
Mapping ESTs by Fiber–FISH
- Nina Horelli-Kuitunen1,6,
- Johanna Aaltonen2,
- Marie-Laure Yaspo3,
- Mervi Eeva1,
- Maija Wessman1,4,
- Leena Peltonen2,5, and
- Aarno Palotie1,5
- 1Departments of Clinical Chemistry and Biomedicine, University of Helsinki and Laboratory, Department of Helsinki University Central Hospital, 00290 Helsinki, Finland; 2National Public Health Institute, Department of Human Molecular Genetics, 00300 Helsinki, Finland and Haartman Institute, Department of Medical Genetics, University of Helsinki, 00014 Helsinki, Finland; 3 Max Planck Institute for Molecular Genetics, D-14195 Berlin, Germany; 4 Department of Biosciences, Division of Genetics, University of Helsinki, 00014 Helsinki, Finland
Abstract
A visual transcript map of six genes was constructed on the chromosome 21q22.3 by high resolution fluorescence in situ hybridization (FISH). Expressed sequence tags (ESTs) from six genes—PWP2, KNP1, AIRE, C21orf3,SMT3A, and C21orf1—were successfully localized by fiber–FISH by use of sensitive tyramide-based detection. The sizes of the ESTs varied between 315 to 956 bp and most of them map within the 3′-untranslated region. The ESTs were assigned to and subsequently ordered within cosmid, PAC, and BAC clones hybridized on DNA fibers. Physical distances between ESTs and known markers were determined. Our results demonstrate the feasibility and accuracy of visual mapping EST sequences in relation to known markers. The main advantage of this approach is that it can be applied to finely map any of the database ESTs for positional cloning efforts. The sensitivity, specificity, and reproducibility of this high-resolution EST mapping technique is evaluated.
Until the complete sequence of the human genome has been revealed, a gene map will have special value for identifying disease-causing genes. The databases of expressed sequence tags (ESTs) are an excellent source of coding sequences and subsequent genes at any genomic region. The EST sequences are short cDNA fragments and a gene may be represented by multiple ESTs that correspond to different parts of the gene. Today, >1,000,000 ESTs of human genes are represented in several databases of ESTs (May 1998;http://www.ncbi.nlm.nih.gov/dbEST/summary.htlm; Boguski and Schuler 1995; Hudson et al. 1995; Hillier et al. 1996; Schuler et al. 1996). These ESTs are mainly derived from the 3′-end sequences of genes. The dbEST database is now estimated to represent >50% of all human genes and >91% of positionally cloned genes mutated in human diseases (http://www.ncbi.nlm.nih.gov/Bassett/dbEST/Posiclon.New.htlm; Banfi et al. 1997).
Chromosome 21 has long been a target for intensive studies mainly because of its small size, its association with several genetic diseases, and its involvement in Down’s syndrome (Korenberg et al. 1997). Although chromosome 21 is small (∼50 Mb), neither all the genes nor the complete sequence are currently known. Several physical maps of chromosome 21 have been constructed, but some gaps still exist in the physical map (Chumakov et al. 1992; Stone et al. 1992; Aaltonen et al. 1997a; Lapenta et al. 1998). Chromosome 21 has proved to be difficult to clone. There are several possible reasons for this difficulty, including a high representation of GC-nucleotides and a high degree of repetitive sequences (Saccone et al. 1992; Gardiner et al. 1996; Lapenta et al. 1998).
Characterization of the complete genetic code requires sequence-ready clone contig construction across the whole genome. Typically, large insert clones such as cosmids, P1s, PACs, BACs, and YACs are utilized in contig construction (Feiss et al. 1982; Burke et al. 1987; Stenberg et al. 1990; Shizuya et al. 1992; Ioannou et al. 1994; Shepherd et al. 1994; Chumakov et al. 1995). This physical mapping approach with different sized clones also enables the assignment of critical landmarks on a restricted genome region. The FISH technology has developed significantly with both improved resolution and sensitivity (Raap et al. 1995; Gijlswijk et al. 1996; Heiskanen et al. 1996) and it has thus become a relevant tool for high-resolution physical mapping. At the same time, there has been increasing development of more sensitive microscopes and digital imaging equipment needed for image acquisition and analysis. FISH-based mapping is one of the few mapping procedures with which there are no requirements for different clones to overlap to be positioned and ordered. To date, physical maps of several disease-linked genomic regions have been constructed, which facilitate disease gene identification utilizing visual high-resolution FISH mapping (Heiskanen et al. 1995; Klockars et al. 1996, 1997;Leppänen et al. 1996; Aaltonen et al. 1997a; Laan et al. 1997;Nikali et al. 1997).
In this study we have combined the high-resolution FISH approach with a sensitive tyramide-based detection method to demonstrate the construction of an EST map of six different genes on the subtelomeric region of the chromosome 21q. A great necessity for such a map was faced during the positional cloning of the APECED disease gene, which has been recently identified (Aaltonen et al. 1997b;Nagamine et al. 1997). The ESTs from the PWP2, KNP1,AIRE, C21orf3, SMT3A, and C21orf1genes were selected corresponding to the 3′ UTR of these genes. The ESTs in this approach were short gene-specific DNA fragments that were amplified by PCR and used as probes for FISH. These small EST fragments from 315 to 956 bp in size were successfully localized in relation to known markers, enabling the construction of a transcript map.
RESULTS
For visual transcript map construction, 10 different ESTs specific to six different genes were mapped in the vicinity of markers D21S1460 and D21S1903 on chromosome 21q22.3 (Fig. 1). ESTs and corresponding cDNAs were hybridized in parallel with long-range clones of known positions on the basis of an earlier constructed clone contig across a 800-kb region on the APECED disease-linked genomic region (Aaltonen et al. 1997a). The contig of clones provided a framework for mapping the ESTs and enabled the assignment of these genes in precise locations in relation to known markers, for example, previously mapped long-range clones and the measurements of distances between these genes.
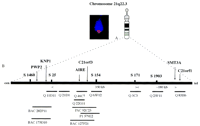
(A) A metaphase chromosome showing a specific hybridization signal of an amplified EST fragment detected by tyramide-based detection (left) and an idiogram of the chromosome 21 (right). (B) A schematic representation of the critical genome region including the markers D21S1460 and D21S1903 on the chromosome 21q22.3. The locations of the genes of interest are positioned according to the known markers and to the clone contig established earlier (Aaltonen et al. 1997a). Symbols above the long solid bar represent polymorphic markers. The short horizontal bars represent clones where Q stands for cosmid clones.
Sensitivity of Visual EST Mapping
To evaluate the sensitivity of hybridization of short DNA sequences in visual EST mapping, 13 clones (cDNAs and amplified PCR fragments) were used as probes for FISH. These included ESTs ranging from 315 to 956 bp in size and three cDNA clones from 1800 to 2700 bp. Metaphase FISH was used to ensure the correct chromosomal location of each EST. The hybridization frequency by tyramide-based detection was <15% with a reasonable signal-to-noise ratio. A specific hybridization signal was most often observed on only one chromatid—double dot signals were rarely observed (Fig. 1A).
The next phase was to hybridize the ESTs and the cDNA clones in parallel with long-range clones on free DNA fibers. The long-range clones provided a scaffold for EST assignment. The ESTs were successfully visualized on free DNA fibers with a hybridization frequency of 25%–45%. The frequency increased with the probe size (Fig. 2). Simultaneous visualization of two to three ESTs was observed with ∼15% hybridization frequency depending on the sizes of the ESTs in question. Thus, in practice, ∼20 images are needed to get reliable assignments if two ESTs are mapped in the same hybridization.
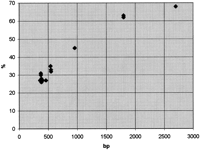
A demonstration of the hybridization frequency when amplified EST fragments are hybridized to free DNA fibers and visualized by sensitive tyramide-based detection method. The hybridization frequency (y) clearly increases along with the increased probe size (x). (♦) Hybridization frequency of one EST or cDNA clone (see also Table2).
Specificity of Visual EST Mapping
To evaluate the specificity and accuracy of the EST mapping, cDNAs for the corresponding genes were mapped by the fiber–FISH procedure. This was done to verify that the ESTs were assigned to the same position as the cDNA clones on DNA fibers and to further evaluate the reliability of this procedure, for example, by determining the rate of false positive signals.
Three different cDNAs and their corresponding ESTs were used for the specificity and accuracy measurements. The acceptable location for an EST signal was determined as the ±1 s.d. distance achieved with the cDNA probe. The specificity was determined as the proportion of EST signals assigned to the acceptable region. Eighty-nine percent (71/80) of the detected EST images fulfilled the criteria (Table1).
The Specificity of Visual EST Mapping on DNA Fibers
Reproducibility and Accuracy of Visual EST Mapping
To determine the reproducibility of visual EST assignments, the variation in the position of the EST signals on the top of a long-range clone was evaluated without prior knowledge of the precise location of the gene. Thus, no information about the corresponding cDNA was used. The accuracy of the EST position is represented by s.d.values in Table 2. The mean value for variation in the position of an EST (the accuracy) was about ±12 kb when positioned on a longer clone such as P1, PAC, or BAC (Fig.3) and ±6 kb on a cosmid clone. Thus, the major contribution of the variation was due to the differences in the length of the long-range clone signal.
The Accuracy of the Visual Assignment of ESTs and cDNAs on Top of a Long-Range Clone
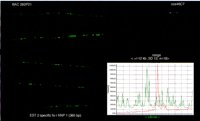
An illustration of the reproducibility of mapping EST on free DNA fibers. The EST (EST 2) (red signal) specific for the KNP1gene was assigned on the BAC clone 282P21 in relation to an orientation marker (cos 46C7). The images represent four consecutive hybridizations. The variation in the position of an EST was determined to be ±12 kb with a s.d. of 12 on top of a long-range clone. The histogram (right) shows the precise location of the EST as a red peak of the intensity.
Construction of a Visual Transcript Map
The specific assignments of the cDNAs and corresponding ESTs on top of different long-range clones are illustrated in Tables 2 and3. The cDNA and the ESTs specific for theAIRE gene were assigned to three different long-range clones; cosmid clone 22G11, the PAC 92C23, and BAC 127P21, separately (Table 2; Fig. 4A). The C21orf3 gene was assigned separately onto three clones; P1 579E2, PAC 92C23, and BAC 127P21 (Table 2; Fig. 4B). The SMT3A gene was positioned earlier between markers D21S154 and D21S171 (Lapenta et al. 1997). The FISH data here demonstrate that the position of the SMT3A gene is telomeric to the marker D21S171 (Yaspo et al. 1998). The SMT3Agene was positioned at the centromeric edge of the cosmid clone 83H6 with both a cDNA clone (L2328) and an EST (EST 8; Fig. 4C). Furthermore, three other genes—the PWP2, the KNP1, and the C21orf1—were mapped and visualized by their specific ESTs on this restricted genomic region on chromosome 21q22.3 (Lalioti et al. 1996; Nagamine et al. 1996; Yaspo et al. 1998). ThePWP2 gene was positioned separately on the BAC clones 175D10 and 282P21 with one EST (EST 1). The KNP1 gene was assigned by two gene-specific ESTs (EST 2 and 3) on the same two BAC clones as thePWP2 gene, the BAC clones 282P21 and 175D10 (Table 2). TheC21orf1 gene was positioned using two ESTs (EST 9 and 10) on the middle of cosmid clone 83H6 (Table 2).
Comparison of Distances of Visualized Genes (by their ESTs and Full-Length cDNAs) from Known Markers on Chromosome 21q22.3
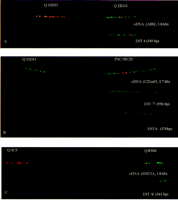
An illustration of the specificity of ESTs mapping procedure. The assignment of ESTs to the same position as the corresponding cDNA is demonstrated. (A) The AIRE gene is visualized with a cDNA clone (B1-1, 1.8 kb) and an EST 4 (549 bp) equally on top of a cosmid clone Q22G11. The cosmid clone Q11D11 (marker D21S25) serves as an orientation marker at top. (B) TheC21orf3 is assigned with a cDNA clone (21919, 2.7 kb) and two ESTs (956 and 376 bp) on a PAC clone 92C23. The cosmid clone Q11D11 serves as an orientation marker. (C) The SMT3A gene was localized with a cDNA clone (L2328, 1.8 kb) and the corresponding EST (541 bp) on a cosmid clone Q83H6 in relation to the cosmid clone Q3C3 (marker D21S171) serves as an orientation marker.
Mapping the ESTs by the fiber–FISH method combined with tyramide-based detection on free DNA fibers resulted in a visual transcript map of six different genes in the vicinity of the markers D21S1460 and D21S1903 on chromosome 21q22.3. The map is illustrated in three parts. Three ESTs—detecting the PWP2 and the KNP1 genes—were assigned to the centromeric part of the region (Fig.5). The PWP2 gene was the most centromeric of these two genes. Both of these genes were located on the centromeric side of cosmid clone 11D11 (the marker D21S25). In the central part of this transcript map, the AIRE gene was positioned centromeric to the C21orf3 gene. Three ESTs and one cDNA—detecting theSMT3A and the C21orf1 genes—were localized distal from the cosmid clone 25F11 (the marker D21S1903) in the third section of the map.
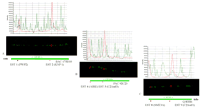
An illustration of a visual transcript map. Red signals are hybridization signals from EST probes, green signals form underlying large insert clones. (A) The centromeric side of the map included ESTs 1 and 2 corresponding to the PWP2 and KNP 1 genes, respectively. (B) The central part of the map included ESTs 4 and 7 representing the AIRE and theC21orf3 genes, respectively. (C) The telomeric part of the map included ESTs 8 and 9 corresponding to the SMT3Aand C21orf1 genes, respectively. In all images, the long-range clone is detected by FITC (green) and the ESTs are detected by biotinylated tyramide with Texas Red (red). The histograms on the upper side of the FISH images show the precise positions of the ESTs as red peak on a long-range clone.
DISCUSSION
The limited sensitivity of detection methods applied in FISH approaches has prevented a comprehensive usage of short clones (<2 kb). The development of the biotinylated tyramide conjugate for FISH has surpassed this limitation (Raap et al. 1995; van Gijlswijk et al. 1996). Here we demonstrate the feasibility of mapping genes via their ESTs by using the fiber–FISH combined with sensitive tyramide-based detection. This study shows that genes can be reliably visualized and localized directly by their amplified EST sequences as small as 300 bp in size. The visual mapping approach for ESTs enables a straightforward use of the increasing database of ESTs and, for example, speeds up the exclusion and localization of disease genes for positional cloning and candidate gene approaches in critical disease-linked genomic regions. This approach is especially useful in genomic regions that are difficult to sequence and clone.
Our data of visualizing ESTs on free DNA fibers showed a hybridization efficiency of 25%–45% in detecting ESTs from 315 to 956 bp in size and a hybridization efficiency of >60% with each cDNA clone. In mapping studies, short probes or DNA fragments have been used earlier, but not by applying the enhanced deposit reaction with tyramide-conjugates (Fan et al. 1990; Viégas-Péquignot et al. 1991; Lemieux et al. 1992; Heppell-Parton et al. 1994; Richard et al. 1994; Korenberg et al. 1995; Florijn et al. 1996). In the visual ESTs mapping procedure, hybridization frequency correlated directly with the size of the probe—the longer probes resulted in higher hybridization frequencies, which has also been reported previously (Florijn et al. 1996). The lower hybridization frequencies detected by mapping ESTs on metaphase chromosomes compared with free DNA fibers was most likely a result of easier accessibility of probe to target along with decreased condensation level of chromatin structure. Tyramide-based detection greatly enhances the specific signals. This enhancement also generates background, creating the need to use stringent posthybridization washing conditions. The long-range clones were necessary to reliably map the ESTs on the same DNA fiber with the help of the anchoring clone and the orientation clone. Therefore, it was possible to measure the correct position of an EST. At the same time, background signals were easily distinguished from specific signals.
The specificity of the ESTs mapping was demonstrated by use of corresponding cDNAs. According to these results, the mapping specificity was sufficient and provided reliable information of the assignments of the genes in question. The variation in the position of the ESTs on the long-range clones may be caused by the differential stretching of the DNA fibers (CV% 20), unequal hybridization and detection efficiency of the whole long-range clone, as well as crossings of the DNA fibers leading to the broken fibers. The resolution of fiber–FISH is currently 1 kb, whereas the detection reaches a sensitivity of 200–300 bp. The fairly low hybridization frequency of the ESTs is compensated by the specificity by which a reasonable number of images can easily be collected from a single preparate. False-positive signals appeared rarely. When presumed false-positive signals appeared, they were on the opposite edge of the anchoring clone, away from the majority of signals. The number of likely false-positive signals were detected to be only 1% or 2% of the total.
The location of the most centromeric gene detected by use of this approach—PWP2—was positioned proximal to the marker D21S25 and proximal to the KNP1 gene. This result is supported by earlier data of the position of the PWP2 gene, which was within a 200 kb range from the PFKL marker (Lalioti et al. 1996). Our results indicate that the next distal gene in this transcript map was the KNP1 gene, which was localized ∼150 kb proximal to the cosmid clone 46C7 (marker PFKL). This gene had been positioned previously within the same region and was also reported to be transcribed in the centromere to telomere direction (Nagamine et al. 1996). Therefore, the presumed location of the 3′ end of this gene corresponds to our visual data for the location of the KNP1gene specific ESTs.
The recently cloned AIRE gene was identified near thePFKL gene by direct sequencing and the gene position was also visualized by the fiber–FISH by use of a full-length cDNA clone as a probe (Aaltonen et al. 1997b; Nagamine et al. 1997). Our results for mapping ESTs specific to the AIRE gene were similar to the previous data, indicating that this gene maps just proximal to thePFKL gene—∼70 kb from the edge of cosmid clone 11D11 (the marker D21S25). In the middle section of this transcript map, a previously cloned gene—C21orf3—was positioned distal to theAIRE gene with two different sized ESTs. The location of this gene corresponds to recently published data by Scott et al. (1998).
The SMT3A and the C21orf1 genes were assigned to the distal part of the transcript map (Lapenta et al. 1997; Yaspo et al. 1998). The SMT3A gene was assigned earlier by Lapenta et al. (1997) between the markers D21S154 and D21S171. Our FISH data indicated a more distal location on the telomeric side of the marker D21S1903, which was also supported and corrected later by Lapenta et al. (1998). Here the results showed that this gene maps ∼170 kb from the marker D21S171 and 80 kb distal to the marker D21S1903, which is in agreement with the most recent results by Lapenta et al. (1998). TheC21orf1 was positioned near and distal to the SMT3Agene on top of the same cosmid clone 86H3, which was in good concordance with recently published data by Yaspo et al. (1998).
The visual EST mapping approach offered higher resolution, for example, than the STS-based mapping, in which the resolution of currently available maps is ∼100 kb (Hudson et al. 1995). The ESTs visual mapping procedure showed that reliable conclusions can be made from the positions of the genes achieved by mapping gene-specific ESTs. The ESTs could be reliably mapped to a precise location within a long-range clone with at least 10 kb resolution. The produced data were similar with previous results or were confirmed during this study (Lalioti et al. 1996; Nagamine et al. 1996; Aaltonen et al. 1997a,b; Lapenta et al. 1997, 1998; Yaspo et al. 1998).
The EST mapping approach is easily established in laboratories experienced in FISH technology. This procedure allows gene assignment at a precise position within a long-range clone using only a short gene-specific DNA fragment as a probe. Although no formal comparison of labor time between restriction fragment-based mapping and high-resolution FISH has been performed, on the basis of our experience, FISH results in considerable time saving in laborious gene-identification projects. Also, the procedure to prepare the fibers from agarose-embedded cells is so robust that essentially every target preparation is successfully applicable for FISH. Another notable advantage of this visual mapping approach is the localization of at least two ESTs at the same time in relation to each other, allowing simultaneous visualization of several genes. This is not possible by other methods currently available. The EST-mapping procedure showed that even small fragments of genes can be visualized by combining the best resolution provided by fiber–FISH with the highly sensitive tyramide-based detection method. New opportunities for visualizing several genes simultaneously has become possible by the development of different tyramide conjugates. The simultaneous signal enhancement of multiple tyramide conjugates will facilitate usage of different colors for detecting small DNA fragments such as ESTs (Speel et al. 1997; van Gijlswijk et al. 1997). In conclusion, the visual ESTs-mapping approach proved to be a straightforward and reliable method that enables the direct usage of the ESTs databases for positional cloning, particularly in the future, for positional candidate gene purposes. This mapping approach is particularly practical in rapidly excluding candidate genes from a well-refined genomic region.
METHODS
Genes and ESTs
This project was first started with the aim to rapidly create a transcript map and identify the defective gene causing the APECED disorder (Fig.1). The genes were selected on the basis of their location to a well-characterized APECED region on chromosome 21q22.3. The genes chosen to this visual mapping approach were theKNP1, the PWP2, the C21orf3 (named C21orf2 by Scott et al. 1998), the AIRE, the SMT3A, and theC21orf1 (Lalioti et al. 1996; Nagamine et al. 1996; Aaltonen et al. 1997b; Lapenta et al. 1997; Nagamine et al. 1997; Yaspo et al. 1998). The C21orf3 and AIRE were novel genes assigned between the markers D21S25 and D21S154 during this approach. TheC21orf3 gene was a candidate gene for APECED disease and was excluded later. The AIRE gene was cloned recently and identified to cause the APECED disease (Aaltonen et al. 1997b; Nagamine et al. 1997).
In the visual mapping procedure, one or two ESTs and cDNA clones (forAIRE, C21orf3, and SMT3A genes) specific to different genes were used (Table 4). ESTs were identified from the NCBI database using the accession numbers of the selected genes (http://www.ncbi.nlm.nih. gov80/cgi-bin/BLAST/nph-blast). The ESTs were selected from the 3′ untranslated region of genes and were amplified by PCR for FISH. Thus, it was known to which large insert clone the EST would hybridize, however, the location within the clone was not known. The ESTs specific for the AIRE gene were generated from the cDNA sequence because no human ESTs corresponding to this gene were available in the databases. The positions in the genes and the sizes of the ESTs and the cDNAs are summarized in Table 4.
cDNAs, ESTs, and Primer Sequences Used in this Study
Long-Range Clones
For transcript map construction, we utilized the long-range clone contig recently constructed across an 800-kb genome region on the chromosome 21q22.3 (Aaltonen et al. 1997a). The clone contig was constructed from different-sized clones including cosmids, P1s, PACs, and BACs. These clones were screened from different libraries by PCR by use of several markers and STSs from this restricted genome region. The sizes of the clones were measured earlier by the fiber–FISH method (Aaltonen et al. 1997a). In the FISH procedure, 12 long-insert clones were used as a framework for mapping ESTs, providing the anchoring and orientation markers for positioning different genes via their EST sequence(s) (Fig. 1).
The genomic region of interest, also called the APECED region, on chromosome 21q22.3 included markers D21S1460, D21S25, D21S154, D21S171, and D21S1903. The centromeric part of this area included markers D21S1460 and D21S25. Long-range clones used for orientation and anchoring markers in this region were BAC clones 175D10, 282P11, and 127P11 and cosmid clones 65F12, 46C7, and 11D11. The middle part of the transcript map contained markers D21S25 and D21S154. The framework of long-range clones used in this region were cosmids 11D11, 21D1, 22G11, and P1 579E2, BAC 127P21, and PAC 92C23. The telomeric part of this region contained markers D21S171 and D21S1903 and clones used were cosmid clones 3C3, 25F11, and 83H6.
In each hybridization, two long-range clones in addition to the ESTs were hybridized as follows: one was used to recognize the expected/known genomic region of the ESTs and the other to orient the clones. The hybridization frequency (expressed in percent) was determined by analyzing the signals from long-range clones and by determining how often a signal from an EST or cDNA probe was observed within the long-range clone. Only clearly visible signals were taken into account and no signal enhancement by the image analysis system was used.
FISH
Target Material
Human peripheral blood lymphocytes were cultured according to standard protocols to achieve metaphase chromosome targets for FISH (Yunis 1976; Lemieux et al. 1992). Free DNA fiber targets were created by applying agarose-embedded human lymphocytes as described earlier (Heiskanen et al. 1994, 1996).
Probe Labeling
Amounts of 100–200 ng of each EST were labeled with biotin 11–dUTP (Sigma) or biotin 16–dUTP (Boehringer Mannheim) by use of random prime labeling (Random Prime DNA Labeling Kit, Boehringer Mannheim). The long insert clones, including cDNAs, were labeled with either biotin 11—dUTP or digoxigenin 11—dUTP by nick translation according to standard protocols (BRL, Nick translation Kit, MD, USA).
Hybridization and Detection Conditions
FISH assays were carried out as described earlier with minor modifications (Pinkel et al. 1986; Lichter et al. 1988; Heiskanen et al. 1996). Amounts of 100–200 ng of labeled long-range clones and 500—1500 ng of each labeled EST clone were used for each slide. Tenfold excess of Cot-1-DNA (GIBCO-BRL, Gaithesburg, MD) was added to suppress repetitive sequences from long-range clones. Three different variables in hybridization conditions were first tested to visualize small clones like ESTs. These tests included formamide concentration in the hybridization mixture (50% and 30%), hybridization time at +37°C (16–72 hr) and the stringency level in posthybridization washings (including temperature variations, 39–43°C and salt concentrations from 2× SSC down to 0.5× SSC). According to the test results (data not shown), the following procedure was chosen: a 30% formamide concentration in the hybridization mixture, 2–3 days hybridization at +37°C. Posthybridization washings were carried out at +39°C in 50% formamide in 2× SSC, three times, for 5 min in each. Biotin-labeled probes were detected with highly sensitive biotinylated tyramide-based detection with few modifications (Raap et al. 1995; Laan et al. 1996). For two-color experiments, the first step was incubation with streptavidin-conjugated horseradish peroxidase, followed by a precipitation reaction of biotinylated tyramide through a peroxidase-catalyzed reaction over a biotin-labeled probe. Texas Red-conjugated streptavidin reacted with the increased amount of biotin molecules, resulting in a bright halo-like signal. Digoxigenin-labeled probes (long range clones) were detected with mouse antidigoxigenin (Sigma) and fluorescein (FITC)-conjugated sheep antimouse antibody (Sigma), and the last layer was FITC-conjugated donkey antisheep antibody (Sigma). DNA-targets were counterstained with DAPI, which was included in the antifading agent (Vectashield, Vector, Burlingame, CA).
Fluorescence Microscopy
The system used for multicolor image analysis for acquisition, display, and quantitative analysis of ESTs mapped on DNA fibers was mainly the same as described earlier by Heiskanen et al. (1996). The data were analyzed by a Macintosh system with the IPLab software (Signal Analytics Corp., Vienna, VA), which included image acquisition, distance measurements, and lane measurement analysis of the position of an EST on the top of a long-range clone showing a two-color distribution in a histogram format. The cosmid clone 65F12 with a known physical size—39 kb—was used as a standard for distance measurements (Aaltonen et al. 1997a).
The position of the signal from the cDNA probe within a long-range clone was measured by use of the line measurement option of the IPLab software much in the same way as the Flpter values are measured (Lichter et al. 1990; Table 2). The position of an EST was determined by measuring the distance of an EST signal from the edge of the underlying large insert probe. The edge closest to the orientation marker was used to measure the EST location. Ten to thirty images were captured from each EST hybridization. The accuracy of the EST position was determined as the variation of the location of the signal and is expressed as s.d..
Acknowledgments
The Maud Kuistila Foundation, the Instrumentarium Foundation, The Research Institute of Helsinki University Central Hospital, the Foundation of Jenny and Antti Wihuri, and the Academy of Finland are thanked for their financial support for this work. We thank Lisbeth Kuitunen for revision of the English language.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵5 Present address: Department of Human Genetics and Department of Pathology, University of California Los Angeles, Los Angeles, California 90095-7088 USA.
-
↵6 Corresponding author.
-
E-MAIL Nina.Horelli-Kuitunen{at}HUCH.fi; FAX 358-9-4714001.
-
- Received June 30, 1998.
- Accepted November 16, 1998.
- Cold Spring Harbor Laboratory Press








