlethwait et al. (1998).
Error Analysis
As one method to assess the error frequency in our data set, we identified double crossovers in short intervals, which occur rarely and are thus likely to represent mis-scored genotypes. Among 18,230 individual genotype assays (an average of 46.9 scorable individuals/locus for the 389 mapped loci), there were only two double crossovers in intervals of 20 cM or less, suggesting the error rate in the data set is <0.1%. There were 20 additional double crossovers in regions >20 cM, an interval large enough to contain bona fide double crossovers. Because each crossover in the data set adds ∼2.1 cM to the length of the map, the 22 total double crossovers have only a modest effect on the length of the map, expanding it by ∼92 cM.
Another measure of reliability derives from a comparison of map assignments for markers common to our map and previous ones. There is very good overall agreement in the marker order shown in Figure 2 and orders reported previously by Postlethwait et al. (1998) and Knapik et al. (1998). For example, 21 of 25 linkage groups show perfect agreement in the order of 214 SSLPs and 50 genes. For two linkage groups (LG 6 and LG 17), two pairs of markers (z1138/z3581 and otx1/pax9, respectively) are inverted with respect to their published positions. These are pairs of tightly linked markers whose assigned positions could be altered by one or a few genotyping errors in one of the data sets. Alternatively, these discrepancies could reflect differences in the strains used for the various maps. The other descrepancies were markers z3054 (LG 1) and wnt4 (LG 11), which were assigned to different positions (LG 10 and LG 15, respectively) in previous maps. We confirmed that wnt4 mapped to LG 11 in our cross by scoring polymorphisms amplified with a total of three independent primer pairs.
Whereas there was good overall agreement of marker order, there were clear differences in distances between our map and others, particularly the microsatellite map (Knapik et al. 1998). This can be seen in total length (2894 vs. 2350 cM) and most strikingly in particular regions. For example, two markers on LG 20, z3211 and z20046, are separated by 39 cM in our map but only 10 cM in the microsatellite map (Knapik et al. 1998). Differences of this magnitude reflect more than statistical variation, because the standard error for markers separated by 10 cM in our map is 4.2 cM and the 95% confidence interval is 2.9–21.3 cM. Sex-based differences in meiotic recombination could be a factor, as the microsatellite map was sex-averaged (Knapik et al. 1998) whereas our haploid map monitors only female meiosis. Whatever the cause of these differences, it is clear that one must be cautious in using marker distances in one cross to infer distances in others, although marker orders are readily comparable between maps.
Comparative Analysis
Previous work identified regions of synteny conserved between zebrafish and mammals (Postlethwait and Talbot 1997; Postlethwait et al. 1998). To investigate conservation of synteny in light of the additional mapped genes, we examined locations of zebrafish genes and their counterparts in mouse and human. Using the BLAST family of search programs (Altschul et al. 1997), we identified mammalian sequences significantly similar to zebrafish genes for which sequence and map location were available from this and previous studies (see Methods for description of search criteria). To determine which homologous sequences are likely to represent mammalian orthologs of zebrafish genes, we constructed phylogenetic trees using the CLUSTALX program (Thompson et al. 1997). Because they are descended from the same gene in the last common ancestor of two species, orthologs share a terminal branch of a phylogenetic tree. For example, in the tree shown in Figure3A, human cadherin 11 is more closely related to zebrafish ventral neural cadherin (vn-cad; Franklin and Sargent 1996) than to any other known human sequence. Thus, human cadherin 11 is the likely ortholog of zebrafish vn-cad. Phylogenetic analysis identified 134 human and 152 mouse genes that are probable orthologs of mapped zebrafish genes (Tables 1 and3). There were 18 cases, such as that shown in Figure3B, in which two zebrafish genes appeared to be orthologous to a single mammalian gene (Table 4; see also Stock et al. 1996; Pfeffer et al. 1998; Sefton et al. 1998; references corresponding to GenBank accession nos. in Table 4). Conversely, we noted three cases in which two mammalian genes appeared to be orthologous to a single zebrafish gene. Some of these cases may be resolved with the discovery of additional genes in zebrafish or mammals, but many are likely to represent true cases of extra genes, as we consider further in the Discussion.
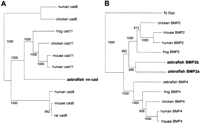
Phylogenetic analysis of zebrafish protein sequences for (A) vn–cadherin and (B) BMP2a and BMP2b. CLUSTALX was used to display the relationships of zebrafish gene products and their homologs in other organisms, as described in the text. Numbers at nodes represent bootstrap support for 1000 replications. NCBI protein sequence ID numbers for the proteins shown are human cad6 (1705545), chicken cad6 (2134302), frog cad11 (3377485), chicken cad11 (3511021), mouse cad11 (1705549), human cad11 (1377894), zebrafish vn-cad (1345125), human cad8 (1705547), mouse cad8 (3023433), rat cad8 (2804294), fly Dpp (118409), chicken BMP2 (2501173), mouse BMP2 (1345611), human BMP2 (115068), frog BMP2 (115070), zebrafish BMP2b (2804175), zebrafish BMP2a (2149148), zebrafish BMP4 (2804177), frog BMP4 (399122, chicken BMP4 (2501175), human BMP4 (115073), and mouse BMP4 (461633).
Orthologs of Other Mapped Zebrafish Genes
Mammalian Genes with Two Apparent Zebrafish Orthologs
By comparing the map locations of orthologous genes, we identified syntenies conserved between zebrafish and mammals (Table 5; Fig.4). Each point in the Oxford grid shown in Figure 4A represents a pair of orthologous genes plotted by map location in zebrafish and human. Bins occupied by more than one point indicate cases in which orthologous genes are syntenic in both species. Of 124 genes mapped in both zebrafish and human, 79 genes are located in a bin containing two or more points, defining a total of 28 conserved syntenic groups. The observed clustering is greater than that predicted by a random Poisson distribution (Fig. 4A; see Methods for calculation). This confirms and extends the observation (Postlethwait et al. 1998) that syntenic genes in zebrafish tend to have orthologs that are on the same chromosome in human. Figure 4B shows a similar analysis of synteny conserved between zebrafish and mouse. This zebrafish–mouse comparison identified 28 conserved syntenic groups involving 73 of 135 genes examined. Figure 4C shows a human–mouse Oxford grid compiled from the same set of genes used for the comparisons with zebrafish in Figures 4A and 4B. Seventy-seven of the 103 genes mapped in both human and mouse fall into 21 conserved syntenic groups.
Summary of Zebrafish–Human Conserved Syntenies
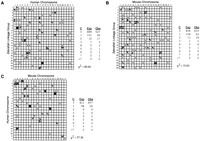
Conservation of synteny among zebrafish, human, and mouse. Each point in the Oxford grids represents an othologous gene pair plotted by map position in the two organisms compared. Conserved syntenies are evident as bins containing more than one point. (A) Comparison between zebrafish and human. (B) Comparison between zebrafish and mouse. (C) Comparison between human and mouse. Orthology relationships and map positions used to construct the Oxford grids are shown in Tables 1 and 3, except for the zebrafish Hox clusters whose orthology and position are from Amores et al. (1998). For tandem clusters of Hox, globin, and MHC genes, each cluster is represented by a single point in the Oxford grid and in the Poisson calculations. Tables associated with each grid evaluate the statistical significance of conserved syntenies. (C) Classes of bins according to the number of genes that occupy the bin; (Exp) the expected number of occurrences of bins in each class (see Methods for Poisson calculation); (Obs) the actual number we observed. To calculate the χ2 value, the Exp and Obs were assigned to three categories, corresponding to classes of bins with 0, 1, or >1 gene occupying the bin. For 1 degree of freedom, a χ2 > 6.64 implies a significant difference at a level P < 0.01.
DISCUSSION
We have localized 104 zebrafish genes and ESTs within a framework map derived from 275 previously mapped genes, STSs, and SSLPs. This work increases the total number of mapped zebrafish genes to >250, representing a majority of zebrafish genes for which full-length cDNA sequences are available. These genes can now be efficiently tested as candidates for mutations. Furthermore, the localization of these genes advances the comparison of the zebrafish genome with those of other vertebrates.
Mapping the Correspondence Between Zebrafish and Human Genomes
By comparing the map locations of zebrafish genes and their mammalian counterparts, Postlethwait et al. (1998) discovered that syntenic genes in zebrafish tend to be syntenic in mammals. We have further analyzed the conservation between zebrafish and mammalian genomes, considering the map locations reported here and in previous work. With the increase in gene number, we have identified eight new syntenic groups conserved between zebrafish and human and added from one to three genes to each of six previously recognized groups. The human–mouse comparison (Fig. 4C) compiled with the same genes used for the zebrafish analysis suggests that conservation between human and mouse extends over larger intervals than between human and zebrafish: The average human–mouse segment contains more genes (3.7) than the average human–zebrafish segment (2.8). Additional mapping experiments are needed to better evaluate parameters important for comparative mapping, such as the size of the average segment and the extent to which gene order is maintained within conserved segments.
Despite the current uncertainties surrounding these parameters, the recent molecular analysis of the you-too (yot) locus illustrates the utility of comparative mapping (Karlstrom et al. 1999). Genetic mapping localized the yot locus to LG 9, which is homologous to human chromosome 2 (Fig. 4A; Postlethwait and Talbot 1997; Postlethwait et al. 1998). This, together with phenotypic analysis of yot mutants, suggested the human chromosome 2 genegli2 as a candidate for yot. As predicted by the zebrafish–human comparison, the zebrafish ortholog of gli2mapped to LG 9 and was found to be disrupted in yot mutants (Karlstrom et al. 1999). This example suggests that comparative mapping will be useful in identifying candidate genes for other zebrafish mutations.
Genome Duplications and Vertebrate Evolution
The important role of genome duplication in the evolution of the vertebrates was recognized by Ohno (1970), who postulated that divergence of duplicated genes resulting from tetraploidization events drives the emergence of new gene functions. At least two genome duplications occurred early in the evolution of vertebrates (for review, see Holland and Garcia-Fernàndez 1996; Sidow 1996). Analysis of chordate genomes reveals a fourfold increase in the number of genes and members of gene families in mammals relative to the cephalochordate amphioxus. For example, tetrapods have four paralogous Hox complexes whereas amphioxus has only one (excluding theParaHox complex, which is thought to have arisen very early in the evolution of animals; Brooke et al. 1998). These paralogous genes derived from genome duplications (rather than multiple independent tandem duplications), because mapping studies show that paralogous genes often reside in duplicated chromosomal segments (Morizot 1990;Lundin 1993; Amores et al. 1998; Postlethwait et al. 1998).
Examination of teleost genomes has provided insight into the timing of these duplications. Mammalian genes and chromosomal segments have identifiable orthologs in zebrafish and other teleosts, indicating that these genes were formed by duplications predating the divergence of ray- and lobe-finned fishes, the lineages leading to extant teleosts and tetrapods, respectively (Morizot 1990; Holland and Garcia-Fernàndez 1996; Sidow 1996; Postlethwait et al. 1998). Unexpectedly, these studies have also revealed that teleost gene families often contain more members than the corresponding families in tetrapods (for review, see Wittbrodt et al. 1998). For example, zebrafish have extra genes in the hox, dlx, msx, engrailed, hedgehog, pax, otx, and bmp gene families (Ekker et al. 1992, 1995,1997; Mori et al. 1994; Stock et al. 1996; Martinez-Barbera et al. 1997; Amores et al. 1998; Pfeffer et al. 1998; Force et al. 1999).
Mapping experiments provide insight into the origins of these extra genes in teleosts. Because extra zebrafish genes are dispersed throughout the genome, rather than clustered with related genes, it seems that zebrafish gene families were not expanded by widespread tandem duplication (Postlethwait et al. 1998; this paper). Instead, there are some cases in which syntenic zebrafish genes have extra paralogs that are also syntenic. For example, bmp2a andsnap25b are located on LG 17, and the corresponding extra genes, bmp2b and snap25a, are both located on LG 20. This supports the view (Postlethwait et al. 1998; Wittbrodt et al. 1998) that these extra genes resulted from a genome duplication. Future mapping studies will address this issue further, as more zebrafish genes in families with expanded membership are localized.
If extra genes in zebrafish are the products of a genome duplication, when did this occur? One possibility is that three genome duplications occurred prior to the divergence of ray- and lobe-finned fishes. According to this model, zebrafish genes outnumber their mammalian counterparts because duplicate genes were lost at a higher frequency in the tetrapod lineage. Alternatively, an extra genome duplication occurred in the fish lineage after it diverged from the tetrapod lineage. Phylogenetic analysis can distinguish between these possibilities, as shown in Figure 5. The products of a duplication after divergence would be equally related to a single element (i.e., a gene or chromosomal segment) in the nonduplicated lineage (Fig. 5A). In contrast, if a shared genome duplication was followed by differential loss of paralogs, then one of the extra paralogs in the high-retention lineage would be more closely related to the surviving paralog in the other lineage (Fig. 5B). Our phylogenetic analysis of expanded zebrafish gene families identified 11 clear cases of mammalian genes equally related to two zebrafish genes (the relationship schematized in Fig. 5A) and only 4 cases in which one of the extra zebrafish genes appeared to be more closely related to the mammalian gene. Thus our comparative analysis of the zebrafish gene map supports the view based on detailed analysis of zebrafish hoxclusters (Amores et al. 1998) that the lineage leading to zebrafish underwent a genome duplication after the divergence of ray- and lobe-finned fishes.
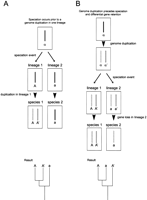
Two models explaining the origin of extra paralogs in one of two related species. (A) If a genome duplication occurs in lineage 1 after its divergence from lineage 2, then the resulting paralogous genomic elements (genes or chromosomes) A and A′ of an extant species from lineage 1 are more closely related to each other than they are to their ortholog a in an extant species from lineage 2. This relationship is evident in the resulting phylogenetic tree, diagramed at the bottom. (B) If a genome duplication produces paralogous genomic elements α and α′, then a subsequent speciation event will produce lineage 1 with paralogs A andA′ and lineage 2 with paralogs a anda′. A and a are orthologs, as they are both directly descended from α, and A′ anda′, descended from α′, are also orthologs. Lineage-specific loss of paralogs (e.g. a′ in lineage 2) results in species 2 having fewer paralogs than species 1. The derivation of A in species 1 and a in species 2 from their common ancestor α is evident in the resulting phylogenetic tree, diagramed at the bottom.
It is likely that this extra genome duplication is not a recent evolutionary occurrence, and it may predate the radiation of teleosts. The genomes of other teleosts, including pufferfish (Fugu rubripes) and medaka (Oryzias latipes), also have extra genes (for review, see Wittbrodt et al. 1998), suggesting that the duplication preceded the last common ancestor of these species. In addition, the fact that zebrafish, medaka, and pufferfish are diploid species argues against a recent genome duplication, which would result in tetraploidy or pseudotetraploidy, as seen, for example, in the African clawed frog (Xenopus laevis) and rainbow trout (Onchorynchus mykiss), respectively (Bisbee et al. 1997;Wittbrodt et al. 1998; Young et al. 1998). Finally, duplicate genes can be expressed in dissimilar patterns, which suggests that the duplication producing these genes was followed by enough time for them to diverge and perhaps acquire different functions. For example, the zebrafish pax2.1 and pax2.2 genes are both apparently orthologous to mammalian pax2. The pax2.1 gene is expressed in the in the presumptive cerebellum earlier thanpax2.2 and only pax2.1 is detected in the developing pronephros (Pfeffer et al. 1998). Furthermore, mutational analysis shows that these expression differences reflect the distinct functions of these duplicate genes. Inactivation of pax2.1 leads to the loss of the cerebellum, indicating that the later expression ofpax2.2 is insufficient for normal development of the CNS (Brand et al. 1996). This and other recent examples (Kishimoto et al. 1997; Feldman et al. 1998; Schauerte et al. 1998) suggest that in many cases duplicate genes closely related in sequence have acquired distinct functions by virtue of their divergent expression patterns.
METHODS
DNA Preparation and PCR
The mapping panel was prepared from 48 haploid individuals obtained from a single female derived from a cross between a Tü strain male and a TL strain female. The Tü and TL strains were described by Haffter et al. (1996). To extract genomic DNA, each embryo was placed in 50 μl DNA preparation buffer [10 mm Tris-HCl (pH 8.3), 1.0 mm EDTA, 12.5 mm KCl, 0.3% Tween 20, 0.3% NP-40] in a microtiter plate well, heated to 98°C for 10 min, and incubated at 55°C overnight with proteinase K (1 mg/ml). The proteinase K was then inactivated by incubation at 98°C for 10 min. For a single PCR assay, DNA from 1/4000 of this DNA preparation was used as a template.
PCR amplification was performed in 12.5-μl reaction mixtures containing 10 mm Tris-HCl, 50 mm KCl, 1.5 mm MgCl2, 0.001% gelatin, 0.1 mg/ml BSA, 100 μm each dNTP, 1 μCi of [α-32P]dATP, 0.25 unit of Taq polymerase, and 100 nm each primer. Thermocycling was done under standard conditions consisting of an initial denaturation at 94°C for 2 min, followed by 45 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec, and a final incubation at 72°C for 5 min.
Gel Electrophoresis
SSLP PCR reactions were diluted 2:1 with loading buffer (80% formamide, 0.1% bromophenol blue, 0.1% xylene cyanol), denatured at 95°C for 5 min, and electrophoresed on a 6% denaturing gel (19:1 acrylamide/bis). SSLP gels were run at 60 W at room temperature for 2.5 to 3 hr, transferred to Whatman filter paper, and exposed to film at −80°C with an intensifying screen.
SSCP PCR reactions were diluted 4:1 with SSCP buffer (80% formamide, 10 mm NaOH, 0.1% bromophenol blue, 0.1% xylene cyanol) denatured at 98°C for 5 min, placed on ice for 5 min, and then immediately loaded on a 4.5% (39:1 acrylamide/bis) nondenaturing gel. SSCP gels were run at 40W at 4°C for 3 hr, transferred to Whatman filter paper, dried, and exposed to film at −80°C with an intensifying screen.
Primer Design
The D. rerio subset of DNA sequence from the nonredundant (NR) database at the National Center for Biotechnology Information (NCBI, Bethesda, MD) was downloaded using Nentrez network tools. TIGR Assembler (Sutton et al. 1995) software was used to cluster the genes and ESTs in this set into contiguous sequence fragments (contigs). Primers used to develop SSCPs were designed from these contigs using Primer 3.0.6 (Rozen and Skaletsky 1997) and synthesized at the Stanford DNA Sequencing and Technology Center as described previously (Lashkari et al. 1997). Primers for SSLP markers were obtained from Research Genetics (Huntsville, AL).
Phylogenetic Analysis
The NCBI NR database was used for searches. Each query sequence designated for ortholog assignment was aligned in a pairwise manner (Needleman and Wunsch 1970; zero end gap penalties, gonnet matrix, gap opening penalty 2.4, gap extension penalty 0.15) with every sequence in the database and the alignments ranked according to their ZEGA similarity scores (ZEGA probability; Abagyan and Batalov 1997). An all-against-all protein domain search of the SCOP database (Murzin et al. 1995) indicated that a ZEGA probability of 0.00001 would not eliminate homologs even in weak identity cases (not shown). This cutoff, or a total of 50 sequences for those cases in which many homologs existed in the database, was used to group the top scoring sequences from the database search. This group of sequences was then multiply aligned, and neighbor-joining trees incorporating evolutionary distances were extracted from the final alignment using the CLUSTALX program (Thompson et al. 1997). Trees were tested by analysis of 1000 bootstrap replicates. The resulting trees were examined to identify orthologs.
Data Collection and Analysis
Data were collected using Map Manager software (Manly 1993;http://mcbio.med.buffalo.edu/mapmgr.html), and analysis was conducted with Map Manager and Map Maker software (Lander et al. 1987). All loci, including published markers, were initially analyzed and ordered independently of published map positions. Each locus was initially placed at the position that maximized its lod score as reported by the “Links” command of Map Manager. Local order was then determined by manually placing the marker at the location that minimized the number of double recombinants. Data were then exported to Map Maker to confirm map order using the “ripple” and “try” features. Data analysis ordered 389 of the 390 loci into 50 multilocus groups that were supported by lod scores >3.0. The 50 groups were then assigned to the standard 25 linkage groups based upon information from previously published map locations. The 50 separate groups were positioned by choosing the order and orientation that maximized lod score and minimized the number of recombinants between the end loci of respective groups. Map graphics were created with Map Maker using the Kosambi mapping function.
For Figure 4, the expected number (Exp) of occurrences of bins of a given class were calculated according to a Poisson distribution: Exp = n{(e−λ)(λC)/C !}, where C = class according to number of genes occupying a bin, and n = number of bins. The parameter λ was estimated as λ = k/n, wherek = total number of data points. For zebrafish–human comparison, n = 600 and k = 124; for zebrafish–mouse comparison, n = 525 and k = 135; for human–mouse comparison, n = 504 and k = 103. The Exp values shown in Figure 4 were rounded to the nearest integer. These calculations make the simplifying assumption that all chromosomes in a given species are of equal length.
Error Analysis
Several steps were taken to monitor the quality of the data, particularly those generated for SSCP markers. The combination of the complex banding patterns possible in SSCP coupled with the occasional ability of some primer pairs to amplify multiple products led us to adopt strict criteria when considering a locus for analysis. Polymorphisms were scored only for the predominant banding pattern on the gel, and we ensured that there were no inconsistencies between the multiple bands that were assumed to represent a given SSCP allele. Finally, putative polymorphisms for which >6% of the individuals (3/48) were assigned different alleles in independent scorings were not further considered for mapping. Discrepant data points were left as unscored. The error rate of mapping is often correlated to the number of double crossovers (particularly in small regions) in the data set. In the first complete evaluation of the data set, there were 27 double recombinants, 7 of which occurred in regions of <20 cM. The genotype assays for all 27 points were repeated and independently rescored. We found that 5 of the 7 small-interval double recombinants were mis-scored in the orignal assays and confirmed the original genotypes of the other 22 double recombinants. Thus the final error analysis left two small-interval double recombinants present in the dataset.
Acknowledgments
We thank the members of the Talbot and Schier laboratories for helpful discussions; R. Burdine, B. Feldman, E. Heckscher, D. Kingsley, and J. Postlethwait for critical comments on the manuscript; and Ron Davis for encouragement and support. We acknowledge fellowship support from the National Institutes of Health (NIH) (H.I.S) and American Cancer Society (S.T.D.). W.S.T. is a Pew Scholar in Biomedical Science. This work was supported by NIH grants R01 RR12349 (W.S.T.) and R21 HG01704 (A.F.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Present address: Department of Developmental Biology, Stanford University School of Medicine, Stanford, California 94305 USA.
-
↵4 Corresponding author.
-
E-MAIL talbot{at}cmgm.stanford.edu; FAX (650) 725-7739.
-
- Received December 18, 1998.
- Accepted February 9, 1999.
- Cold Spring Harbor Laboratory Press








