
Endonucleolytic cleavage is the primary mechanism of decay elicited by C. elegans nonsense-mediated mRNA decay
- 1Department of Molecular, Cellular, and Developmental Biology, University of California at Santa Cruz, Santa Cruz, California 95064, USA;
- 2RNA Center, University of California at Santa Cruz, Santa Cruz, California 95064, USA;
- 3Genomics Institute, University of California at Santa Cruz, Santa Cruz, California 95064, USA
Abstract
Premature stop codon–containing mRNAs can produce truncated and dominantly acting proteins that harm cells. Eukaryotic cells protect themselves by degrading such mRNAs via the nonsense-mediated mRNA decay (NMD) pathway. The precise reactions by which cells attack NMD-target mRNAs remain obscure, precluding a biochemical understanding of NMD and hampering therapeutic efforts to control NMD. Here, we modify and deploy single-molecule nanopore mRNA sequencing to clarify the route by which NMD targets are attacked in an animal. We obtain single-molecule measures of splicing isoform, cleavage state, and poly(A) tail length. We observe robust endonucleolytic cleavage of NMD targets in vivo that depends on the nuclease SMG-6. We show that NMD-target mRNAs experience deadenylation and decapping, similar to that of normal mRNAs. Furthermore, we show that a factor (SMG-5) that historically was ascribed a function in deadenylation and decapping is in fact required for SMG-6-mediated cleavage. Our results support a model in which NMD factors act in concert to degrade NMD targets in animals via an endonucleolytic cleavage near the stop codon, and we suggest that deadenylation and decapping are normal parts of mRNA (and NMD target) maturation and decay rather than unique facets of NMD.
Nonsense-mediated mRNA decay (NMD) is a post-transcriptional surveillance system that targets and degrades mRNAs containing premature termination codons (PTCs), which could otherwise produce truncated and/or toxic proteins (for review, see Kurosaki et al. 2019). NMD plays a crucial role in human health by degrading mRNAs containing PTCs, which drive diverse diseases (Mort et al. 2008). NMD also regulates the abundance of ∼5%–20% of endogenous transcripts in animals, making NMD broadly relevant for gene expression (Mendell et al. 2004; Rehwinkel et al. 2005; Wittmann et al. 2006; Ramani et al. 2009; Muir et al. 2018; Kim et al. 2022).
Multiple competing models exist to describe how NMD-targeted mRNAs are degraded by cellular machinery in animals, featuring endonucleolytic cleavage, deadenylation, and/or decapping. In the endonuclease model, the metal-dependent endonuclease domain of SMG-6 cleaves NMD targets (Gatfield et al. 2003; Glavan et al. 2006; Eberle et al. 2009). The essential role of SMG-6 in NMD function is supported by several studies (Colombo et al. 2017; Alexandrov et al. 2017; Baird et al. 2018; Zhu et al. 2020; Boehm et al. 2021; Zinshteyn et al. 2021; Kim et al. 2022) and is challenged by others (Jonas et al. 2013; Loh et al. 2013; Metze et al. 2013; Huth et al. 2022). In an alternative to SMG-6-mediated cleavage, deadenylation of NMD targets occurs via SMG-5 and SMG-7 (Chen and Shyu 2003; Lejeune et al. 2003; Yamashita et al. 2005; Loh et al. 2013). SMG-7 associates with the deadenylase complex factor CNOT8 (previously known as POP2) and elicits mRNA decay in tethering assays (Loh et al. 2013), and NMD-targeted mRNAs experience deadenylation in mammalian cells (Chen and Shyu 2003; Lejeune et al. 2003; Yamashita et al. 2005). SMG-5 plays an additional role by promoting deadenylation-independent decapping through recruitment of the decapping complex and its enhancer PNRC2 (Lykke-Andersen 2002; Lejeune et al. 2003; Cho et al. 2009, 2013; Lai et al. 2012). Any one of these degradation methods—decapping, deadenylation, or cleavage—would be sufficient to destabilize targeted mRNAs and mitigate truncated protein production. The presence of independent degradation pathways conflicts with genetic evidence supporting the requirement of all of SMG-1 through SMG-7 in Caenorhabditis elegans’ NMD (Hodgkin et al. 1989; Cali et al. 1999; Anders et al. 2003).
A contemporary SMG-5/6/7 study by the Gehring group supports a single pathway of decay in human cells, more closely aligned with the initial C. elegans genetics (Boehm et al. 2021). In the work of Boehm et al. (2021), loss of any one of SMG-5/6/7 stabilized a similar set of target mRNAs in human cells. Their work challenges the idea that SMG-5/7 target one set of mRNAs, and SMG-6 a separate set, and concludes that SMG-5/7 somehow license mRNAs for cleavage by SMG-6. These results raise a number of questions: Are similar observations true across animals? Given the findings in human cells, do SMG-5/7 function in deadenylation/decapping, SMG-6-mediated decay, or both? The genetics clearly substantiate a requirement for SMG-5/7 in SMG-6-mediated decay, but two models could explain the data: (1) SMG-5/7 deadenylate and/or decap NMD targets, which is a prerequisite for SMG-6-mediated cleavage; and (2) SMG-5/7's main biochemical function is to stimulate SMG-6-mediated cleavage. As Boehm et al. examined RNA levels and cleavage events, but not poly(A) status, either model remains possible. If it turns out that SMG-5/7 do not deadenylate NMD targets, what explains the existing literature around deadenylation in NMD?
The understanding of poly(A) tails, deadenylation, and decapping in normal (non-PTC-containing) mRNA metabolism improved over the past several years (for review, see Zhang et al. 2023). Contemporary studies suggest that deadenylation is a part of normal mRNA maturation as well as normal mRNA decay (Chang et al. 2014; Lima et al. 2017; Yi et al. 2018; Eisen et al. 2020; Tudek et al. 2021; Alles et al. 2023; Park et al. 2023). Nascent mRNAs have a long poly(A) tail that is partially deadenylated to an approximately 30–70 poly(A) tail as the mRNA matures. Another set of reactions can shorten the poly(A) tail and expose the 3′ end of the message, facilitating decapping at the 5′ end and exonucleolytic degradation of the mRNA. How these two periods of deadenylation relate is unclear, but existing data support both occurring widely. In mouse fibroblasts, normal mRNAs’ deadenylation rates correlate well with their stability (Eisen et al. 2020). However, the authors of that study briefly note that NMD targets do not display this same correlation between deadenylation and mRNA stability. This observation indicates that NMD targets may be degraded independent of deadenylation and/or decapping, conflicting with the extant literature on deadenylation in NMD. This observation has not been addressed in the NMD literature, again confounding a mechanistic understanding of mRNA degradation during NMD.
Several technical challenges complicate analysis of the effects of deadenylation, decapping, endonucleolytic cleavage, and exonucleolytic decay during NMD. A common technique to study NMD is short-read RNA sequencing. Short-read sequencing provides highly sensitive ensemble measures of fragments of mRNAs produced from a gene. Because of the short nature of reads, isoform assignment is impossible for many reads, limiting most analyses to the gene level. Most short-read RNA-seq techniques lose poly(A) tail information. For short-read RNA sequencing techniques that retain poly(A) tail information (such at PAL-Seq [Yu et al. 2020] and TAIL-Seq [Chang et al. 2014]), it is difficult or impossible to assign most poly(A) tail-containing reads to NMD isoforms as the PTC-introducing event is typically hundreds (or thousands) of bases away from the poly(A) tail. Short-read sequencing techniques also require RNA fragmentation, which obscures endogenous cleavage information. Although gel-based techniques (e.g., northern blots) circumvent some of these issues, they are also low throughput, yield comparatively little sequence information, and are still ensemble-based measurements of gene expression. However, recently developed long-read, whole-transcript, single-molecule mRNA sequencing techniques promise to circumvent these issues (for a more complete discussion of short and long-read sequencing, see Hu et al. 2021).
Despite the important insights provided by foundational genetic evidence and recent compelling work (e.g., Boehm et al. 2021), the role of deadenylation, decapping, and endonucleolytic cleavage and the roles of SMG-5/6/7 during NMD remain unclear. As evidence of this, a number of contemporary studies and reviews describe NMD as driven by multiple, parallel degradation routes—often invoking deadenylation, decapping, and endonucleolytic cleavage in combination (for primary articles, see Sanderlin et al. 2022; Sarkar et al. 2022; Wallmeroth et al. 2022; Nasif et al. 2023; Steiner et al. 2023; for literature reviews, see Lejeune 2022; Carrard and Lejeune 2023; Nagar et al. 2023; Sun et al. 2023). To resolve the confusion around routes of NMD mRNA target degradation, here we investigate the relative contributions and roles of deadenylation, cleavage, and decapping in C. elegans NMD. We leverage mutants that ablate NMD, the sensitivity of short-read RNA sequencing, and the unique information captured by long-read, nanopore direct RNA sequencing. Using a modified nanopore sequencing protocol, we capture isoform, decapping/cleavage site, and poly(A) tail information for individual, native mRNA molecules as they exist in vivo. With these data, we set out to address the relative contributions of decapping, deadenylation, and endonucleolytic cleavage, as well as their genetic dependency on SMG-5 and SMG-6.
Results
C. elegans SMG-2 (UPF1), SMG-5, SMG-6, and SMG-7 repress a common set of mRNAs
SMG-5, SMG-6, and SMG-7 (Fig. 1A) target a similar set of mRNAs in human cells (Boehm et al. 2021), and we set out to determine if this was also true in C. elegans. We elected to measure NMD activity genome-wide with short-read RNA-seq. Because ∼5%–20% of the genome is targeted by NMD, short-read RNA-seq measures NMD function across a diverse array of endogenous NMD targets. We generated short-read RNA-seq libraries from smg-5, smg-6, and smg-7 mutants. As a control, we analyzed a smg-2 mutant, which is deficient in SMG-2 (UPF1), the central, most highly conserved NMD factor (Hodgkin 1986). Differential expression analysis identified genes with mRNAs significantly upregulated in each mutant: 389 genes (smg-2), 363 genes (smg-5), 160 genes (smg-6), and 16 genes (smg-7; Methods) (Fig. 1B).
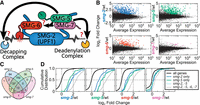
C. elegans UPF1, SMG-5, and SMG-6 repress a common set of endogenous mRNAs. (A) Model of decay step(s) of NMD on an mRNA target. (B) Differential expression analysis (DESeq2) of biological replicates of RNA-seq from wild type, smg-2 (UPF1), smg-5, smg-6, and smg-7. Genes with mRNAs that are upregulated in the indicated mutant (adjusted P ≤ 0.01) are colored. (C) A four-way Venn diagram indicating overlap of genes with mRNAs upregulated in the indicated mutants. The slight difference in numbers of targets compared with A (e.g., 16 vs. 15 smg-7 targets) is caused by taking the intersection of all genes included by DESeq2 across all four smg mutants. (D) Expression of all genes’ mRNAs (black) across smg mutants and grouped by whether the genes’ mRNAs are upregulated in smg-2 only (blue line; 84 genes), smg-5 only (green line; 56 genes), smg-2 and smg-5 (light blue dotted line; 152 genes), or smg-2, smg-5, and smg-6 and/or smg-7 (gray dotted line; 150 genes). All changes are statistically significant relative to all genes (KS test P <1 × 10−5).
We noticed substantial overlap between the mRNAs upregulated in each smg mutant. For example, of the 389 genes with upregulated mRNAs in the smg-2 mutant, all but 86 were also identified as upregulated in the smg-5 mutant (Fig. 1C). Notably, however, the 86 genes that were not identified by DESeq2 in smg-5 were all upregulated in smg-5 (P <1 × 10−5 KS test) (Fig. 1D), suggesting that they represent false negatives from DESeq2 rather than bona fide UPF1-only NMD targets. Such false negatives would be expected given the limited power of RNA-seq (Schurch et al. 2016). We noted a similar overlap for each of the smg-2, smg-5, and smg-6 target lists (Fig. 1C,D). The list of mRNAs in smg-6 was smaller than those of smg-2 and smg-5, the result of decreased sequencing depth in one smg-6 library (Supplemental Table S1). Our results support the idea that SMG-5 and SMG-6 regulate a common set of target mRNAs with UPF1 in C. elegans, similar to findings in human cells (Boehm et al. 2021).
Mutation of smg-7 had a markedly different effect and had comparatively subtle effects on the transcriptome (Fig. 1B). This result is aligned with prior descriptions of C. elegans smg-7, showing that smg-7 is not required for NMD at lower temperatures (16°C, 20°C) (Cali et al. 1999). As our RNA-seq was performed on animals grown at 16°C, the expected result in a smg-7 animal would be a minimal effect on the NMD pathway. Despite the smaller changes in the smg-7 transcriptome, we identified 16 targets, 13 of which were upregulated in all of smg-2, smg-5, and smg-6 (Fig. 1C). Furthermore, when taken as a group, the other smg target lists were all significantly upregulated in smg-7 animals (P <1 × 10−5 KS test) (Fig. 1D). This result is consistent with the idea that smg-7 regulates a similar set of mRNAs to the other SMG proteins but that SMG-7's requirement in the NMD pathway is not as strict as the other SMG proteins at low temperatures. We note that in other animals, SMG7 has also proven exceptional among the SMG proteins. In humans, SMG7 can be more easily mutated than the other SMG genes, and NMD function can be restored to SMG7-mutant cells via high expression of SMG5 (Boehm et al. 2021). In Drosophila melanogaster, SMG7 is absent (Gatfield et al. 2003).
Taken together, our results support the model that SMG-5, SMG-6, and SMG-7 work together to repress a common set of endogenous mRNA targets in C. elegans. Given that only SMG-6 is thought to function biochemically in endonucleolytic cleavage, we next aimed to better understand the contributions of deadenylation and decapping to mRNA target decay during NMD.
Nanopore degradome sequencing captures full-length mRNAs and degradation intermediates
To assess the roles of deadenylation, endonucleolytic cleavage, and decapping on NMD targets, we turned to whole-transcript, native RNA sequencing. Long-read sequencing offers key advantages over short-read sequencing, namely, (1) the ability to sequence full-length mRNA molecules, producing single-molecule descriptions of poly(A) length, splicing and isoform status, and 5′ ends and (2) the ability to avoid artifacts of short-read sequencing introduced during library preparation (e.g., fragmentation, PCR) that can skew the observed RNA populations. The tradeoff of long-read sequencing is depth: At the time that we performed these direct RNA experiments on R9 chemistry minion flow cells, libraries typically yielded a million or so reads and thus would only report on the most abundant mRNAs and NMD targets. Despite this concern, we reasoned that long-read sequencing would prove insightful to understand what reactions NMD targets experience in vivo.
Prior to long-read sequencing, we captured full-length and degradation intermediate mRNAs using a degradome sequencing protocol (5TERA-seq; for further protocol description, see Methods) (Fig. 2A; Ibrahim et al. 2021). Briefly, we annealed and ligated an Oxford Nanopore Technologies (ONT) 3′ adapter using an oligo(dT) splint, which captures mRNAs preferentially and enables targeting to nanopores. The oligo(dT) splint requires a minimum of 10 adenosines on the 3′ end of an RNA molecule; mRNA poly(A) tails are more than 25 adenosines in length (Lima et al. 2017; Eisen et al. 2020). We performed no additional poly(A) selection as poly(A) selection can irreproducibly skew the view of the transcriptome (Viscardi and Arribere 2022).
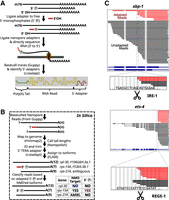
Workflow for the capture of mRNA degradation intermediates. (A) Strategy to identify RNAs with a 5′ monophosphate using direct RNA sequencing of ONT. For details, see Methods. (B) In silico read processing workflow for degradome sequencing. For details, see Methods. (C) RNAs with 5′ monophosphate ends produced from known endonuclease targets xbp-1 and ets-4. (Top) Red lines indicate molecules that contained the 5′ adapter sequence and were thus derived from 5′ monophosphate–containing RNAs. Black lines indicate molecules that did not contain the 5′ adapter sequence. Thick lines indicate aligned sections of the RNA sequences, and thin lines indicate alignment gaps of RNA reads, which span annotated introns. The isoform annotations for xbp-1 (and ets-4) are indicated in blue. The thickest sections of the isoform annotations indicate the coding sequences. (Bottom) A zoomed-in window showing reads with 5′ ends near the known endonuclease cleavage sequence and site (indicated with dotted line and scissors).
To clearly identify RNAs experiencing decay, we also performed a 5′ monophosphate–specific ligation on our total RNA (Fig. 2A; Ibrahim et al. 2021). 5′ Monophosphates are produced via decapping, 5′→3′ exonucleolytic decay, or metal-dependent endonucleolytic cleavage (e.g., SMG-6). The strategy is similar to other degradome sequencing methods (Schmidt et al. 2015; Ottens et al. 2017; Won et al. 2020), with the following important differences: (1) we performed full-length RNA sequencing without PCR, circumventing PCR-dependent biases in molecule capture; (2) the 3′ ONT adapter is added at the end of the poly(A) tail, thus retaining information on the entirety of the RNA molecule from its poly(A) tail through its 5′ end; and (3) the library contains both unadapted and adapted mRNAs, with the latter identified in silico via the presence of the 5′ adapter (Fig. 2B). To enhance the stability of mRNAs with exposed 5′ ends, we also performed a knockdown of the primary 5′→3′ exonuclease xrn-1 via RNAi (Supplemental Fig. S1A). The technique yields reproducible libraries (Supplemental Fig. S1B,C) of similar depth to each other (Supplemental Fig. S1D,E), of similar depth to published nanopore direct RNA-seq data sets (Legnini et al. 2019; Roach et al. 2020; Viscardi and Arribere 2022), and of lower depth than short-read RNA-seq (Supplemental Fig. S1E). Overall, reads with the 5′ adapter were low abundance (∼2%–5% of libraries) (Supplemental Table S2), as expected, because degradation intermediates are transient species and a minority of the overall RNA pool.
We performed nanopore direct RNA degradome sequencing with C. elegans total RNA and validated the technique's performance using known endogenous endonuclease targets: xbp-1 (cut by IRE-1) and ets-4 (cut by REGE-1) (Shen et al. 2001; Habacher et al. 2016; Arribere and Fire 2018; Kim et al. 2022). For both ets-4 and xbp-1, we observed adapted 5′ ends at and downstream from the previously identified cleavage site. The assay provided a high degree of specificity as some RNA molecules had 5′ ends at the known cleavage site with single-nucleotide precision (Fig. 2C). We also observed full-length molecules spanning the cleavage site, as expected from the recovery of uncleaved mRNAs. The unadapted population includes some shorter mRNAs resulting from incomplete adapter ligation, in vitro RNA hydrolysis, and/or pore sequencing dropoff.
Visual inspection of the library identified abundant adapted 5′ ends within several additional endogenous genes, including rps-15A, rpl-30, and rps-27A. Each of these genes contains an mRNA isoform encoding a premature termination codon (PTC), namely, an NMD target. Indeed, many ribosomal protein genes produce NMD-eliciting splicing isoforms (Mitrovich and Anderson 2000). Given the high expression of ribosomal protein genes, their PTC-containing isoforms would be expected to be a significant proportion of the NMD targets captured with the limited depth of long-read sequencing.
In the cases of rps-15A and rpl-30, the splicing event that creates the PTC enabled the unique assignment of unadapted and adapted mRNA molecules to either the NMD isoform or the non-NMD isoform (Fig. 2B). By assigning reads to isoforms, we observed that the vast majority of adapted mRNAs derived from rps-15A and rpl-30 were made from the PTC-containing isoform (Fig. 3A; Supplemental Table S3). Conversely, most unadapted mRNA molecules corresponded to the non-PTC-containing isoform. In the case of rps-27A, unique isoform assignment is possible for near full-length molecules but not mRNAs with 5′ ends downstream from the PTC owing to a lack of unique splice information; we classify such molecules as “ambiguous” (Fig. 3A).
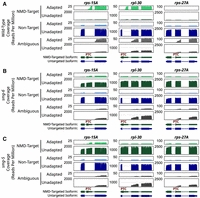
Nanopore degradome sequencing captures smg-6- and smg-5-dependent degradation intermediates on NMD targets. (A) Coverage plots of loci (rps-15A, rpl-30, and rps-27A) in wild-type animals that produce NMD-target and nontarget mRNAs. Read coverage (y-axes) is shown in reads per million. From top to bottom, coverages are for the following categories: adapted NMD isoforms (light green), unadapted NMD isoforms (dark green), adapted non-NMD isoforms (light blue), unadapted non-NMD isoforms (dark blue), adapted ambiguous isoforms (light gray), and unadapted ambiguous isoforms (dark gray). Annotations at the bottom indicate the NMD-targeted isoform and the non-NMD-targeted isoform. (PTC) Location of the NMD-eliciting stop codon. (B) Coverage plots of the indicated loci in smg-6-mutant animals. (C) Coverage plots of the indicated loci in smg-5-mutant animals.
The low abundance of unadapted, full-length PTC-containing isoforms (rps-15A, rpl-30, and rps-27A) suggests that such isoforms are unstable, which is further supported by the abundant degradation intermediates captured on the same isoforms. These observations demonstrate that 5TERA-seq can capture and study NMD products in vivo. The most abundant degradation products are NMD targets cleaved near their stop codon. These results support a central role for endonucleolytic cleavage in NMD-target decay.
Degradome sequencing captures smg-6-dependent degradation intermediates on NMD targets
We next determined if these were NMD degradation fragments, which should be SMG-6 dependent (Gatfield et al. 2003; Glavan et al. 2006; Eberle et al. 2009; Kim et al. 2022). SMG-6's PIN nuclease domain is required for NMD and mRNA cleavage (Gatfield et al. 2003; Glavan et al. 2006; Eberle et al. 2009). We performed degradome sequencing of RNA isolated from animals carrying a previously validated mutant of an active site residue on SMG-6 (referred to as smg-6 animals) (Kim et al. 2022), and determined isoform identity and 5′-end identity of RNA molecules. The abundance and distribution of reads on non-PTC isoforms of rps-15A, rpl-30, and rps-27A were similar in the smg-6 mutant (Fig. 3, cf. A and B, dark blue). However, PTC-containing isoforms were affected in two key ways: (1) the smg-6 mutant exhibited loss of mRNA fragments with adapted 5′ ends at and downstream from the PTC (Fig. 3, cf. A and B, light green, “adapted NMD-targets”), and (2) the smg-6 mutant exhibited high levels of unadapted, full-length mRNAs on the PTC-containing isoform (Fig. 3, cf. A and B, dark green, “unadapted NMD-targets”). Thus, the nuclease activity of SMG-6 is required for PTC-proximal mRNA cleavage, aligned with our prior work (Kim et al. 2022) and studies across experimental systems (Gatfield et al. 2003; Glavan et al. 2006; Eberle et al. 2009). Our results also establish that the mRNA products and degradation intermediates, as well as their genetic dependencies, can be studied with nanopore sequencing.
We reasoned that the pattern of adapted and unadapted reads in wild-type and smg-6 animals could expand our NMD-target list. We performed a simple statistical analysis to identify additional genes with a population of SMG-6-dependent degradation fragments (Methods) (Supplemental Fig. S2; Supplemental Text). The approach identified 25 genes; all 25 were previously identified by at least one NMD study (Supplemental Table S4), showcasing the robustness of the approach. We are able to clearly differentiate a PTC-containing isoform for 17 loci (allowing for analyses similar to that seen in Fig. 3). These 25 NMD targets provide a useful list of genes with sequencing depth sufficient to be included in subsequent analyses.
smg-5 is required for smg-6-dependent cleavages on NMD targets
SMG-5 is required for NMD across animals (Hodgkin et al. 1989; Alexandrov et al. 2017; Baird et al. 2018; Nelson et al. 2018; Zhu et al. 2020; Boehm et al. 2021; Zinshteyn et al. 2021; Huth et al. 2022). Our work in C. elegans (Fig. 1) and others’ work in human cells (Boehm et al. 2021) suggest that SMG-5 supports SMG-6's role in some way. We therefore utilized our degradome sequencing method to examine the cleavage events in smg-5-mutant animals.
Upon mutation of smg-5, we noted loss of degradation intermediates as well as an increase in full-length RNAs mapping to NMD isoforms (Fig. 3C) similar to the loss seen in smg-6 animals (Fig. 3B). The effect was specific to NMD targets; non-NMD-target isoforms (Fig. 3C) and other endonuclease targets (ets-4 and xbp-1) were unaffected in the mutants (Supplemental Fig. S3). A biological replicate of these experiments reproduced our observations in our example genes and other NMD targeted loci (Supplemental Fig. S4).
Our data support the idea that SMG-5 and SMG-6 target a similar set of mRNAs (Fig. 1), with SMG-5 stimulating SMG-6-mediated cleavage (Fig. 3C) in C. elegans, as occurs in humans (Boehm et al. 2021).
NMD-target mRNAs and their degradation products have poly(A) tails that are as long as, or longer than, non-NMD-target mRNAs
Prior work advocated both for (Chen and Shyu 2003; Lejeune et al. 2003; Yamashita et al. 2005; Loh et al. 2013) and against (Eisen et al. 2020) a role for deadenylation in NMD. Given this literature, we next turned our attention to the poly(A) tail. We called poly(A) tails for each read via Nanopolish (Methods) (Workman et al. 2019). Analysis of unadapted mRNAs genome-wide revealed a distribution of poly(A) tail lengths with a median length of about 52 adenosines (Fig. 4A), consistent with others’ measurements of poly(A) tails in C. elegans via long-read sequencing (Legnini et al. 2019; Roach et al. 2020), short-read sequencing (Lima et al. 2017), and gels (Nousch et al. 2017). The distribution of poly(A) tails on 5′-adapted mRNAs resembled that of unadapted mRNAs (Fig. 4A). RNA standards spiked into the library showed the technique's ability to capture poly(A) tails as short as 10 adenosines and to identify differences as short as five adenosines (Fig. 4A; Supplemental Fig. S5A,B). Thus, our libraries recovered poly(A) tail information in line with prior measurements and synthetic standards.
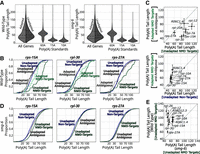
NMD-target mRNAs and their degradation products have poly(A) tails that are as least as long as normal mRNAs. (A) Violin plots for all mRNAs in wild-type and smg-6 animals. The left side of each violin (in light gray) shows the distribution of adapted reads’ tail lengths, and the right (in dark gray) shows unadapted reads. The three violins to the right show the tail length distributions for three spike-in RNA standards. Long dashed lines indicate the means, and short dashed lines indicate first and fourth quartile boundaries. (B) Poly(A) tail length cumulative distribution function (CDF) plots of example genes (rps-15A, rpl-30, and rps-27A) in wild-type animals. The same color scheme is used here as in Figure 3: adapted NMD isoforms (dashed light green), unadapted NMD isoforms (dark green), adapted non-NMD isoforms (dashed light blue), unadapted non-NMD isoforms (dark blue), adapted ambiguous isoforms (dashed light gray), and unadapted ambiguous isoforms (dark gray). For each plot, only categories that had at least 10 poly(A) tail-called reads are shown. For statistical analysis, see also Supplemental Table S4. (C) Scatter plot comparisons of mean poly(A) tail lengths between adapted versus unadapted NMD targets and adapted NMD-target reads versus unadapted nontargets for genes in which we could identify the NMD-target isoform. The dashed line indicates the diagonal where X = Y. Error bars indicate the SEM. Only genes with at least 10 reads in each category are shown. (D) Poly(A) tail length CDF plots of example genes in smg-6 animals (as in B). Low read counts caused adapted, PTC-mapping RNA species to fall below the 10 read cutoff. For statistical analysis, see also Supplemental Table S4. (E) Scatter plot comparison of poly(A) tail lengths between unadapted NMD targets in wild-type animals versus smg-6 animals.
To better understand the relationship between poly(A) tails and NMD, we focused on mRNA molecules produced from the exemplary genes rps-15A, rpl-30, and rps-27A (Fig. 4B). Adapted NMD-target mRNAs exhibited poly(A) tail lengths similar to the genome-wide distribution and similar to unadapted normal mRNAs produced from the same genes. Thus, at (or immediately after) the time that cleavage occurs, NMD targets have a poly(A) tail length similar to that of non-NMD targets. Put another way, the existence of polyadenylated, cleaved NMD targets is inconsistent with complete deadenylation as a prerequisite for cleavage.
To gain further insight into the life cycle of poly(A) tails during NMD, we examined unadapted NMD-target mRNAs (Fig. 4B). Unadapted NMD-target mRNAs would be expected to include mRNAs that have not yet entered the translational pool and are yet to be targeted by NMD (i.e., recently transcribed mRNAs). Unadapted PTC-containing mRNAs exhibited significantly longer tails than either adapted, PTC-containing mRNAs or unadapted, non-PTC-containing mRNAs. The effect was seen for PTC-containing isoforms in each of rps-15A, rpl-30, and rps-27A (Fig. 4B; for accompanying statistical analysis, see Supplemental Table S5), as well as each of the other 17 genes in which we could identify the NMD-eliciting isoform with sufficient depth (Fig. 4C; Supplemental Figs. S6, S7).
Our analysis of poly(A) tail lengths revealed that NMD targets’ poly(A) tails are as long as, or longer than, normal mRNAs’ poly(A) tails in NMD-competent animals. NMD targets’ poly(A) tails also shorten between their transcription and their NMD targeting, but only to a length similar to that of non-NMD targets.
NMD-target and nontarget mRNAs have similar poly(A) tail lengths upon smg-6 mutation
We considered two possible models to explain NMD-target mRNAs’ long poly(A) tails in wild-type animals:
-
Maturation-dependent deadenylation. Full-length NMD targets’ long poly(A) tails are owing to their relative youth among cellular mRNAs. As NMD targets are targeted for degradation early in their life span, a higher proportion of full-length NMD-target mRNAs will be nascent. As nascent mRNAs’ poly(A) tails are longer than those of mature mRNAs, NMD targets’ poly(A) tails will be longer on average. In this model, the increased poly(A) tail lengths of full-length NMD targets are a side-effect of the absence of a stable, mature mRNA population.
-
NMD-dependent deadenylation. NMD targets experience deadenylation during their decay. In this model, NMD-target mRNAs have longer poly(A) tails, and in the course of NMD, their poly(A) tails are shortened toward a length that resembles normal mRNAs. In this model, deadenylation is central to the decay reaction(s) of NMD. If true, such a model could offer a simple route to reconciliation for SMG-5's requirement for SMG-6 cleavage despite SMG-5's association with deadenylation.
As an initial test between these models, we examined mRNA molecules in smg-6-mutant animals. In smg-6 animals, NMD targets are not cleaved, and unadapted mRNA targets accumulate (Fig. 3A,B). The maturation-dependent deadenylation model predicts that poly(A) tails of NMD targets will resemble those of normal mRNAs in an NMD-mutant background. In contrast, the NMD-dependent deadenylation model predicts that NMD targets will maintain their longer poly(A) tails owing to the loss of NMD and associated deadenylation. Our results show that the poly(A) tails of NMD-target mRNAs are significantly shorter in smg-6 animals relative to wild-type animals and are similar to that of normal mRNAs (Fig. 4D,E; Supplemental Figs. S6, S7). This result is consistent with the maturation-dependent deadenylation model.
NMD-target and nontarget mRNAs have similar poly(A) tail lengths upon smg-5 mutation
The existing literature suggests that the heterodimer SMG-5/7 promotes deadenylation during NMD. Because this role for SMG-5/7 would fit into the NMD-dependent deadenylation model, we entertained a variation to this model that could fit the data shown thus far: SMG-5/7 deadenylate NMD targets before cleavage via SMG-6. In this model, unadapted, full-length NMD targets would accumulate with a shorter poly(A) tail in smg-6 animals compared with the wild type, as is the case (Fig. 4D,E). To distinguish between the maturation-dependent and the revised NMD-dependent (SMG-5/7-elicited) deadenylation models, we considered the expectations of a smg-5 mutant. Under the revised model, we would expect full-length NMD targets to accumulate in smg-5 animals with long poly(A) tails. However, if the maturation-dependent deadenylation model was correct, in smg-5 animals we would again expect NMD targets to accumulate with normal-length poly(A) tails.
To distinguish between the models, we analyzed poly(A) tail lengths in smg-5 animals. Normal mRNAs’ poly(A) tails were comparable to the wild type, and spike-in standards were unchanged (Fig. 5A). However, unadapted NMD targets’ poly(A) tails were notably shorter than in the wild type and were similar to normal mRNAs’ poly(A) tail lengths (Fig. 5B,C; Supplemental Figs. S6, S7). This result is similar to results in smg-6 animals (Figs. 4D, 5D; Supplemental Figs. S6, S7) and is consistent with the maturation-dependent deadenylation model but would not be expected under the NMD-dependent deadenylation model.
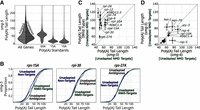
NMD-target poly(A) tail lengths resemble normal mRNAs in smg-5 animals. (A) Violin plots for all mRNAs in smg-5 animals. The left side of the violin (in light gray) shows the distribution of adapted reads’ tail lengths, and the right (in dark gray) shows unadapted reads. The three violins to the right show the tail length distributions for three spike-in RNA standards. Long dashed lines indicate the means, and short dashed lines indicate first and fourth quartile boundaries. (B) Poly(A) tail length cumulative distribution function (CDF) plots of example genes (rps-15A, rpl-30, and rps-27A) in smg-5 animals (as in Fig. 4B). The same color scheme is used here as in Figures 3 and 4: unadapted NMD isoforms (dark green), unadapted non-NMD isoforms (dark blue), and unadapted ambiguous isoforms (dark gray). For each plot, only categories that had at least 10 poly(A) tail-called reads are shown. For statistical analysis, see also Supplemental Table S4. (C) Comparison of poly(A) tail lengths between unadapted NMD targets in wild-type animals versus smg-5 animals (similar to Fig. 4E). The dashed line indicates the diagonal where X = Y. Error bars indicate the SEM. Only genes with at least 10 reads in each category are shown. (D) Comparison of poly(A) tail lengths between unadapted NMD targets in smg-6 animals versus smg-5 animals. A larger number of genes are shown compared with C because of more genes passing the cutoff of 10 reads in each category. Within the dashed box, there are nine unlabeled genes: rps-27A, rps-15A, rpl-30, rpl-12, rpl-3, C53H9.2, K08D12.3, rpl-26, and rpl-7A.
Our analysis of poly(A) tail lengths in wild-type, smg-6, and smg-5 animals is consistent with the idea that the extent of deadenylation experienced by NMD targets is within the scope of that experienced during normal mRNA maturation. In the Discussion, we expound on this idea and relate it to the extant literature on deadenylation and NMD.
NMD-targeted mRNAs exhibit substantial decapping only upon mutation of smg-5 or mutation of smg-6
Decapping is also reported to contribute to NMD. Specifically, SMG-5 has been implicated in recruiting the decapping complex and its enhancer PNRC2 to target mRNAs (Lykke-Andersen 2002; Lejeune et al. 2003; Cho et al. 2009, 2013; Lai et al. 2012). We utilized our degradome sequencing method to explore the contribution of decapping to decay of NMD-targeted mRNAs in C. elegans.
To assess decapping, we categorized the 5′ ends of RNA reads based on the proximity of their 5′ end to the trans-splicing site (TSS; as many C. elegans gene mRNAs are trans-spliced after transcription, the 5′-most nucleotide of the spliced mRNA differs from the transcription start site). Decapping leaves a 5′ monophosphate (Wang et al. 2002), and decapped mRNAs would thus appear as full-length adapted RNA reads (Fig. 6A). We note that we do not know the background of the technique for generation of adapted reads on a pure population of capped mRNAs, but because in vitro sample degradation products from RNA handling (heat, alkaline) would yield a 5′-hydroxyl and would not be expected to preferentially occur near the TSS, we expect that most of the adapted RNAs with 5′ ends near the TSS represent the products of mRNA decapping.

NMD-targeted transcripts exhibit low levels of decapped RNAs in wild-type animals. (A) Diagram illustrating how near trans-splicing site (TSS) 5′ ends are defined. As in Figure 3B, the coverage plots show read pileups on the gene rps-15A. Reads with their 5′ ends within 50 nucleotides of annotated TSSs are labeled as “near TSS.” This process was carried out for NMD-target and nontarget isoforms. (B) Box and strip plots of the fraction of all reads with 5′ ends spanning to the TSS for each isoform. Red solid lines indicate the median. The inner box denotes the inner quartile range (IQR) from the 25th to the 75th percentiles; whiskers span out to any points within 1.5 times the IQR. Each point indicates the mean for each isoform across replicates. Within each strain (x-axis), reads were separated based on isoform identity as non-NMD-targets (left; blue) or NMD-targets (right; green). Example genes shown in other figures are indicated by red points (rpl-30, rps-15A, and rps-27A) and labeled where not tightly clustered. Only isoforms that contained at least one adapted read in at least two replicates were considered. Dashed lines between points connect isoforms within strains from the same gene loci. Box and strip plots (as in B) of the fraction of all adapted reads near the TSS.
In wild-type animals, NMD targets produced 10-fold lower levels of full-length RNAs (irrespective of adapter ligation status) compared with nontargets (Fig. 6B). Upon mutation of smg-5 or mutation of smg-6, NMD-target isoforms accumulate full-length reads at a level similar to that of nontarget isoforms. These results are consistent with the idea that NMD targets are stable full-length mRNAs only after loss of smg-5 or smg-6.
Finally, we examined the fraction of degradation intermediates with 5′ ends located near the TSS to quantify how much decay at each locus is attributable to decapping (Fig. 6C). In wild-type animals, nontarget mRNAs showed a substantial decapping signal (a high fraction of adapted 5′ ends near the TSS, median 67.33%), whereas NMD-target isoforms exhibited a much lower decapping signal (median 0.17%). Upon disruption of NMD via smg-5 mutation or smg-6 mutation, NMD-target isoforms transitioned to an RNA degradation landscape similar to that of nontargets.
Our analysis of decapped RNAs indicates that decapping is a significant decay mechanism for nontarget RNAs, but we observed no evidence for an NMD-dependent decapping pathway on uncleaved NMD-targeted species. In wild-type animals, NMD-targeted isoforms primarily undergo endonucleolytic cleavage rather than decapping. Decapping does not appear to be a substantial route of NMD-dependent degradation as loss of SMG-6 increases NMD-target mRNAs to a level similar to that of non-NMD-target mRNAs (Fig. 3). This effect has also been noted previously, with NMD-reporter constructs being completely stabilized by mutation of smg-2 (UPF1), smg-5, and/or smg-6 (Hodgkin et al. 1989; Cali et al. 1999; Kim et al. 2022). These results cannot rule out the presence of an NMD-elicited decapping process concurrent to or after SMG-6-mediated cleavage. A decapping process coupled to endonucleolytic cleavage could help to additionally destabilize the 5′ cleavage fragment for NMD-target mRNAs.
Discussion
Our results suggest a single primary route of PTC-mediated degradation through endonucleolytic cleavage mediated by SMG-5/6 and suggest that deadenylation and decapping are not significant modes of decay in animal NMD. Our results represent an important step forward in delineating the NMD mechanism in animals.
The data we present are consistent with a model with the following steps of NMD in animals: (1) like normal (PTC-lacking) mRNAs, nascent, PTC-containing mRNAs are produced with long and heterogeneous poly(A) tails; (2) like normal (PTC-lacking) mRNAs, by the onset of translation, PTC-containing mRNAs’ poly(A) tails are shortened to a length similar to that of mature mRNAs; (3) after the onset of translation, PTC-containing mRNAs are targeted by NMD machinery and cut near their PTC in a reaction that requires SMG-5 and SMG-6 and that is enhanced by SMG-7; and (4) the downstream mRNA cleavage fragment is then cleared via XRN-1. We propose that NMD targets’ deadenylation is part of mRNA maturation rather than a consequence of decay.
The model is based on a detailed long-read sequencing–based molecular analysis of the polyadenylation, cleavage, and capping status of mRNA molecules produced from approximately two dozen endogenous C. elegans NMD-target genes. We expect that a similar model will hold true for many NMD targets across animals given the substantial overlap of SMG-5/6/7 targets in C. elegans (Fig. 1) and humans (Boehm et al. 2021) as assessed by short-read sequencing.
The model is consistent with existing data on deadenylation of normal mRNAs. Normal mRNAs emerge from the nucleus with long and heterogeneous poly(A) tails and, subsequently, experience a rapid period of deadenylation (Sawicki et al. 1977; Eisen et al. 2020; Tudek et al. 2021). Normal mRNAs can also experience a more variable (and often much slower) deadenylation over their lifetime that is associated with translation and eventual decay (Yi et al. 2018; Eisen et al. 2020; Tudek et al. 2021; Park et al. 2023). NMD targets are only recognized as such during translation, predicting that PTC-containing nascent mRNAs will also experience the initial period of deadenylation. Because NMD targets are not found in the translationally stable pool of mRNAs, they do not experience the second period of deadenylation. Although both periods of deadenylation are thought to occur after nuclear export, their precise relationship to translation and translation termination remains unclear. Future studies in this area will prove informative, for understanding both normal mRNA metabolism and the metabolism of PTC-containing mRNAs.
The model is also consistent with existing data on deadenylation during NMD, although our interpretation of prior data differs. Of three studies that directly examined poly(A) tails on NMD targets, each observed that NMD targets experience partial deadenylation to some nonzero poly(A) tail length prior to loss of the mRNA (e.g., Yamashita et al. 2005, Fig. 3B; Lejeune et al. 2003, Fig. 6B,C; Chen and Shyu 2003, Fig. 1C). At the time these studies were done, it was generally thought that longer poly(A) tails were associated with more stable mRNAs, and so the deadenylation seen on NMD targets was invoked as a mechanism of NMD. Knowing now that mRNAs are born with long poly(A) tails that are subsequently shortened, the deadenylation signature in these studies may be attributable to nascent mRNA maturation. Deadenylation appears heightened (and is more easily visualized) on NMD targets because of the lack of a stable, translationally mature, partially deadenylated mRNA population. Thus, we reason that the deadenylation reported in these studies is not directly causal to NMD-target mRNAs’ accelerated decay. Again, a better understanding of the deadenylation experienced by nascent mRNAs will reveal the extent to which deadenylation is similar or different on normal mRNAs and NMD targets.
PTC-associated 5′ monophosphate ends are, in principle, consistent with either SMG-6-mediated endonucleolytic cleavage or decapping followed by 5′ > 3′ exonuclease degradation, but we favor the former. In prior work (Kim et al. 2022), we captured RNA 3′ ends at and upstream of the stop codon, a result expected from endonucleolytic cleavage near the PTC but not expected from decapping. We note a requirement for at least 10 3′-terminal adenosines for capture with our nanopore strategy, preventing the detection of RNAs with poly(A) tails shorter than 10 or RNAs with nonadenosine terminal residues. Untemplated uracils can occur on mRNAs with poly(A) tails with fewer than 25 adenosines (Eisen et al. 2020), although such tails are a late-stage product of deadenylation, and thus, we are not concerned with their capture here. The capture of unusually tailed mRNAs (and their relationship to NMD) will require protocols different from those used here.
We noticed that some NMD targets exhibited slightly longer poly(A) tails even in NMD mutants, suggesting that PTC-containing mRNAs are mildly defective in deadenylation. For example, rps-15A PTC-containing mRNAs exhibited longer poly(A) tails compared with rps-15A non-PTC-containing mRNAs in the wild type (Fig. 4B), smg-6 (Fig. 4D), and smg-5 (Fig. 5B). The magnitude of the difference was smaller in smg animals (a median difference of about 10 adenosines compared to about 45 adenosines in the wild type), although still statistically significant (Supplemental Table S5). Thus, even in the absence of NMD, NMD targets experience weaker deadenylation compared with nontargets. This result may prove mechanistically informative for the understanding of deadenylation as it relates to translation and termination; perhaps PTCs or their resulting long 3′ UTRs alter ribosome dynamics in such a way that NMD targets inefficiently recruit deadenylases compared with normal mRNAs.
Despite a commonly held view that SMG-5/7 and SMG-6 target distinct mRNAs, our short-read and long-read work supports the idea that all three factors (SMG-5/6/7) regulate the same set of mRNAs; these mRNAs are also regulated by UPF1. Our work thus supports the idea that SMG-5/6/7 work with UPF1 to bring about target repression. Among these factors, SMG-7 appears to play a stimulatory rather than essential role: In the absence of SMG-7, regulated transcripts were still repressed, albeit less efficiently so. We speculate that SMG-7 enhances the biochemical activities of the other SMG proteins in some way. We expect that a better understanding of the SMG proteins (in particular, SMG-5/6) will help understand SMG-7's role on the common set of SMG-regulated mRNAs.
To reconcile the molecular function of SMG-5/7 with genetic data, we arrived at a model in which SMG-5 promotes RNA cleavage by SMG-6 rather than RNA deadenylation in C. elegans. This model is consistent with data from human cells (Boehm et al. 2021), although our capture and analysis of poly(A) tails and decapping products more directly refute a role for either in the endonuclease reaction. Our model is based on observations of a handful of highly expressed NMD targets. Although it remains possible that NMD mechanisms will diverge among more lowly expressed genes, our short-read sequencing data suggest not. Thus, the SMG-6-mediated endonuclease reaction is quite broadly conserved in animal NMD. We imagine that a better understanding of the endonuclease reaction will clarify how SMG-5 stimulates it and how it is coupled to early stop codon recognition.
Here, we showcase the ability to attain novel mechanistic insight into mRNA metabolism from long-read sequencing technology. Sequencing entire mRNA molecules from poly(A) tail through isoform/splicing information and 5′ end/cleavage/decapping status allowed us to deconvolve properties of different mRNA populations that are difficult, or impossible, to study with other approaches. Coupled with the genetic tractability of C. elegans, this approach yielded novel insight into the NMD pathway. We expect similar approaches to be informative for various mRNA metabolism pathways across animals.
Methods
Strains
All novel C. elegans strains were made in N2 background animals (VC2010) (Thompson et al. 2013), and a list of strains is available (Supplemental Table S6).
Sample collection for short-read RNA sequencing and library preparation
Animals were bleached to obtain a synchronous population of eggs and then grown at 16°C until the L4 stage on OP50. Animals were washed off the plate the N50 (50 mM NaCl), passed through a 5% sucrose cushion in N50, and washed once with N50. Aliquots of animals were snap-frozen in liquid nitrogen. Animals were lysed by grinding with a mortar and pestle cooled with liquid nitrogen. Total RNA was TRIzol-extracted, and a custom C. elegans ribosome subtraction protocol was performed as previously described (Monem et al. 2023). RNA-seq libraries were made with the NEBNext Ultra II directional RNA library prep kit for Illumina sequencing.
Short-read RNA-seq analysis
Short-read RNA-seq libraries were processed essentially as previously described (Monem et al. 2023). Briefly, reads were trimmed with cutadapt v3.5 (Martin 2011), requiring a minimum length of 50 nt. Reads were mapped to the C. elegans genome (WBCel235) with STAR RNA-seq aligner (v2.7.3a) (Dobin et al. 2013), allowing for two mismatches. Stranded-read counts were tabulated with the Arribere laboratory pipeline (https://github.com/arriberelab/arriberelab) and fed into DESeq2 (v1.42.1) (Love et al. 2014) for differential expression analysis. Genes were considered as upregulated in a given smg mutant if the log2 fold change was positive and the adjusted P ≤0.01.
RNA interference knockdown of xrn-1 and sample collection for degradome sequencing
All animals used for degradome sequencing were grown under RNAi knockdown conditions of the primary 5′-to-3′ endonuclease, xrn-1, based on the methodology previously described (Pule et al. 2019). Briefly, an RNAi feeding strain was made via transformation of an xrn-1 targeting RNAi plasmid from Kamath and Ahringer (2003) into ht115(de3). Bacteria were grown overnight at 37°C to an optical density of 4.0 in 2 × YT medium containing carbenicillin (25 µg/mL). IPTG was added to a concentration of 100 µg/mL to induce dsRNA expression, and growth was continued for 4 h. Bacteria was then spread on NGM plates containing carbenicillin (25 µg/mL) and IPTG (100 µg/mL). The RNAi-expressing lawns were grown overnight at room temperature prior to the addition of C. elegans eggs (next).
Animals were bleached to obtain a synchronous population of eggs and then grown at 20°C until the L3/L4 stage on the above-described RNAi plates. Animals were collected on a sucrose cushion (5%) to minimize bacterial contamination, pelleted, and washed with EN50 and M9. Pellets were resuspended in TRIzol reagent (Ambion 15596026) and lysed by freeze-cracking, and total RNA was isolated by chloroform extraction. Total RNA integrity was assessed with the Agilent high-sensitivity RNA system for TapeStation. Only total RNA samples with RNA integrity number equivalent (RINe) values greater than 7.0 were used for sequencing.
5′ Adapter ligation, spike-in poly(A) standards, and direct RNA sequencing (together encompassing degradome sequencing)
To identify RNAs containing 5′ monophosphates, we used 5TERA (Ibrahim et al. 2021) with modifications. Briefly, total RNA was subjected to a T4 RNA ligase reaction with an RNA adapter (JA-MV-25, /5Biosg/rArArUrGrArUrArCrGrGrCrGrArCrCrArCrCrGrArGrArUrCrUrArCrArCrUrCrUrUrUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArNrNrN) for 3 h at 37°C. Ligated RNA was cleaned with Zymo Research RNA Clean & Concentrator Kits (R1019) using manufacturer specifications to remove RNA species <200 nt, including unligated adapters. Differing from the 5TERA protocol, we did not enrich for adapted RNAs.
RNA standards of known poly(A) tail length were spiked into each prep to make up 5% of the library by mass. Briefly, RNA standards were prepared as follows: a Saccharomyces cerevisiae ENO2 PCR product was used as a template for overhang PCR. The overhangs included a T7 transcription site, barcodes, and poly(A) tails of known lengths. After in vitro T7 transcription, RNAs were pooled, and the pool was aliquoted to ensure minimal differences in the spike-ins for each library.
For nanopore sequencing, 5 μg of ligated total RNA with standards was used as input for ONT's direct RNA sequencing kit (dRNA-seq, SQK-RNA002). dRNA-seq libraries were prepared according to the manufacturer's instructions (protocol version as released on September 13, 2021) with the following modification: (1) increased input, (2) use of total RNA as input, and (3) use of SuperScript IV (Invitrogen 18090010) rather than the ONT's specification of SuperScript III (Invitrogen 18080051).
Degradome (nanopore) sequencing software, basecalling, and alignment
All raw voltage traces were collected as FAST5 files using ONT's software MinKNOW (Core versions 4.3.4 to 5.3.1). To minimize variability in basecalling and downstream analyses based on software versions, all libraries were reprocessed from FAST5s with the same pipeline as follows. Raw FAST5 files from MinKNOW were basecalled with Guppy (v6.5.7 + ca6d6af) in GPU mode using parameters: guppy_basecaller -c rna_r9.4.1_70bps_hac.cfg. Basecalled reads were aligned to the C. elegans genome (WBCel235) using minimap2 (v2.17-r941) (Li 2018) with recommended settings for dRNA-seq: minimap2 -x splice -uf -k14. Additionally, the parameter –junc-bed was used with a BED genome annotation file to provide minimap2 with splice junction information.
Additional postprocessing for degradome sequencing
To identify reads containing the 5′ adapter sequence (derived from RNAs that had 5′ monophosphates and underwent ligation), we utilized cutadapt (Martin 2011) as previously specified (Ibrahim et al. 2021). FASTQ comments containing information from cutadapt were integrated into post-alignment BAM/SAM files using the pysam library (https://github.com/pysam-developers/pysam) as SAM tags.
Nanopolish (Workman et al. 2019) was used with default parameters to assess poly(A) tail lengths in all sequenced libraries. For plots utilizing mean tail lengths as a summary statistic, we restricted analyses to genes with 10 or more reads. Information from basecalling, adapter identification, alignment, gene assignment, and tail-length calling was consolidated into extended SAM format files as additional tags. Reads were required to have successful mapping and gene assignment in order to be used for downstream analysis.
De novo endonuclease target identification
To identify NMD targets de novo, we quantified adapted (cleaved) and unadapted (mostly full-length) read counts in wild-type and smg-6 (or smg-5) animals in a 2 × 2 contingency table. We then used SciPy's implementation of Fisher's exact test (Virtanen et al. 2020) with a Bonferroni-corrected P-value cutoff to identify genes at which the fraction of adapted RNA reads decreased in mutant animals. To ensure adequate statistical power, we also imposed read count cutoffs of 100 reads per gene per library (Supplemental Fig. S1B,C).
An important limitation is that this analysis was performed at the gene level and will miss many examples in which individual isoforms change but not substantially enough to produce a detectable effect at the gene level. Although we attempted to run the analysis at the isoform level, issues with annotations and accurate/unambiguous isoform assignment limited success. We found that manual identification of the NMD-eliciting isoforms was necessary to identify PTC sites. This is because of errors in systematic annotation software that will (for example) annotate the longest possible coding sequence that could be produced by an mRNA rather than what is actually translated, which is often much shorter, more 5′, and more likely to generate a PTC. Additionally, annotations are built from wild-type transcriptomes, and thus, the unstable mRNA isoforms found in NMD-mutant animals are often absent. For previously identified NMD targets, we produced our overlap table by relying on each publication's reported target list (Supplemental Table S4).
Isoform-level tail, decapping, and coverage plots
Because of the above-noted limitations, only a subset of NMD-targeted genes had sufficient information to distinguish between NMD-targeted and untargeted isoforms. We utilized this list of 17 genes (rpl-30, rps-15A, rps-27A, rpl-7A, rpl-3, rpl-10A, rpl-12, hel-1, aly-3, rsp-6, K08D12.3, R06C1.4, C53H9.2, rsp-5, ZK228.4, rpl-26, and pqn-70), for which we could unambiguously identify the NMD eliciting isoform, and distinguish it from any other isoforms at the locus. All genes in this list were identified de novo (Supplemental Fig. S2), as well as in multiple (two or more) previously published data sets (Supplemental Table S4).
Visualizations
All plot visualizations were produced with Python using publicly available libraries: plotly (v5.11.0), seaborn (v0.12.1), pandas (v1.5.2), and Matplotlib (v3.6.2; the axes join function needed by the coverage plotting scripts is depreciated and will be removed in v3.8+).
Software availability
All custom scripts and software utilized for the manuscript are freely available as Supplemental Code and at GitHub (https://github.com/MViscardi-UCSC/2025_GenomeResearchPaperCode) for academic and nonprofit use.
Data access
All sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1010807.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Arribere laboratory for feedback, and Melissa Jurica, Manny Ares, Fadia Ibrahim, and Robert Hogg for comments on the manuscript. This work was supported by a T32 training grant (National Institute of General Medical Sciences [NIGMS], 5T32GM133391) to M.J.V., Searle Scholars Award (Kinship Foundation) to J.A.A., and an R01 grant (NIGMS, R01GM131012) to J.A.A.
Author contributions: M.J.V. and J.A.A. conceived the project and designed the study. E.S. performed the molecular experiments for the short-read sequencing libraries with analysis and guidance by J.A.A. (Fig. 1). M.J.V. performed the molecular experiments and analysis with assistance and guidance from J.A.A. M.J.V. and J.A.A. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280046.124.
- Received September 19, 2024.
- Accepted March 29, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








