
Multiplex Sequencing of 1.5 Mb of the Mycobacterium leprae Genome
- Douglas R. Smith1,2,
- Peter Richterich2,
- Marc Rubenfield2,
- Philip W. Rice2,
- Carol Butler2,
- Hong-Mei Lee2,
- Susan Kirst2,
- Kristin Gundersen2,
- Kari Abendschan2,
- Qinxue Xu2,
- Maria Chung2,
- Craig Deloughery2,
- Tyler Aldredge2,
- James Maher2,
- Ronald Lundstrom2,
- Craig Tulig2,
- Kathleen Falls2,
- Joan Imrich2,
- Dana Torrey2,
- Marcy Engelstein2,
- Gary Breton2,
- Deepika Madan2,
- Raymond Nietupski2,
- Bruce Seitz2,
- Steven Connelly2,
- Steven McDougall2,
- Hershel Safer2,
- Rene Gibson2,
- Lynn Doucette-Stamm2,
- Karin Eiglmeier5,
- Staffan Bergh5,
- Stewart T. Cole5,
- Keith Robison4,
- Laura Richterich4,
- Jason Johnson4,
- George M. Church1,3,4, and
- Jen-i Mao2
- 2Genome Therapeutics Corporation, Collaborative Research Division, Waltham, Massachusetts 02154; 3Howard Hughes Medical Institute and 4Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115; 5Unite de Genetique Moleculaire Bacterienne, Institut Pasteur, 75724 Paris CEDEX 15, France
Abstract
The nucleotide sequence of 1.5 Mb of genomic DNA fromMycobacterium leprae was determined using computer-assisted multiplex sequencing technology. This brings the 2.8-Mb M. leprae genome sequence to ∼66% completion. The sequences, derived from 43 recombinant cosmids, contain 1046 putative protein-coding genes, 44 repetitive regions, 3 rRNAs, and 15 tRNAs. The gene density of one per 1.4 kb is slightly lower than that ofMycoplasma (1.2 kb). Of the protein coding genes, 44% have significant matches to genes with well-defined functions. Comparison of 1157 M. leprae and 1564 Mycobacterium tuberculosisproteins shows a complex mosaic of homologous genomic blocks with up to 22 adjacent proteins in conserved map order. Matches to known enzymatic, antigenic, membrane, cell wall, cell division, multidrug resistance, and virulence proteins suggest therapeutic and vaccine targets. Unusual features of the M. leprae genome include large polyketide synthase (pks) operons, inteins, and highly fragmented pseudogenes.
[The sequence data described in this paper have been submitted to GenBank under accession nos. L78811–L78829,U00010–U00023, U15180–U15184, U15186, U15187, L01095, L01536, L04666, and L01263. On-line supplementary information for Table 1 is available at http://www.cshl.org/gr.]
Despite improved medical care and large vaccination programs, infectious organisms are still the leading cause of death, worldwide, and the pathogenic mycobacteria are among the worst offenders. There are estimated to be ∼5 million cases of leprosy, globally, while tuberculosis kills ∼3 million persons per year. The frequent occurrence of multidrug resistantMycobacterium tuberculosis and the documented appearance of dapsone resistant Mycobacterium leprae are reminders that current therapies may not always be effective and that we should continue to search for and develop new antiinfective agents.
M. leprae is one of the few bacterial pathogens that infects humans and cannot be cultivated outside of animals. The organism is an intracellular parasite that grows extremely slowly (generation time, 14 days). A number of immunodominant protein antigens have been identified and characterized in M. leprae (Murray and Young 1992), but few metabolic enzymes have been studied. This combination of urgent problems and difficulties with classical biological approaches have made the mycobacteria prime candidates for comparative genome sequencing. This approach promises to aid in the identification of targets for vaccine and therapeutics development, possible regulatory elements and mechanisms, and will help us to understand the unique biochemistry of microbial intracellular parasites. The recent construction of a cosmid-based genome map for M. leprae has facilitated study of the genome by molecular biological techniques. This report summarizes DNA sequencing results on 43 cosmids selected from this set.
Advances in large-scale sequencing driven by the Human Genome Project have stimulated sequencing projects on a variety of small genomes. For example, at least six microbial genomes and one fungal genome have now been sequenced, ranging in size from 0.58 to 12 Mbp and representing all major phylogenetic kingdoms. These include Haemophilus influenzae (Fleischmann et al. 1995), Mycoplasma genitalium (Fraser et al. 1995), Saccharomyces cerevisiae(Dujon 1996), Methanococcus jannaschii (Bult et al. 1996),Methanobacterium thermoautotrophicum (Smith et al. 1996),Synechocystis sp. 6803 (Kaneko et al. 1996), andMycoplasma pneumoniae (Himmelreich et al. 1996). Thirty-seven other small genome sequencing projects are now reportedly under way (Gaasterland and Sensen 1996). Thus, there is considerable biological and economic motivation for the development of more rapid DNA sequencing technologies that offer high accuracy and lower cost than current methods.
Multiplex sequencing is a rapid sequencing approach based on sample tagging, mixing, and molecular decoding by oligonucleotide hybridization (Church and Kieffer-Higgins 1988). The approach is compatible with a variety of sequencing strategies, including transposon-ordered and whole genome shotgun sequencing (Church and Kieffer-Higgins 1988). The potential throughput is very high, because all of the “front-end” steps, from DNA amplification and isolation through gel electrophoresis, are performed on mixtures of plasmid clones. Using pools of 20 plasmid clones (each clone provides two sequences), these front-end steps are facilitated by a factor of 40 compared to M13-based methods. Sequencing patterns are generated by32P-labeled film-based detection, by chemiluminescence (Richterich and Church 1993), by direct fluorescence, or by enzyme-linked fluorescence detection (Cherry et al. 1994). Digitized images of the sequencing patterns are then processed on computer workstations using automated image analysis and sequence reading software. These techniques have allowed the generation of significant volumes of sequencing data over the past few years of development onEscherichia coli (Church and Kieffer-Higgins 1988),Salmonella typhimurium (Roth et al. 1993), Helicobacter pylori, M. tuberculosis, Staphylococcus aureus, Streptococcus pneumoniae, Clostridium acetobutylicum, M. thermoautotrophicum(Smith et al. 1996), Arabidopsis thaliana, Pyrococcus furiosus, and Homo sapiens (Cawthon et al. 1990). Nevertheless, this is the first publication describing the application of the technology on a megabase scale. The sequences described here were generated over a 3.5-year period as the technology was developed and optimized.
Sequencing Strategy and Accuracy
The cosmids used in this study (Fig. 1) were constructed from M. leprae DNA isolated from armadillo liver infected with the dapsone-resistant Tamil Nadu strain of a clinicalM. leprae isolate (Eiglmeier et al. 1993). The DNA sample has been shown to be heterogeneous, at least with respect to one putative transposon (Fsihi and Cole 1995). Cosmids were sequenced by a shotgun strategy at 5- to 10-fold redundancy followed by fragment assembly and primer-directed finishing to bridge contigs and eliminate single-stranded regions. The individual fragment sequences were proofread to correct obvious errors as the data were entered, and the contigs were proofread after assembly to correct errors detectable as discrepancies between individual fragments. The data were analyzed to identify errors resulting in frameshifts, and these were also corrected, wherever possible. The shotgun data were derived almost exclusively by chemical sequencing, which produced satisfactory data with very even band intensities although it suffered somewhat from a lack of reproducibility.

M. leprae genome map indicating regions discussed in the text. Cosmid clone names (starting with B or L) follow the M. lepraemap (Eiglmeier et al. 1993). Cosmid sequences described here are indicated by yellow boxes (see Table 3, below). Red and blue boxes indicate cosmids sequenced by the Institut Pasteur (IP) and other members of M. leprae World Health Organization (WHO) genome consortium, respectively. Unboxed cosmids are mapped but not sequenced. Eighteen of the cosmids sequenced in this study overlapped to form contigs (eight contigs, averaging 67 kb in size). Most of the gaps remaining between adjacent sequenced cosmids were small, and many could be bridged by long-range PCR.
The average G + C content of the cosmids sequenced was 58%. This resulted in a significant electrophoretic gel compression every 200 bp or so, on average. Difficult compressions were resolved by careful analysis of reads from both strands, and by electrophoresis of the products of primer-directed cycled sequencing reactions on formamide gels, which were capable of resolving all compressions encountered. This worked well enough that in some of the later sets of cosmids, formamide gels were routinely used to generate ∼30% of the shotgun coverage. This up-front measure significantly reduced the need for special-case compression resolution, although it often led to a reduction in gel resolution and a reduction in read length of ∼10%. The insertion/deletion (indel) error rate after contig proofreading was estimated to average 1.8 × 10−4, based on ∼67 kb of overlapping sequence between pairs of cosmids that were finished independently. The frequency of missense errors was similar. Gel traces from all genes with potential frameshift errors were carefully examined for errors, and additional sequences were generated in ambiguous regions. After such homology-based frameshift editing, the indel error rate was reduced to 1.0 × 10−4 for sequences with database homologies (53 likely frameshift errors remaining in 31 genes out of a total of 562 with database homologies spanning a total of ∼542 kb). Overall, the raw data indel error rate was considerably higher than that associated with ABI dye-terminator chemistry. The lack of an equivalent chemistry is one of the current limitations of multiplexing sequencing. Other limitations in comparison to ABI technology are the shorter read lengths and lower overall data quality.
Identification of Potential Gene Sequences
The sequences were analyzed for open reading frames (ORFs) using a set of computer programs unified through a single platform, GenomeBrowser (Robinson et al. 1994; Robinson and Church 1995). The programs identify all possible ORFs larger than a specified size (60 codons) and parse them to the National Center for Biotechnology Information (NCBI) network BLAST server to identify database homologies. They also search for tRNAs (Fichant and Burks 1991), perform codon usage analysis, and perform a nucleotide BLAST search. The results were displayed using the GCG Figure program, or the Belmont Tool Kit, an interpretive object-oriented graphical environment. This provided a graphical representation of each cosmid displaying the locations of putative reading frames with corresponding BLAST homologies displayed above each frame. Below each frame was displayed a series of dots (which may merge into a solid line) if the dicodon usage matched an M. leprae gene-specific dicodon usage table.
Reading frames with dicodon usage similar to previously identifiedM. leprae genes were analyzed further for the presence of translation initiation sites. Acceptable sites were selected from a comprehensive list for each reading frame and contained an ATG or GTG initiation codon preceded by an optional spacer (0–8 nucleotides) and a sequence complementary to at least 4 out of 11 nucleotides from the 3′ terminus of M. leprae 16S rRNA (Shine and Dalgarno 1975; Liesack et al. 1990). Alignments with the amino termini of homologous proteins were also used to select translational start sites, in some cases. Possible coupled translation signals (an initiation codon within 20 nucleotides of a stop codon, characteristic of many bacterial operons) were also accepted as putative start sites. The positions of all putative genes meeting one or more of these criteria were recorded, together with the nature of the initiation site or operon linkage.
A list of putative M. leprae genes identified in this study and sorted by function is provided in Table 1 [a more comprehensive list is available on the Genome Research Web site (http://www.cshl.org/gr)]. Functional designations and gene names were assigned to genes with homologs having BLAST scores over 100; otherwise a name beginning with the letter “y” was assigned. We stress that the functional assignments must be viewed as provisional because of the inherent uncertainties in assigning gene function by sequence similarity. Gene names are based on existing mycobacterial names, where acceptable. Otherwise, names are based on E. coli nomenclature rules corresponding to the closest bacterial homologs in the following order of priority: E. coli, S. typhimurium, Bacillus subtilis, Streptomyces species, and other bacteria. In many cases, new names were assigned. A more extensive table of interpretations, including accession numbers, is available from http://www.cric.com/ and from MycDB, a database of mycobacterial mapping and sequence information (Bergh and Cole 1994) based on the acedb (Durbin and Thierry-Meig 1991–1995). The following sections describe some of the more striking findings from the data. We stress that owing to the limitations imposed by an incomplete geneome sequence, it is not possible to make definitive conclusions concerning the unique nature of mycobacterial metabolism relative to other organisms.
List of 1064 Putative M. lepraeGenes Identified in This Study
Repetitive Sequences and DNA Duplications
The M. leprae genome was found to contain several types of repetitive sequences by cross-searching for homology between different cosmids (precise location and size of repeats are given in Table 2). The most common repeats were a large family of 70- to 80-bp sequences, which we have called REP1 elements. The functional significance of these elements is unknown, but some of them were found to be located near the beginnings of genes. We found several RLEP elements (originally described as near-perfect 700-base repeats (Woods and Cole 1990). These occurred in cosmids where they had been previously located by physical mapping techniques (Eiglmeier et al. 1993). However, the RLEPs do not appear to encode any proteins. Of particular interest is the DNA polymerase I gene in cosmid L247 (Table3), which is closely flanked by two inverted RLEP elements. This arrangement is reminiscent of certain composite transposons and provides a possible explanation for the origin of RLEPs as “IS-like” elements (Fsihi and Cole 1995). The only clearly identifiable IS element, the 1051-bp REP13 element, was found in cosmid B1620, and this shows 65% identity at the DNA level with IS1081 from the M. tuberculosis complex (Poulet and Cole 1995).
Strong DNA Sequence Matches and Repetitive Elements
M. leprae Cosmid Sequence Accession Numbers
Some smaller direct repeats were also seen. For instance, two identical copies of the 309-bp REP9 element were contained in cosmids B2168 and B1790 (Honoré et al. 1993). The 52-bp REP14 element with 69% identity between two copies in B1790 also detected 12- to 18-bp stretches in several other cosmids (data not shown). Simple sequence repeats, including 6-copy CAC and 21-copy TTC tandem trinucleotide repeats, are longer than those in E. coli.
Several apparent gene duplication events were evident. One of these is a 1.6-kb sequence that recurs in several members of a family of polyketide synthase (pks) genes (including four within a single large operon in cosmid L518). The 1.6-kb repeat is composed of two segments, 120 bp and 1385 bp (separated by a 120-bp spacer), which are virtually identical between repeats pksA and pksC (Table 2). These two repeats, separated by 3.8 kb, are contained in two adjacent polyketide synthase genes encoded by the L518 operon (discussed in more detail below). The overall identity of repeatspksA and pksC, including the 120-bp spacer, is 95%. The polypeptide encoded by the repeat contains an acyltransferase consensus sequence, VVGHSMGESAAAVVAGAL, near its center. The repeats inpksD, pksE, and pksX share 68%, 66%, and 55% identity to the pksA DNA sequence.
Cosmid B2126 contains a duplicated 1.5-kb segment that encodes an amino acid transport gene similar to aroP (Tables 1 and 2). The two identical copies of this segment are arranged in tandem with a 122-bp spacer. The perfect nature of this repeat indicates an evolutionarily recent duplication or gene conversion.
Split and Fragmented Genes
At least three genes in M. leprae are likely to encode proteins that undergo autocatalytic splicing reactions to remove an intein (protein intron) from a nascent precursor molecule. The corresponding genes are believed to have been “invaded” by a DNA sequence, coding for a homing endonuclease, that is inserted in-frame in a protein-coding gene. These are gyrA (Fsihi et al. 1996),xheA, and recA (the sequence of M. lepraecosmid B2235 contained a recA gene andrecA-associated ORF). There is a relationship between theM. leprae and M. tuberculosis recA genes (Davis et al. 1994). The sequences of the intein and the insertion points are different in the two organisms. In contrast, the recA exteins are 92% identical. Such divergence among inteins is common even among inteins targeting the same gene (Pietrokovski 1996). Features shared by the inteins include two homing double-stranded DNA (dsDNA) endonuclease motifs (LAGLIDGDG also found in introns and in HO endonuclease) separated by 80–121 amino acids plus protein-splicing catalytic sites at the intein amino terminus (Cys) and carboxyl terminus (His–Asn). The M. leprae recA intein has a match to intron-encoded DNA–endonucleases/RNA–maturases (e.g., P03873 | Cybm_Yeast, P = 4.5e-05 overall, P = 0.36 for the segment below), which is not detected in other intein sequences.
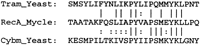
The intein found in an 870-codon ORF, xheA in cosmid B1496, shows significant similarity to the protein-splicing and homing endonuclease domains of the vent polymerase intein and yeast HO proteins, respectively (Fig.2). The part of the gene flanking this potential intein (codons 1–202, 581–870) corresponding to the N and C exteins, is homologous to ORFs from three major kingdoms—eukaryotes (Antithamnion, Plasmodium), archaebacteria (Methanobacterium), and eubacteria (Synechocystis andShigella). The significance of the homing endonuclease motifs may be to target conserved gene sequences as hypothesized for thymidylate synthase (Sherman et al. 1995). Of the reported 20 gene families targeted by inteins, all 17 with homologs of known function are involved in metabolism of phosphorylated compounds.

Analysis of the xheA intein. The tightly coupled operon shown is right-to-left 5′ to 3′: ybhF, xheA, ybhE, abcA, nifS, nifU shown in GenomeBrowser format. ORFs longer than 50 codons (blue horizontal lines) have stop codons indicated by short vertical black lines. Magenta horizontal lines above each ORF indicate matches to the NR database (Altschul et al. 1990) with significant BLASTP scores (P < 0.001), where the vertical displacement indicates the percent amino acid identity for that sequence segment. The red lines below the ORFs indicate quality of dicodon usage. Frame number, accession numbers, and gene names based on sequence similarity are in the text below the red lines. The xheA gene is located in M. leprae cosmid B1496 from nucleotide position 2020 to 9152. The amino- and carboxy-terminal regions have strong matches with eukaryotic, prokaryotic, and archaebacterial URFs (unknown function readingframes), including sp | P51240 | YC24_PORPU, and gi | 1742763 (E. coli) at 30%–42% identity (P < 1E-22) as does the central intein region (where intein BLASTP segments are in green to contrast with the normal magenta). The paralogous (intragenomic M. leprae) xheA–ybhF (gi | 466874) duplication is 24% identity, P = 2E-15. The numerals in parentheses represent the ORF numbers for a related cyanobacterial gene cluster (D64004). The sequence alignments (below) indicate the shift in amino acid identity pattern and the conserved motifs at the intein boundaries and internally.
About 3.5% of possible coding regions in the 1.5 Mbp described here appeared to contain multiple (three or more) frameshifts and/or in-frame termination codons relative to strongly similar, known genes. Reinspection of the raw data in these regions (from data on both strands) did not support the multiple changes that would be required to generate functional coding sequences. Of the total of 39 such regions, an average of 9 and as many as 21 changes per gene would be required. Highly fragmented gene sequences such as these were assumed to represent nonfunctional pseudogenes and were therefore not annotated as putative coding sequences. One possible explanation for their abundance is that strains of M. leprae, being slow-growing, obligate intracellular pathogens, have accumulated mutations in certain genes that are not essential for their survival in, or for transmission between, humans. It is even possible that there is a selective advantage associated with the loss of certain functions. No homologs of genes considered essential for all organisms (Mushegian and Koonin 1996) were found to be disrupted.
An alternative source of fragmented genes might be gene duplication and subsequent inactivation of one copy, possibly by repeat induced point mutagenesis (Ozer et al. 1993; Singer et al. 1995). However, in no case can a normal copy of a scrambled gene be found elsewhere in the genomic sequence (which now covers about two-thirds of the genome). Other possible explanations for highly fragmented genes should also be considered. Among these are mutations occurring during bacterial strain isolation and recombinant cloning. Biological processes have been described that can counteract insertions or frameshifts at the DNA, RNA, or protein levels at rates compatible with selective advantage for retaining such genomic regions. Such processes include cryptic genes (Hall and Sharp 1992; Hall and Xu 1992), which can easily switch via one or two mutations to a state expressing enzymatically active products at a high level, RNA splicing and editing (Bechhofer et al. 1994; Belfort et al. 1995), ribosomal reprogramming (Gesteland et al. 1992), and protein splicing (Davis et al. 1994; Perler et al. 1994;Belfort et al. 1995).
Figure 3 illustrates an extreme case of gene fragmenting, where high amino acid sequence conservation within short blocks is seen. The sequence is derived from cosmid B2235 (4387–5673) and is homologous to ythY, an M. tuberculosis gene described in SWISSPROT and EMBL databases as encoding a putative thymidylate synthase. This assignment is probably inaccurate, as there is no significant similarity with the large thymidylate synthase (TYSY) family and there is no published evidence supporting it (the M. leprae thyA gene is on cosmid B1554). It is interesting to note that a ythY homolog on M. tuberculosis cosmid Y154 (Smith et al. 1996), which contains several genes in common with M. leprae cosmid B2235, is also fragmented. Alignment of the M. leprae and M. tuberculosis ythY-coding sequences revealed that the nucleotide spacing was identical at the position of each frameshift in the M. tuberculosis sequence. This suggests that loss of function preceded the divergence of these M. leprae and M. tuberculosisorthologs. However, this situation does not hold for all fragmented genes. For example, the pyc gene of M. tuberculosisis intact, whereas the M. leprae pyc homolog has 21 frameshifts.


An extreme example of gene mangling in M. leprae: A region of cosmid B2235 homologous to M. tuberculosisTYSY_MYCTU. (Top line) The asterisks indicate positions of identity between the M. tuberculosis TYSY amino acid sequence (TYSY_MYCTU) and the conceptual translation of bases 4387–5673 of M. leprae cosmid B2235 (Translate). The PairWise (Birney and Thompson 1995) nucleotide triplets are displayed under the corresponding M. leprae amino acids. This represents only one possible ythY reconstruction involving 10 frameshifts (^) and 12 stop codons (#) using the results of analysis with Detect43 and PairWise programs. Additional identities covering the VGQG and AIPVQ sequences can be obtained with different hypothetical mutations. The TBLASTN probability for all GenBank nonredundant (NR) protein sequences at 376, 511, 003 amino acid residues is 6.4 e-67.
Polyketide Synthase Operons
A large number of putative operons were identified in the sequences reported here based on functional relationships, collinearity, and possible translational coupling. A consistent feature of such putative operons is translational coupling between adjacent genes. A particularly long example, the polyketide operon in cosmid L518, contains at least 10 genes spanning 30 kb, most of which appear to be translationally coupled (the first gene begins at the end of the cosmid, so there may be additional genes at the 5′ end of the operon). Five genes from this operon contain a possible start codon overlapping the stop codon of the previous gene but shifted back by 1 nucleotide. In one gene the putative start is shifted back from the previous stop by 11 nucleotides, and in three others the start is shifted forward by 3, 12, and 30 bases.
The overall structure of this operon is interestingly similar to the putative mycocerosic acid synthase (mas) operon on cosmid B1170. The L518 operon contains six pks genes encoding large proteins (>2000 amino acids) of modular organization followed by three genes encoding components of an ABC transporter similar to the daunorubicin resistance system of Streptomyces (P32011) and a gene encoding a homolog of BCG (Bacillus Calmette-Guerin) masORFII. The B1170 operon includes one large pks gene and genes encoding homologs of surfactin synthase (D13262) and a Streptomycesantibiotic transporter gene (C40046). However, there does not seem to be any translational coupling in this operon, with the downstream genes starting ∼50 nucleotides after the stop codon of the previous gene. Figure 4 shows the relationship of the putative pks’s from M. leprae to other members of this protein family. The M. leprae proteins contain some, or all, of the modules that are commonly found in polyketide or fatty acid synthesis that are known to effect the various functional and catalytic steps in pks genes (Fig. 4). Although the actual function of these pks genes is uncertain, it seems likely that they will be involved in the biosynthesis of cell wall components, like mycocerosic acid, as these often belong to the polyketide family.

Modular organization and enzymatic motifs in synthetases for fatty acids (fas), mycocerosic acid (mas), methylsalicylic acid (msas), erythromycin (eryA), actinorhodin (act), tetracenomycin (tcm), frenolicin (fren), griseusin (gris), and avermectin (avr) from the following organisms: M. leprae (Mle, this work),Saccharopolyspora erythraea (Ser), Rattus norvegicus(Rno), Penicillum patulum (Ppa), Streptomyces antibioticus (San), S. avermitilis (Sav), S. coelicolor (Sco), S. glaucescens (Sgl), and S. griseus (Sgr). The motifs are abbreviated as follows: (A) acyl transferase; (C) acyl carrier protein; (D) dehydratase; (E) enoyl reductase; (L) chain length factor; (M) aromatase; (N) aromatase/cyclase; (O) O-methyltransferase; (R) ketoreductase; (S) ketoacyl–ACP synthase; (T) thioesterase; (Y) cyclase. Lowercase letters indicate uncertainty in functional assignment (Donadio et al. 1991; Mathur and Kolattukudy 1992). The 5′ end of Mle pksAis missing from our current sequence. Underlines indicate all of the protein domains ending in translational terminators, except for Sav avr modules, where the sequence data are incomplete. The CLF domain appears closer to pksC (P = 8.9e-13) than to masA (P = 0.0018).
Sequence Relationships Between M. leprae and M. tuberculosis
Approximately half of the M. tuberculosis genome is now available for comparison to M. leprae (2 Mbp of unique sequence comprised of 19 cosmids from our group and 49 from the Sanger Centre) (Barrell et al. 1996; Smith et al. 1996). Regions of similarity at the DNA level can be readily detected with an average identity of ∼78%, and extending over a total of 411,800 nucleotides. These matches occur in short blocks of ∼1400 nucleotides, on average, which extend over larger genomic regions (10 kb for a given pair of cosmids, on average).
The results of DNA-based cross-genome comparisons between two selectedM. leprae cosmids and the available M. tuberculosiscosmids (as of October 1996) are shown in Figure 5. In the example of M. leprae cosmid L471 (Fig. 5A) and M. tuberculosis cosmids MTCY130 and MTCY373, there is a high degree of collinearity between the sequences over a 23.3-kb region. The twoM. tuberculosis cosmids map directly adjacent to one another. A ribosomal operon has been mapped to the region containing MTCY130 (Philipp et al. 1996) but was not annotated on the sequence. The sequence beyond the argS gene on L471 (∼7 kb) does not appear to contain any genes and is not conserved in any sequencedM. tuberculosis cosmids. In the second example (Fig. 5B) withM. leprae cosmid B32, large blocks of matching sequences occur on two M. tuberculosis cosmids, MTCY427 and MTCY338, which are ∼650 kb apart on the genome (some of the coding sequences in the B32ftsY region appear to be truncated, or frameshifted, relative to those on MTCY338). In this example, the region encoding thehspD gene on B32, which would be expected to occur on MTCY338, occurs instead on M. tuberculosis cosmid MTCY339 (which is located adjacent to, but not overlapping, MTCY427). Thus, there appears to have been a significant amount of gene shuffling between these two closely related species.

DNA-level matches between two M. leprae cosmids from this study and M. tuberculosis cosmids (Sanger Center, GenBank). The alignments show two particular examples from an exhaustive comparison of the set of M. leprae cosmids reported here against all available M. tuberculosis cosmids (see text). The shading indicates regions of significant similarity between each pair of cosmids. The alignments for cosmid L471 spanned a total of 26,302 nucleotides with 75%–94% identity; those for cosmid B32 spanned 24,009 nucleotides with 71%–85% identity. The M. lepraeORFs are color coded according to function as indicated. The sequences were compared and aligned using Cross_match, an implementation of the Smith–Waterman algorithm developed by P. Green (University of Washington, Seattle). Alignments with >60% identity were sorted using Matchtable (P. Richterich, unpubl.) and examined using a Web browser. A table summarizing the positions of aligned segments between each pair of cosmids was assembled; it was read by a Perl-tk script, Cosmid_map (R. Gibson, unpubl.) in conjunction with two other tables similar to Table 1 (but sorted by cosmid) summarizing the positions of coding frames and functional information (if available) for the M. leprae and M. tuberculosis cosmids.
Another example of apparent gene shuffling between mycobacterial species involves the mas genes and associated ORFs of M. leprae and the close M. tuberculosis relative,Mycobacterium bovis BCG. In BCG these genes are in the orderorfII, orfI, mas, and orfIII, with no more than 400 bp separating adjacent genes. In M. leprae, the apparent homologs are spread out over three regions. An M. leprae mashomolog with 58% identity to the BCG gene is located in cosmid B1170. A gene homologous (59% identity) to BCG orfIII(Q02278 | YMA2_MYCBO) occurs ∼7.5 kb away as the fourth gene in a putative operon that is transcribed from the opposite strand as mas. A gene homologous to BCG orfII(Q02279), which shows 81% identity over 349 codons, occurs in cosmid L518 as the terminal gene in a 30-kb large pks operon. The closest homolog to mas in this putative pks operon is 8.5 kb away. Although it is not certain that these mas-related genes ofM. leprae are orthologous to the BCG genes, it is quite clear that they are all members of a multigene (pks) family that may have arisen through gene duplication events.
At the protein level there are many strong similarities between M. leprae and M. tuberculosis gene products. We performed a cross comparison generating Smith–Waterman alignments between 1157M. leprae proteins (reported in this study and elsewhere) and 1564 M. tuberculosis protein sequences reported in public databases. A plot of the percent identity for the best alignment of each M. leprae protein against the M. tuberculosisdatabase is shown in Figure 6 (the percent identity values from long and short alignments were normalized by multiplying by the fraction of query amino acids represented in each alignment). Approximately one-quarter of the alignments (to the left of the vertical line in Fig.6) have normalized matches ranging from ∼40% to 87% identity. Most of these are likely to represent orthologous pairs, as at least 40% of the total M. tuberculosis proteins were represented in the target database. Most of the remaining proteins have matches ranging from 10% to 30% identity with at least one other M. tuberculosis protein in the data set. Although the stronger matches in this second group may represent alignments between paralogous members of protein families, the weaker ones are likely to represent only conserved motifs.
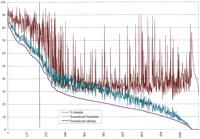
Summary of alignments from similarity searches between 1157 M. leprae proteins (including all of the gene products from this study) and 1564 M. tuberculosis proteins from GenPept. Each of the M. leprae proteins was searched against the set of M. tuberculosis proteins using an implementation of the Smith–Waterman algorithm with default parameters on a Biocellerator (Compugen) in conjunction with the GCG Wisconsin Package. The Normalized Similarity and % Identity values were obtained from the best alignment for each M. leprae protein by multiplying by the fraction of query amino acids represented in each alignment (no. of query residues in alignment/total query length). This was done to provide a better indication of the overall similarity of each M. leprae protein to the best M. tuberculosis homolog. The resulting values were termed Normalized Identity and Normalized Similarity. The pairs were sorted according to the Normalized Identity values in descending order, and the normalized values were plotted together with the raw percent identity values (for comparison) on a graph.
Examples of parologous mycobacterial genes include a DnaJ homolog on cosmid B1937, which shares 40% identity with the M. tuberculosis DnaJ protein and 38% identity with another, previously sequenced M. leprae DnaJ homolog that itself is 87% identical with the M. tuberculosisprotein. Similarly, a Chaperonin 60 homolog in the overlapping cosmids B229/B1620 is 61% identical to a previously sequenced M. leprae Ch60 gene and 61% identical to an M. tuberculosisCH60 gene, whereas the latter two are 94% identical to each other. The lack of a complete genomic data set from either organism precludes a definitive analysis of orthologs and gene families.
Relationships to Other Bacterial Genomes
The current M. leprae genome map and sequence were examined for collinearity of genes with E. coli, H. influenzae, M. genitalium, and B. subtilis. Although patterns possibly indicative of genome duplications conserved from B. subtilisto E. coli have been described (Kunisawa 1995), the M. leprae data only support limited clustering at the operon level of related functions. Such clustering may be advantageous for gene transfer or gene regulation and, hence, convergent. A similar observation of widespread scrambling but consistent clustering is seen in comparison of large operons in S. typhimurium andPseudomonas denitrificans (Roth et al. 1993). The clustering of genes in adjacent operons may reveal selective pressures to maintain proximity, for example, for recombination, coassembly, or coregulation. A cluster of seven genes in M. leprae are related to carbohydrate catabolism via the glycolytic and pentose phosphate pathways. Although no two of these genes is directly adjacent to each other in E. coli, two of them (tpi and pgk) are in the same order and orientation in B. subtilis. The gene order for the two transcriptionally converging operons in M. leprae is 5′ (tkt tal zwf) 3′ and 3′ (ppc tpi pgk gap) 5′, and the corresponding order inB. subtilis is 3′ (eno pgm tpi pgk) 5′. At least two similar clusters, including some of these genes, exist inE. coli; they are 5′ (gapB pgk fda) 3′ and 5′ (zwf edd eda) 3′.
The largest M. leprae region without identified genes covers ∼7 kb (on cosmid L471, mentioned above). Such regions are rare in bacteria, are generally <2 kb (Daniels et al. 1992; M. Raha, M. Kihara, I. Kawagishi, and R.M. Macaab, unpubl.), and are often later found to contain genes (Robison et al. 1994). This particular region does not appear to have any problems with data quality, yet it contains few ORFs over 100 codons, and none over 170 codons. The ORFs have poorM. leprae-specific codon usage and no significant database matches.
Genes with Similarities to Eukaryotes
A number of putative genes listed in Table 1 (including xheA, dhaP, leuA, udp, glyS, pepN, ctrA, hup, nakA, ybqR, ycjC, andybtY) have significant homology to known and hypothetical proteins from distant species (e.g., yeast, plants, human, and other eukaryotes). These sequences contain regions that could be called ancient conserved regions (ACRs) (Green et al. 1993), although a better term might be phylogenetically diverse conserved regions. The latter term has the advantage of avoiding the implication that such regions were actually maintained over the entirety of the time separating the most distantly related species, when instead they might represent remnants of more recent acquisitions by horizontal transfer. This possibility holds even in cases where the exact level of ancient in ACRs is specified (Doolittle et al. 1996).
METHODOLOGY
Genome Sequencing
Cosmids were selected from a mapped, overlapping set comprising four large segments of the M. leprae genome (Eiglmeier et al. 1993). The cosmid clones that were sequenced in this study were derived from three of these segments (contigs), and included cosmids depicted in Figure 1. High purity DNA preparations were made from each clone [using anion exchange columns with a procedure recommended by the manufacturer (Qiagen, Inc.) followed by equilibrium CsCl density gradient centrifugation]. The DNA was sheared by sonication or nebulization to an average size of 1–2 kb and fractionated on an agarose gel. A 1.5-kb average-size fraction was treated with T4 DNA polymerase, capped with BstXI linkers (5′-TCTAGACCACCTGC and 5′-GTGGTCTAGA in 100- to 1000-fold molar excess), gel purified, and ligated to one of a set of 20 uniquely tagged BstXI-cut plex vectors (Church and Kieffer-Higgins 1988) (M. Rubenfield, P. Rice, and D. Smith, unpubl.) to construct a series of shotgun subclone libraries. Each pool of 20 clones was picked using a 100 μl glass capillary attached to a light vacuum source to touch sequentially 20 colonies from different libraries. The capillary was then placed into a flask with growth medium to rinse out the cells. DNA was purified from a sufficient number of clones (Roach 1995) to obtain 5- to 10-fold sequence redundancy with 250- to 350-base average read lengths (typically 12 sets of 96 pools per cosmid).
DNA samples were chemically sequenced, separated on polyacrylamide gels, and transferred onto nylon membranes by electroblotting (Church and Kieffer-Higgins 1988) or by direct transfer electrophoresis from 40-cm gels (Richterich and Church 1993). In some cases, cycle sequencing reactions using Sequitherm polymerase were used. The DNA was covalently bound to the membranes by exposure to ultraviolet light and hybridized with labeled oligonucleotides complementary to tag sequences on the plex vectors (Church and Kieffer-Higgins 1988). The membranes were washed to remove nonspecifically bound probe and exposed to X-ray film to visualize individual sequence ladders. After autoradiography, the hybridized probe was removed by incubation at 65°C, and the hybridization cycle was repeated with another plex oligonucleotide until the membrane had been probed 25–41 times (depending on the number of templates present). Thus, each gel produced a large number of films, each containing new sequencing information. Whenever a new blot was processed, it was initially probed for an internal standard sequence added to each of the pools.
Digital images of the films were generated using a laser-scanning densitometer (Molecular Dynamics, Sunnyvale, CA). The digitized images were processed on computer workstations (VaxStation 4000’s) using the program REPLICA (Church et al. 1994). Image processing included lane straightening, contrast adjustment to smooth out intensity differences, and resolution enhancement by iterative gaussian deconvolution. The sequences were then automatically picked in REPLICA and displayed for interactive proofreading before being stored in a project database (each cosmid was saved in a separate project directory). The proofreading was accomplished by a quick visual scan of the film image followed by mouse clicks on the bands of the displayed image to modify the base calls. For most sequences derived by chemical sequencing, the error rate of the REPLICA base calling software was 2%–5%; a smaller percentage of samples had higher error rates, particularly near the end of the sequence read. Each sequence automatically receives a number corresponding to the blot number (microtiter plate and probe information) and lane set number (corresponding to microtiter plate columns). This number serves as a permanent identifier of the sequence so it is always possible to identify the origin of any particular sequence without recourse to a specialized database.
The sequences were assembled using the programs GTAC and FALCON (Church et al. 1994; Gryan 1995). These programs have proven to be fast and reliable for cosmid sequences. The assembled contigs are displayed using a modified version of GelAssemble, developed by the Genetics Computer Group (GCG) (Devereux et al. 1984) and modified by G. Church and P. Richterich to interact with REPLICA. This provides an integrated editor that allows multiple sequence gel images to be instantaneously called up from the REPLICA database and displayed to allow rapid scanning of contigs and proofreading of gel traces where discrepancies occur between different sequence reads in the assembly. Any ambiguous regions or regions with low coverage that required more coverage were resequenced by primer-directed cycled sequencing using commercially available kits and cosmid or multiplex pool templates. Each assembly was analyzed for regions with only single-strand coverage using the program SECSO (A. Graf, unpubl.).
Some of the cosmids—L518, B2126, L247, and B1170—contained large repeats that required additional analysis. In these cases, positional information associated with sequences derived from the two ends of each plasmid insert (which should have opposite orientation with respect to each other and should be separated by the average insert size of 1.5 kb) was used to remove misassembled sequences and align contigs in the proper order. This was done using the program CHECKMATES (R. Lundstrom and C. Tulig, unpubl.) which provides information on the location, spacing, and orientation of sequence pairs (mates) that do not fall within the normal range. (Comparison of cosmid restriction digests with two enzymes against predicted fragment sizes from the assembled sequence was used to verify correct assembly.) All contigs were analyzed using GenomeBrowser, and the output was examined to identify tRNAs, repetitive elements, and potential coding regions.
Acknowledgments
This work was funded by National Institutes of Health grants R01HG00520 and P01HG01106 to D.R.S. and J.M. and Department of Energy grant DE-FG02-87ER60565 to G.M.C.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding authors.
-
E-MAIL church{at}salt2.med.harvard.edu; smith{at}cric.com; FAX (617) 432-7663.
-
- Received February 13, 1997.
- Accepted June 10, 1997.
- Cold Spring Harbor Laboratory Press








