
Large-Scale Comparative Sequence Analysis of the Human and Murine Bruton’s Tyrosine Kinase Loci Reveals Conserved Regulatory Domains
Abstract
Large-scale genomic DNA sequencing of orthologous and paralogous loci in different species should contribute to a basic understanding of the evolution of both the protein-coding regions and noncoding regulatory elements. We compared 93 kb of human sequence to 89 kb of mouse sequence in the Bruton’s tyrosine kinase (BTK) region. In addition to showing the conservation of both position and orientation of the five functionally unrelated genes in the region (BTK, α-d-galactosidase A, L44L, FTP-3, and FCI-12), the comparison revealed conservation of clusters of noncoding sequence flanking the first exon of each gene. Furthermore, in the sequence comparison at the BTK locus, the conservation of clusters of noncoding sequence extends throughout the locus; the noncoding sequence is more highly conserved in the BTK locus in comparison to the flanking loci. This suggests a correlation with the complex developmental regulation of expression of btk. To determine whether a highly conserved 3.5-kb segment flanking the first exon of BTK contains transcriptional regulatory signals, we tested various portions of the segment for promoter and expression activity in several appropriate cell lines. The results demonstrate the contribution of the conserved region flanking the first exon to the cell lineage-specific expression pattern of btk. These data show the usefulness of large scale sequence comparisons to focus investigation on regions of noncoding sequence that play essential roles in complex gene regulation.
[The sequence data described in this paper have been submitted to GenBank under accession nos. U78027 and U58105.]
One analytical approach to functional characterization of both coding and noncoding DNA sequences is through comparison of sequence of syntenic regions in multiple species. Conserved noncoding sequence has been hypothesized to be important in regulating gene expression, maintaining the structural organization of the genome, and contributing to yet unknown functions of the chromosome (Koop and Hood 1994).
Sequence data from murine and human 5′ flanking or promoter regions is available for a substantial number of loci but no systematic comparison has yet been performed. Little data are available for sequence comparisons of intronic regions, as a relatively small number of genes have been sequenced in their entirety from more than one species. The few comparative sequence studies initiated between human and various species have revealed varying degrees of conservation. Comparisons of the β-globin gene cluster in both human and mouse (Collins and Weissman 1984; Sheehee et al. 1989) and the excision repair cross-complementing rodent repair group 2 (ERCC2) region in human, mouse, and hamster (Lamerdin et al. 1996) revealed little sequence conservation in the intergenic regions. A comparison of the X-ray repair cross-complementing rodent repair group 1 (XRCC1) between human and mouse revealed 22 individual conserved segments, some of which were as highly conserved as exons (Lamerdin et al. 1995). Most striking, however, was the alignment of nearly 100 kb of contiguous sequence from the α/δ T-cell receptor loci in human and mouse. Approximately 94% of the intergenic sequence was conserved allowing for the overall alignment of the entire two sequences. This alignment showed 71% similarity between the two sequences. In focusing on known regulatory elements the researchers concluded that these sequence comparisons will sometimes illuminate the existence of regulatory elements through the identification of highly conserved regions (Koop and Hood 1994).
X-linked agammaglobulinemia (XLA) was first described by Bruton in 1952 and is characterized by a severe deficiency of circulating mature B cells and serum immunoglobulins. Patients suffer from recurrent infections but can be maintained with immunoglobulin therapy (Bruton 1952; Rosen et al. 1984). The mutated gene in XLA, Bruton’s tyrosine kinase (BTK), was identified by a combination of positional cloning (Vetrie et al. 1993) and candidate gene approaches (Tsukada et al. 1993) and is a member of a subfamily of src-related nonreceptor tyrosine kinases (for review, see Rawlings and Witte 1995).
The BTK locus consists of 19 exons (Hagemann et al. 1994; Ohta et al. 1994; Rohrer et al. 1994) covering ∼40 kb. Sixty-nine kilobases of contiguous genomic sequence in the region reveals three genes within the 35-kb 5′ to BTK and an average of 0.7 Alu repeat elements per kilobase (Oeltjen et al. 1995). The availability of the genomic structure has permitted extensive mutation analysis. One hundred seventy-five unique mutations distributed throughout the BTK gene have been identified (Vihinen et al. 1995).
The expression of btk has been investigated using Northern analysis, Western blotting, and immunoprecipitation in a variety of lymphoid cell lines. btk is expressed in very early stages of B-cell differentiation before immunoglobulin heavy- or light-chain rearrangements and continues throughout B-cell maturation. Upon maturation to a plasma cell, btk expression is down-regulated. btk is also expressed in several different myeloid lines including myeloblast and mast cells, but is not expressed in T cells (deWeers et al. 1993; Smith et al. 1994). Although btk is expressed in various myeloid cell lines, the skewing of X-inactivation exclusively in the B-cell lineage found in female carriers of XLA (Allen et al. 1994) provides strong evidence that dysfunction of btk only affects the growth of early B-cell precursors.
Recently two different studies began to delineate the promoter, enhancer, and silencer elements important in this specific expression pattern of btk. In the first study, 310 bp of sequence 5′ to the transcription start site were shown to contain promoter activity in DG75 (B cell) and K562 (erythroleukemia) cell lines (Sideras et al. 1994). The second study examined promoter activity of a series of constructs from 50 to 910 bp 5′ to the transcription site in the HS Sultan plasmacytoma cell line. Expression of the most active construct, −450 bp, was also observed in the K-562 erythroleukemia cell line, but was down-regulated in the Jurkat T-cell line. Further analysis revealed the importance of the specific binding sites for transcription factors Sp1 and PU.1 within the 200-bp upstream of BTK for the expression activity of the −450 bp construct (Himmelmann et al. 1996).
The data reported here illustrate the use of large-scale interspecies sequence comparison whose primary aim is the identification of required regulatory elements. In principle, highly conserved noncoding DNA should contain elements important to the normal function of the locus. A direct comparison of 93 kb of human and 89 kb of mouse genomic sequence flanking the BTK locus reveals areas of high intronic or noncoding sequence conservation in all five loci in the region including BTK. Concentrating on a region of conservation extending 0.9 kb upstream and 2.6 kb downstream of the first exon of BTK, we demonstrate that these sequences function in lineage-specific transcriptional control of expression.
RESULTS
Sequencing of the Human and Murine Loci
Human genomic sequence of the region flanking BTK was obtained through random shotgun sequencing of an additional cosmid that overlapped the sequence reported previously from two other cosmids (Oeltjen et al. 1995). Sequencing of the three cosmids resulted in nearly 93 kb of contiguous human genomic sequence, which includes the entire BTK genomic locus (GenBank accession no. U78027). In sequencing the murine genomic Btk locus, we applied the same methods of random shotgun sequencing to sequence a single P1 clone spanning the same distance. Sequencing of the larger clone was accomplished through increases in the number of “random” sequencing reactions performed to gain sufficient coverage of the P1 before the “directed” phase of sequencing, and smaller adjustments to account for the increased data management and assembly. In a comparison of the number of reads required to complete the human sequence, the increase in clone size decreased the number of sequencing reactions needed to sequence completely the murine Btk locus by nearly one-third. This increase in efficiency is primarily attributable to a reduction in redundancy; >14 kb of human genomic sequence was resequenced as overlap between the two cosmids. Sequencing of the single P1 clone resulted in nearly 89 kb of contiguous murine genomic sequence including the entire Btk genomic locus (GenBank accession no. U58105).
Comparison of Coding Regions
Mapping of a murine P1 clone by hybridization showed the presence of three of the four genes previously mapped to the BTK region by sequencing the human region (Oeltjen et al. 1995). Sequencing demonstrated the presence of all four of these genes in the mouse, and another gene immediately 3′ of BTK, which was also revealed by further sequencing of a human-derived cosmid. All five genes are conserved between the two species in both orientation and exon organization (see Fig. 1A) of intergenic and intronic distances showed no gross (>5 kb) differences; however, in general, the murine locus has larger introns and greater intergenic distances than the human locus. Repeats in the region (as detailed below) did not fully account for the observed differences in intron and intergenic lengths between human and mouse.
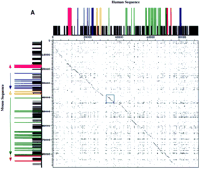
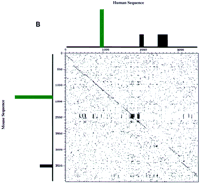
DOTTER dot matrix comparisons of human and mouse sequence in the BTK region. (A) Comparison of the entire genomic sequence in the region. Conservation in the dot blot is represented on a gray scale with black dots representing the highest conservation. Each axis has a graphical representation of the corresponding sequence in which all sizes are relative to each other and between species. Black bars represent all repeat sequences found. Individual exons are represented by coloration. Fuchsia represents FTP-3, blue represents α-d-galactosidase A, orange represents the L44L ribosomal gene, green represents BTK, red represents FCI-12, and purple represents the sequence with similarity to the L21 ribosomal gene. Direction of transcription is indicated by arrows on the mouse sequence and is the same for the human sequence. The region boxed in light blue is expanded in B and represents the conserved region flanking the first exon of BTK (green) complete with repetitive elements (black boxes) in both mouse and human.
Comparisons of the nucleic acid and predicted amino acid sequence of the five genes in the region are summarized in Table1. As expected, the amino acid sequence of the five genes is more highly conserved than the nucleic acid sequence. Further database analysis of the two genes in the region with unknown function, FTP-3 and FCI-12, revealed several points. In a BLAST (Altschul et al. 1990) search, FTP-3 matched expressed sequence tags (ESTs) from several different libraries including pancreas, parathyroid, melanocyte, retina, fetal heart, fetal brain, and aorta. This supports previous Northern blot analysis showing widespread expression (Vorechovsky et al. 1994). Because of the high similarity with an hnRNA-binding protein F, FTP-3 has been hypothesized previously to be an RNA-binding protein (Vorechovsky et al. 1994). This proposed function is further suggested in extensive database analyses with BEAUTY (Worely et al. 1995) which show 83% amino acid identity with the hnRNA-binding protein H (GenBank accession no. L22009) and identity with annotated RNA-binding domains in several other proteins from different species. In a BLAST search of the EST database, FCI-12 matched several clones from several different libraries. Predicted translation of the cDNA in the orientation published previously (Vorechovsky et al. 1994) revealed no extensive open reading frame that was conserved between the two species. Translation in the reverse direction, however, revealed a reading frame of 97 amino acids that spanned the two predicted exons in both mouse and human. Furthermore, AG/GT exon/intron borders were only found in the reverse complement of the FCI-12 cDNA in both species. Last, in BEAUTY (Worely et al. 1995) analysis, the reverse complement of FCI-12 showed similarity to aSchizosaccaromyces pombe hypothetical 11.4-kD protein on Chromosome 1 (GenBank accession no. Q09783).
Gene Comparisons
Database searches further downstream from FCI-12 in the human sequence revealed a region with identity to an L21 human ribosomal protein cDNA (GenBank accession no. U14967). Searches also showed two Alurepetitive elements in the region; a more detailed analysis showing that the repetitive elements had interrupted predicted exon/intron boundaries. Comparisons between the L21 sequence and the sequence downstream of BTK demonstrated 89.7% identity with several insertions and deletions. Predicted translations showed termination codons in the sequence. From this we conclude that this sequence downstream of BTK likely represents an archaic copy of L21 that has been rendered nonfunctional by Alu insertions.
Comparison of Repetitive Elements
Repetitive elements in both the murine and human sequence were localized and identified using CENSOR (Jurka et al. 1996). Both human and mouse show a high density of short interspersed repetitive elements (SINEs). In human, the average of 0.86 Alu repetitive elements per kilobases is fourfold higher than the estimated genome average of 0.2 Alu repetitive elements per kilobase (Deininger 1989;Jurka et al. 1993). In mouse, the average of 1.1 B1/B2 repetitive elements per kilobase is about 20-fold higher than that expected from estimates of the total number of such elements in mouse (Deininger 1989). Although the Alu repetitive element density is lower than corresponding B1/B2 repetitive element density, the total number of elements is similar in both human and mouse, 124 and 116, respectively. A comparison of the percentage of sequence occupied by repetitive elements in the human and mouse, 31.22% and 16.49%, respectively, demonstrates a substantial difference between the two species. This is accounted for, in part, by the shorter average length of the B1/B2 repetitive elements in comparison to Alurepetitive elements. The human sequence proceeds further downstream of BTK into a region of ∼8 kb in length composed nearly entirely of long interspersed nuclear repetitive elements LINEs.
Direct Comparison of the BTK Region in Human and Mouse
As shown in Figure 1A, a direct comparison of the sequence represented by a DOTTER dot plot analysis reveals high conservation in the region. Although the coding sequence in the region is conserved, much of the noncoding sequence flanking individual coding regions is also conserved. Straying of the diagonal corresponds with the location of interspersed repetitive elements in the sequence. These elements also account for the high background attributable to matching of the poly(A) tracts. A comparison of each genomic sequence to itself [which revealed several internal repeats in the human T-cell receptor β-chain locus (Rowen et al. 1996)] did not reveal any internal repeats in either the human or mouse sequence (data not shown).
As accommodated by DOTTER (Sonnhammer and Durbin 1995) and illustrated in Figure 1B, an expanded visualization of the “identity line” that extends through the sequence comparison reveals clustering of smaller individual stretches of conserved sequence. To visualize better both the patterns of and sequence contained within the smaller regions of conserved sequence, the entire two sequences were aligned. Alignments of the sequences were prepared using the program SIM (“similar”) (Huang et al. 1990) with default parameters and with SINE and LINE elements masked (see Methods). Visualization of the patterns of sequence conservation throughout the loci was accomplished by transforming the alignments into percent identity plots relative to positions in human sequence. The resulting data were drawn with a modified version of the local alignment to postscript (LAPS) program (Schwartz et al. 1991). To further analyze the sequence contained within the conserved segments of the SIM alignment, we wrote a program called CONSERVED to extract individual gap-free alignments. Parameters were set to limit the extracted sequence segments to a minimum of 50 bp with 60% sequence identity.
The resulting identity plot from the sequence alignment is shown in Figure 2. In examining the plot, several different features are evident. First, the 2.5-kb gap-free alignment has a 92% identity at the FTP-3 locus. This is consistent with the putative coding region of 449 amino acids within the single exon; however, when compared to the remainder of the coding regions in the locus and their associated conservation, the conservation at the FTP-3 locus is striking. Throughout the remainder of the locus, the conservation of the coding sequence, as individual exons, is variable.
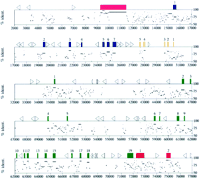
Identity plot of the sequence in the BTK region. The mouse and human genomic sequence were aligned with SIM and the identity between the conserved segments are displayed in relation to the human sequence by a modified version of LAPS. As in Fig. 1, individual exons are represented by coloration. Exon numbers are displayed above the exon blocks. Triangles represent the location and orientation of the humanAlu repetitive elements in the region.
The most interesting feature illustrated by the identity plot is the patterns of conservation of noncoding sequence in the region. In comparing the sequence conservation between the two ubiquitously expressed genes L44L and α-d-galactosidaseA (GLA) to the more specifically expressed BTK locus, the noncoding sequence is apparently more conserved within the BTK locus than within the L44L locus and especially within the GLA locus. Noncoding sequence conservation in both the L44L and GLA loci is confined primarily to the region flanking the first exons. Specifically within the GLA locus, the sequence from 15018 to 15318, which corresponds with a CpG island, aligns as a gap-free match with 87% identity. This 300-bp region is as conserved as the exons within GLA. Within the BTK locus, there are strong matches throughout the locus, particularly in the regions upstream of the first exon, at both ends of the first intron, within the fourth and fifth introns, between the eighth and tenth exons, and between the thirteenth through sixteenth exons.
In extracting the smaller individual alignments from the overall alignment using CONSERVED, other trends in the conservation are apparent. In the alignment presented here, a total of 179 individual alignments were localized with >60% sequence identity, including the 34 coding exons mentioned previously (see Fig. 2). These alignments vary in size from 50 to 2199 bp and demonstrate identities ranging from 60% to 95% between the human and mouse sequence. In the human sequence, a total of 18,634 bp of conserved sequence was identified representing 25% of the total sequence compared.
Further distinctions can be made between the coding and noncoding sequence that is conserved (see Table 2). In the human sequence, a total of 11,193 bp of noncoding sequence is conserved representing 16% of the total human noncoding sequence compared. In comparing the percent identity of the conserved noncoding sequence with the percent identity of the conserved coding sequence, the coding sequence is more conserved than the noncoding sequence. A weighted percent identity can be calculated by ΣC i F i/N, where C i is the length in base pairs of each individual aligned sequence of either mRNA coding or noncoding sequence, F i is the percent identity of the aligned sequence, and N is the total number in base pairs of compared coding or noncoding sequence. The weighted percent identity yields an average of 87% identity for the human coding sequence and an average of 73% identity for the human conserved noncoding sequence.
Summary of Sequence Conservation in the BTK Region
The percent identity plot shows that of the four loci in the BTK region that contain introns, there are differences in the amount of conserved noncoding sequence. When the total amount of conserved noncoding sequence for each locus is calculated, the human BTK locus has 22% of the total noncoding sequence conserved, the human GLA locus has 12% of the noncoding sequence conserved, the human FCI-12 locus has 16% of the noncoding sequence conserved, and the human L44L locus has 5% of the total noncoding sequence conserved. Thus, the BTK locus has the highest amount of noncoding sequence conserved of all five loci in the region. In particular, the BTK locus has nearly 10% more of its noncoding sequence conserved as compared to the other disease-related gene in the region, the GLA locus.
Conservation of the noncoding sequence in the GLA locus is restricted primarily to upstream and downstream of the first exon. The conservation of several regions downstream of the first exon is comparable to the conservation of the first exon of GLA. The restriction of noncoding sequence conservation between human and mouse to upstream and downstream of the first exon is more apparent in the L44L locus. A 133-bp region within the first intron of L44L is conserved with 62% identity. Within the FCI-12 locus, sequence is also conserved between human and mouse upstream and downstream of the first exon. Within the first intron, a 58-bp region is 83% identical, as conserved as the entire cDNA between the two species. In addition, a 226-bp region downstream of the second exon is 78% identical.
Upstream of the BTK locus, four different regions totaling 377 bp are conserved between the two species. Within this region, a 94-bp region of human sequence is conserved with 90% identity, again as conserved as the coding exons. Three regions downstream of the first exon also demonstrate this trend, a 62-bp region with 85% identity, a 59-bp region with 86% identity, and a 53-bp region with 87% identity. Within the first intron, a total of 2836 bp of sequence is conserved that can be divided into 35 individual alignments. As with the remainder of the conserved regions in the BTK locus, breaks between these individually aligned regions often correspond with the insertion of repetitive elements in either or both of the species.
The second cluster of noncoding sequence conservation in the BTK locus spans the fourth and fifth introns encompassing a total of 1824 bp in human. As with the region flanking the first exon of BTK, this region can be subdivided further into 23 individual alignments. These alignments range in size from 50 to 229 bp and in identity from 63% to 90%. Of note, within the fifth intron is a 229-bp region with 90% identity between human and mouse. With conservation similar to that of surrounding exons, this region could be hypothesized to contain coding sequence. Attempts to translate the sequence, however, identify at least two stop codons in all six potential reading frames, and BLAST searches against protein and EST databases do not show any matches.
In introns 9 and 10, a total of 417 bp of sequence is conserved and can be divided into five individual alignments ranging in size from 72 to 91 bp and in identity from 64% to 75%. In introns 13-15, a total of 1126 bp of sequence is conserved between human and mouse and can be divided into 15 individual alignments ranging in size from 50 to 156 bp and in identity from 65% to 81%.
Nucleotide Divergence Rates in the Region
In reviewing the human and rodent DNA sequence comparisons available to date, Koop (1995) hypothesized that large portions of the genome evolve at different rates. This conjecture was based on differences seen in the conservation between several different human and mouse loci. The wide variation suggests that not all coding sequence diverges at the same rate (Koop 1995).
Because the noncoding sequence in the BTK region appears to be conserved differentially, a constant that could be used to compare the evolutionary divergence across the region was sought. Synonymous nucleotide substitution rates in coding sequence should provide insight into the rates of evolutionary divergence, (i.e., mutation rates) in this segment of the X chromosome. If the nucleotide mutation rates across the region are constant and not mosaic, synonymous nucleotide substitution rates within all five loci should be similar.
Several methods of nucleotide distances in synonymous and nonsynonymous substitutions within coding regions have been derived that attempt to estimate the different variables affecting nucleotide substitutions including transition and transversion rate differences and species-specific codon usage preferences. The most recent methods ofIna (1995), as shown through computer simulation comparisons with previously proposed methods of calculation, appear to be the most robust.
Nucleotide substitution rates were calculated using both methods for estimating the numbers of synonymous and nonsynonymous substitutions for all five loci in the BTK region (Ina 1995) and the estimate of human and rodent divergence of 80 million years (Li and Graur 1991). As shown in Table 3, the nonsynonymous substitutions differ greatly between the different genes, from 0.00 for the L44L protein, which is 100% identical, to 8.17 × 10−10substitutions per nucleotide per year for the GLA gene. These figures parallel the conservation shown in comparing the percent identity between human and mouse for all five loci. In contrast, the synonymous substitution rate is not nearly as variable. Although these values are very similar, the outlying values calculated by Ina’s method 1 (and nearly by method 2) are more than two standard deviations apart from each other. This would suggest that the mutation rate, even across such a small region, is variable. Statistically substantiating this conclusion, however, is difficult because of both the small number of loci and the small number of individual codons examined. If, however, the synonymous nucleotide substitution rates can be considered as indications of the rates of divergence across the locus, the substitution rates do not correlate with the observed patterns of noncoding sequence conservation. The locus that demonstrates the highest amount of noncoding sequence conservation, BTK, does not have the lowest synonymous nucleotide substitution rate in the region.
Nonsynonymous and Synonymous Mutation Rates of the Five Loci in the BTK Region
Conservation of Potential Transcription Factor-Binding Sites
The conserved sequences mentioned above that correspond with the first exons of each of the five loci and with the fifth exon of BTK were analyzed for the presence of potential transcription factor-binding sites using TFSEARCH. TFSEARCH uses the TFMATRIX matrix table of the TRANSFAC database. The searches were performed on both human and mouse sequence and then compared manually. Conservation of sequence, orientation, and location were accounted for.
Four different clusters of potential transcription factor-binding sequences are observed. The first is upstream of GLA and the other three are within the BTK locus: one upstream of the first exon, one downstream of the first exon, and the third downstream of the fifth exon.
The identities of the transcription factors associated with the conserved potential-binding sites, although varied, provide some possible insight into the regulation of btk. Of the different potential transcription factor-binding sites conserved in the regions flanking the first and fifth exons of BTK, several are associated with transcription factors that have been shown previously to be either expressed in or to be important in the development of the hematopoietic lineages and, more specifically, the lymphoid lineages. These include Ap1-b, CCAAT/enhancer-binding protein β (C/EBPβ), c-ETS-1, c-Rel, GATA1, GATA2, GATA3, necrosis factor-E2 (NF-E2), and, NF-κB (for review, see Kerhl 1995). Furthermore, both the PU.1 and Sp1 sites that were shown previously to be important in btk expression (Scott et al. 1994) were also conserved between the two species. The previously noted inverted CCAAT box (Scott et al. 1994) and potential retinoic acid receptor-binding sites (Rohrer et al. 1994) in the upstream region of BTK were not conserved between the two species.
Promoter and Enhancer Activity of the Conserved Region Flanking the First Exon of BTK
As the main goal of this comparative analysis was to illuminate conserved regions that might function as regulatory elements of BTK, we focused on the conserved sequence flanking the first exon of BTK. Shown in the direct comparison in Figure 1B, the sequence 900 bp upstream and ∼2.5 kb downstream of the first exon is highly conserved. At the sequence level, the dark box in the middle of the dot matrix represents a GA-rich simple repeat region that is present in both species. A direct alignment of the sequence over the entire region represented in Figure 1B demonstrates a 70.89% identity (data not shown).
To investigate the potential biological activity of this conserved region in the expression of btk, we analyzed its ability to act as a promoter and enhancer in cell lines representing three relevant developmental lineages (deWeers et al. 1993; Smith et al. 1994). We examined the interaction of the previously determined promoter (−350 bp) (Sideras et al. 1994; Himmelmann et al. 1996) to interact with the 2.5 kb of conserved intron sequence. Four constructs were generated to compare the promoter and enhancer activity of this conserved region that used the splicing of the first exon to the second exon of BTK with luciferase inserted into the second exon before the start translation signal. These constructs were transfected into the K-562 (myeloid), HS Sultan (B), and CEM (T) cell lines.
The results of transient transfection assays with these constructs are represented in Figure 3. Each construct showed differing expression both within and between each cell line. In the K-562 cell line, promoter activity within the first 360 bp upstream of the first exon was enhanced by the addition of 530 bp further upstream of the first exon. Addition of the conserved 2540 bp of the first intron acted as a silencer in both constructs in the K-562 cell line. In the HS Sultan cell line, the promoter activity within the first 360 bp upstream of the first exon was attenuated partially by the addition of an additional 530 bp upstream of the first exon and no similar activity was observed with the first intron segment. In the CEM cell line, promoter activity of 360 and 890 bp upstream of the first exon was observed. This promoter activity was silenced to levels equivalent to the negative control construct (data not shown) by the addition of the 2540 bp of the first intron. In summary, the conserved sequence in the first intron of BTK represses the expression from the BTK promoter in both myeloid and T cells but not in B cells. We conclude that elements within the first intron contribute to the lineage specific down-regulation of btk expression in T cells and myeloid cells.
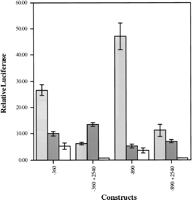
Luciferase assay of the promoter and enhancer activity of the conserved sequence flanking the first exon of BTK. Pictured is the luciferase expression observed in K562 myeloid cells (light shaded bar), HS Sultan B cells (dark shaded bar), and CEM T cells (open bar), with four different constructs containing portions of the sequence flanking the first exon of BTK as stated in base pairs. Each construct was coelectroporated a total of nine times (three sets of three) with CMVβgal and results were pooled and compared directly using β-galactosidase expression as a control.
DISCUSSION
We undertook the comparison of the human and mouse genomic sequence flanking BTK to investigate conserved elements that could be responsible for the specific expression patterns of btk. The study revealed a markedly conserved region containing five different genes. In addition to conserved exons lengths and orientation of the five genes, intronic and intergenic lengths are also conserved, and both the human and mouse sequences are rich in repeats. At the DNA level, sequence conservation extends beyond the exon/intron borders in all five genes in clusters interrupted primarily by repetitive element insertions.
A more detailed examination of the conservation reveals 179 individual conserved regions including the 34 coding exons. Between the amount of human sequence comprised of repetitive (34%) and conserved (25%) sequence, nearly 60% of the entire human sequence in the region is accounted for. Although less conserved than the 34 coding exons, the 145 noncoding aligned segments are substantially more conserved than the flanking sequence, as illustrated by the weighted percent identity. These aligned segments are nonrandomly distributed being clustered around the first exon of each gene and within the fourth through fifth, eighth through ninth, and thirteenth through sixteenth introns of BTK. Transcription factor-binding site database searches revealed several different conserved sites in sequence, orientation, and location within these aligned regions. Specifically within the BTK locus, several potential transcription factor-binding sites of these factors, which have been demonstrated previously to be important in hematopoietic cell lineage development, are conserved.
Within the BTK locus, conservation of noncoding sequence points to several different regions that are highly conserved and are hypothesized to be important in the expression and structural organization of BTK. Focusing on the region flanking the first exon of BTK reveals a 3.5-kb region of 71% identity. Transient expression analysis in relevant cell lines indicates that this 3.5-kb region contributes to the lineage-specific expression pattern of btk. The specific expression pattern mimics that observed for the expression of btk in which expression is silenced in T cells (Tsukada et al. 1993).
Numerous precedents for control elements within the first intron of genes have been described, but comparative sequence data are rare. One example is provided by another gene important in hematopoietic development, adenosine deaminase (ADA). ADA is essential for purine catabolism and mutations in ADA result in a failure to develop cortical thymocytes. A 2.3-kb human intronic thymic locus-controlling region (LCR) has been shown to include a 200 bp DNase I hypersensitive region flanked by extended segments, which are required for insertion site-independent and copy number-proportional transgene expression (Aronow et al. 1989, 1992, 1995). Further analysis revealed a 28-bp core within the 200-bp region, which specifically binds the transcription factor c-Myb (Ess et al. 1995). In comparison to the murine sequence, a 236-bp first intron segment with 71.1% identity to the human ADA thymic enhancer was found. Four highly conserved regions within this segment were further delineated, including a 72-bp region with 83.6% identity to the 28-bp human core enhancer sequence. The conserved 236-bp region demonstrated only weak activation as compared to corresponding human sequence (Brickner et al. 1995) suggesting that other elements outside of the smaller controlling region are important. Further investigation of the conserved first intron sequence in BTK is also expected to reveal both the binding of specific transcription factors and flanking sequence that contribute to the expression of BTK.
In an overview of the conservation observed in the BTK region, one general theme arises. As compared to the other genes in the region, the noncoding sequence within the BTK locus is more conserved and contains more conserved potential transcription factor-binding sites. This suggests the hypothesis that the special regulation of expression of btk, as compared to the other ubiquitously expressed genes in the region, simply requires more regulatory elements. In both human and mouse this region of the genome has been invaded extensively by repetitive elements serving as a manner of naturally saturating insertional mutagenesis. The positions of the insertion of these elements are nearly identical between the two species suggesting permissive and nonpermissive segments. In addition, other mutational mechanisms have caused essentially complete divergence of more than half of the remaining noncoding sequence. It is interesting, however, that conservation in the BTK locus of up to several kilobases of sequence flanks the 10- to 20-bp consensus sequence for the transcription factor-binding sites. Although the binding of transcription factors represents a fundamental mechanism for controlling transcription, the conservation of large regions flanking these binding sites suggests that a higher order DNA structure needs to be preserved. This preserved structure must be dependent on the specific sequence rather than on spacing alone. Whether this structure is preserved for the binding of transcription factors, transcription of the region by RNA polymerase, or even splicing of the transcript is still speculative. Although it has been suggested that some sequence conservation can be attributed to pure chance (Koop and Hood 1994), including variations in the evolutionary divergence of the sequence (Koop 1995), the length of the conserved segments and the differences in conservation noted between the different loci in the BTK region suggest that apparent conservation of sequence (homoplasy) attributable to stochastic effects or even convergence play little role in the conservation observed. This is further substantiated by an inability to demonstrate a correlation between coding nucleotide substitution rates and the conservation of flanking noncoding sequence. It would appear that the conserved blocks of sequences are constrained by functional selection. The inability to measure a functional activity of one or more of these conserved segments by conventional transgenic or targeted mutagenesis experiments would more likely reflect a limitation of analytical method. Therefore, we hypothesize that all of the conserved noncoding segments are (or were) required functionally for the proper expression of the btk locus.
This analysis represents the second largest comparison of human and mouse DNA sequence and the largest representing a region of diversely expressed and functionally unique genes. We believe that evolutionary sequence comparison will prove to be both the most robust and the fastest way to identify functional noncoding sequence, especially in very large loci, with complex developmental regulation as with BTK. In using the large-scale sequence comparison as the primary means of identifying gene regulatory elements, we revealed the contribution of the intronic sequence downstream of the first exon to the expression pattern of btk. Future work will focus on both further delineating the sequences flanking the first exon that control btk expression and examining the contribution of other conserved sequences in the locus to btk expression.
METHODS
Isolation of Cosmid and P1 Clones
The human genomic sequence was obtained through sequencing three different cosmids from the Lawrence Livermore X-chromosome specific library. Isolation and sequencing of the first two cosmids, U230D1 and U237D10, was reported previously (Oeltjen et al. 1995). The third cosmid, U166B1, was isolated using a BTK exon 19-specific probe generated by PCR to probe the X-chromosome library. Positive clones were further mapped by PCR to obtain a single clone with minimal overlap. Cosmid DNA was isolated using a combination of Qiagen maxiprep purification protocols (Qiagen Inc., Chatsworth, CA) and equilibrium centrifugation in CsCl gradients (Sambrook et al. 1989).
The mouse genomic sequence was obtained through sequencing a single P1 clone isolated from the Genome Systems Inc. (St. Louis, MO) C-129 murine P1 library. The library was screened by Genome Systems with two sets of PCR primers. The first set was specific for the second exon of murine α-galactosidase A (CgggATgCAggTTATgACTA, ggACgTAATTTgCgAggTgT). Positive clones were rescreened with a set of primers that amplify intron 11 of BTK sequence (CCgTgTCTgTgTTTgCTAA, ggTAATACTggCTCTgTgg). The single positive clone P1-6186 was then mapped further by hybridization and found to span the entire BTK locus through the FTP-3 gene 5′ of BTK. P1 DNA was isolated using a modified Qiagen protocol (Cheryl Ericsson, Dr. Martin’s Laboratory, Humane Genome Center, Lawrence Berkeley Laboratory, pers. comm.) in combination with equilibrium centrifugation in CsCl gradients (Sambrook et al. 1989).
Sequencing of Cosmid and P1 Clones
A random shotgun sequencing library was generated for both the cosmid and P1 clones using the ΔM13 adaptor-based strategy described (Andersson et al. 1994). Sequencing templates were prepared by a modified glass fiber filter (GFC) capture method as described (Kristensen et al. 1987). Clones were sequenced using both random and directed sequencing protocols as described (Civitello et al. 1993). Reverse templates were generated from M13 supernatants using a modified asymmetric PCR protocol (Muzny et al. 1994). Dye primer sequencing reactions were assembled using a Biomek 2000 workstation (Beckman Instruments Inc., Fullerton, CA) and cycled on Perkin Elmer Cetus (PEC) 9600 thermocyclers. For sequencing reagents, Perkin Elmer TaqFS was used with a combination of standard fluorescein- and rhodamine-labeled dye terminator and dye primers purchased from Perkin Elmer and dipyrrometheneboron difluoride (BODIPY) labeled dye primers (Metzker et al. 1996). Sequencing reactions were electrophoresed on Applied Biosystems (ABI) 370, 373, 373A, or 377 sequencers.
Sequence Assembly and Gap Closure
Raw sequence data were transferred to a UNIX platform for editing and assembly. Vector and low-quality data were removed using SEQPREP software developed by the Molecular Biology Computational Resource Center at Baylor College of Medicine. Cosmid and P1 clone sequence assemblies were performed using Staden XGAP software (Dear and Staden 1991). Gap closure was achieved through a mapgap strategy as described (Richards et al. 1994). Regions resilient to cloning in M13 vectors were amplified by PCR, cloned into pGEMT-vector (Promega Corp., Madison, WI), and sequenced with dye-terminator chemistry. Sequencing was completed on both strands except within the human regions of high repeat density in cosmid U166B1 where at least four reads in a single direction using both dye terminator and dye primer chemistry were used to resolve the sequence.
The random shotgun strategy used in sequencing cosmids was adapted to the sequencing of a single P1 clone simply by increasing the number of “random” forward and reverse sequencing reactions (Richards et al. 1994). In comparing the final sequencing read statistics, a greater efficiency was accomplished in sequencing the larger clone primarily attributable to elimination of overlapping sequence. If the overlap between the cosmids is accounted for (the total length of sequence generated from three different cosmids is divided by the total number of reads for all three cosmids), an average of 26.21 reads per kilobase of sequence were required to obtain the same sequence at a cost of 16.41 reads per kilobase of sequence using the P1 as a template. This decrease in number of reads per kilobase is accompanied by an overall decrease in the number of primers generated to finish the double-stranded sequence.
Sequence Analysis and Comparison
The sequences were analyzed with a variety of computer software programs including BLAST (Altschul et al. 1994), XGRAIL 1.2 (Uberbacher and Mural 1991), BEAUTY (Worely et al. 1995), and GCG (sequence analysis software package, v. 8, Genetics Computer Group, Madison, WI). Repetitive elements in both human and mouse were localized and identified by CENSOR (Jurka et al. 1996). Comparative sequence analysis was dependent primarily on the June 1996 update of DOTTER (Sonnhammer and Durbin 1995). DOTTER window size was set at 100. For Figure 1A, the gray amp tool was set at a minimum of 3 and a maximum of 24. For Figure1B, the gray amp tool was set at a minimum of 26 and a maximum of 39. These programs were accessed through the Baylor College of Medicine genome informatics core (http://kiwi.imgen.bcm.tmc.eud:8088/search-launcher/launcher.html).
Entire sequence alignments with the SINE and LINE repeats masked were prepared using the program SIM (Huang et al. 1990) with default parameters. That is, matching nucleotides scored 1, mismatches scored −1, a gap of length k was penalized 6 + 0.2k, and we retained only the alignments scoring above the 95% significance level, as described in Schwartz et al. (1991). A few high-scoring but spurious alignments (e.g., involving dinucleotide repeats) were discarded. The remaining alignments were transformed into percent-identity information relative to positions in the human sequence. To do this, the mouse coordinates of each gap-free segment of an alignment were replaced by the percent of identical nucleotides in that segment. The resulting data were drawn with a modified version of the LAPS program (Schwartz et al. 1991). Individual noncoding gap-free alignments with >60% identity for longer than 50 bp were extracted from the SIM alignment with the CONSERVED program. All 34 coding exons were isolated manually from both genomic sequences and aligned using the GCG BESTFIT program.
Synonymous and nonsynonymous substitution rates were calculated using the two methods DISTS1 and DISTS2 from Ina (1995).
Transcription factor-binding sites were located using TFSEARCH (1995, Yutaka Akiyama, Kyoto University, Japan) accessed through the World Wide Web site http://www.genome.ad.jp/SIT/TFSEARCH.html. TFSEARCH searches highly correlated sequence fragments versus TFMATRIX transcription factor-binding site profile database, TRANSFAC MATRIX TABLE (Rel.2.5 17-11-1995). Searches were restricted to the vertebrate database.
Construction of Promoter Constructs
Four different constructs were generated containing the base pairs −360/+16, −360/+2537, −887/+16, and −887/+2537 in respect to the first exon of BTK. All four fragments were isolated using combination of long PCR (Cheng et al. 1994) Perkin Elmer XL PCR kit (Perkin Elmer Corp.) and restriction enzyme digests. All four fragments were cloned by blunt ligation into the BamHI site of a modified Bluescript (BS) SK− vector (Stratagene). The modified BS vector contained luciferase excised from RSVLuc2 (deWet et al. 1987) and blunt ligated into the EcoRI site downstream of a fragment, which contained 311 bp of genomic sequence upstream and the first 19 bp of the second exon of BTK, blunt ligated into the SmaI site of BS. This second BTK fragment was also isolated by a combination of PCR and restriction digest to generate a fragment containing the splice acceptor of exon 2 without the start translation codon of BTK. All PCRs generating genomic fragments from the BTK locus used cosmid U237D10 DNA as a template. Control constructs cytomegalovirus (CMV) β-galactosidase (CMVβgal) and CMV luciferase (CMVLuc) were kindly provided by Dr. Gretchen Darlington’s laboratory (Baylor College of Medicine, Houston, TX). Constructs were verified by sequencing and were isolated for electroporation by a combination of Wizard maxiprep purification protocols (Promega Corp., Madison, WI) and equilibrium centrifugation in CsCl gradients (Sambrook et al. 1989).
Transient Transfection and Luciferase Assays
HS Sultan, K562, and CEM cells were maintained in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS) at 37°C in 5% CO2 and synchronized by replating at recommended densities 24 hr before electroporation. For electroporation, cells were harvested and resuspended in either PBS (K-562) or RPMI-1640 (HS Sultan and CEM) medium supplemented with 10% FCS and 10 mm HEPES buffer (pH 7.5). Six micrograms (HS Sultan and K-562) or 24 μg (CEM) of luciferase reporter construct and 1 μg of CMVβgal control construct were added to 2.5 × 106 cells in 125 μl of medium and incubated 5 min at room temperature. The DNA was transfected into the cells through electroporation using Bio-Rad (Hercules, CA) Gene Pulser cuvettes (0.2 cm) and a BTX Electrocell Manipulator 600 (San Diego, CA) at 200 V/1000 μF (K-562) and 125 V/3000 μF (HS Sultan and CEM). Transfected cells were incubated for 24 hr, harvested, and lysed with Promega Reporter Lysis Buffer. Aliquots of the cell lysate were assayed for β-galactosidase activity, with Clonetech (Palo Alto, CA) Luminescent β-galactosidase Genetic Detection Kit II, and luciferase activity, with Promega Luciferase Assay Systems, in a Turner Designs TD-20e Luminometer (Mt. View, CA). Comparisons between different cell lines and electroporations used CMVβgal expression as the constant.
Acknowledgments
We thank M. Ali Ansari-Lari and Dr. Gretchen Darlington’s laboratory for their assistance. This work was supported by National Institutes of Health (NIH) grant R01HL51232 (J.W.B.), NIH grant R01HG01459 (R.A.G.), National Library of Medicine grant LM05110 (W.M.), and NIH Medical Scientist Training Program training grant (J.C.O.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL belmont{at}bcm.tme.edu; FAX (713) 798-5386.
-
- Received December 16, 1996.
- Accepted February 12, 1997.
- Cold Spring Harbor Laboratory Press








