
A unified analysis of atlas single-cell data
- 1Ray and Stephanie Lane Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA;
- 2Department of Computer Science, University of Illinois Chicago, Chicago, Illinois 60607, USA;
- 3Machine Learning Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
Abstract
Recent efforts to generate atlas-scale single-cell data provide opportunities for joint analysis across tissues and modalities. Existing methods use cells as the reference unit, hindering downstream gene-based analysis and removing genuine biological variation. Here we present GIANT, an integration method designed for atlas-scale gene analysis across cell types and tissues. GIANT converts data sets into gene graphs and recursively embeds genes without additional alignment. Applying GIANT to two recent atlas data sets yields unified gene-embedding spaces across human tissues and data modalities. Further evaluations demonstrate GIANT's usefulness in discovering diverse gene functions and underlying gene regulation in cells from different tissues.
Several recent efforts have attempted to generate atlas-scale single-cell data. Examples include The Human BioMolecular Atlas Program (HuBMAP) (HuBMAP Consortium 2019; Jain et al. 2023), the Human Cell Atlas (HCA) (Rozenblatt-Rosen et al. 2017), the Cellular Senescence Network (SenNet) (Lee et al. 2022), the Human Lung Cell Atlas (HLCA) (Sikkema et al. 2023), and many more. In all of these efforts, researchers aim to collect and analyze data from multiple tissues, modalities, conditions, and more. Although the initial phases of the programs often focused on the coverage of different tissues and on the separate, modality-specific analysis of each data set, we are now at a point at which, for the first time, we can perform joint cross-tissue and cross-modality analysis, on single-cell data at the atlas scale.
The joint analysis is challenging for many reasons. First, one needs to decide on a common reference unit for the analysis. Although most of the prior methods for atlas analysis focused on cells (Butler et al. 2018; Stuart et al. 2019; Hao et al. 2021; Domínguez Conde et al. 2022), such analyses often fail to combine data across modalities. In many cases, the large technical variation between data modalities can lead to inappropriate matching cells being selected between data sets, which results in ineffective integration (Haghverdi et al. 2018). In addition, cell-based analysis makes it more difficult to identify pathways and genes that are active across multiple cell types, in part because it can severely distort or lose the original gene space (Argelaguet et al. 2021). Another issue is the type of information the method can handle. Cell embedding and clustering based on gene expression similarity, which has been commonly used (Kiselev et al. 2017; Hasanaj et al. 2022; Quah and Hemberg 2022), may not be appropriate for spatial technologies, which provide information on cell relationships in space (Yuan and Bar-Joseph 2020; Li et al. 2021; Teng et al. 2022). Finally, different signal-to-noise levels across data sets (Argelaguet et al. 2021) and the large volume of data in atlas studies create considerable challenges for joint analysis.
To date, there have been some attempts at a large-scale joint analysis of data from different modalities (not just in biology). The main approach used for such integration utilizes alignment-based multiview learning (Welch et al. 2017; Butler et al. 2018; Hie et al. 2019; Nguyen and Wang 2020). The goal is to transform multiview (multi–data set) inputs, represented as a multimodal distribution, into a homogeneous unimodal distribution for downstream learning tasks. The key idea of these methods is to project the manifolds of multiple data sets into a common space such that both the locality of each data set is preserved and the distance between the same entities in different data sets (e.g., the same individuals in different social networks) is minimized. Although this may work for some biological data sets, we often observe overalignment problems with this approach (Argelaguet et al. 2021). Some other methods require paired data (multimodal measurements derived from the same set of samples) to learn the mapping between different modalities (Argelaguet et al. 2020; Ashuach et al. 2023), which are usually not available in real cases.
To improve on these issues and to enable large-scale joint analyses of atlas-level data, we developed a new method, gene-based data integration and analysis technique (GIANT), that focuses on genes rather than on cells. Genes provide a level of granularity, which, on the one hand, can serve as anchor points common to all modalities but, on the other hand, make it easy to perform higher-level analysis. We first convert cell clusters from each data set and each modality into gene graphs based on the specific characteristics of that modality. Genes are more basic building blocks for joint analysis than cells or pathways, and as we show, such graphs can be extracted from different high-throughput methods that are being profiled in large atlas studies. To integrate these graphs across modalities and tissues, we project genes from all the graphs into a latent space (embedding) using recursive projections. In recursive projection (Gopal and Yang 2013; Zitnik and Leskovec 2017), a dendrogram is used to enforce similarity constraints across graphs while still allowing genes with multiple functions to be projected to different locations in the embedding space. In other words, the recursive method can be seen as a hierarchical alignment along embedding, in which graphs nearby in the tree structure are embedded closely in the latent space.
We applied the method to the HuBMAP data from 10 different tissues, three different major modalities, and nine different single-cell platforms. We also validated it on another human development cell atlas data set (Cao et al. 2020; Domcke et al. 2020). As we show, for both data sets, despite the considerable heterogeneity, our joint processing and analysis method was able to successfully group genes based on their functions and pathways, to associate specific pathways with tissues, to discover new functions for a large number of genes, and to identify tissue-specific regulations of genes based on their cross-modality clustering.
Results
We developed GIANT to construct a unified embedding for genes from 10 different tissues, including the heart, kidney, large intestine, liver, lung, lymph node, pancreas, small intestine, spleen, and thymus, and three different data modalities, which include single-cell RNA-seq (scRNA-seq), single-cell ATAC-seq (scATAC-seq), and spatial transcriptomics (Slide-seq). Figure 1, A through C, presents an overview of GIANT. We first construct gene graphs for cell clusters from each tissue and data modality based on expression or epigenetic correlations (Methods). Next, all gene graphs are connected in a cell cluster dendrogram. A recursive graph-embedding method that takes into account both the dendrogram and gene identity connections is applied. The final result is an embedding (a location in a latent space) for each gene in each of the graphs. For details, see Methods.
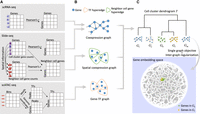
Schematic illustration of the method. (A) Cell clusters are identified from each tissue of each data modality. For each scRNA-seq cell cluster, a gene coexpression matrix is constructed for all the gene pairs in the cell cluster. For each Slide-seq cell cluster, the same gene coexpression matrix is also constructed, and another spatial coexpression matrix is built for gene pairs between the cells of this cell cluster and their neighboring cells. For each scATAC-seq cell cluster, identified peaks from the bin matrix are associated with TFs and genes, which forms a gene–TF matrix. (B) We obtain gene coexpression or coregulation graphs for each of these modalities based on the specific matrices computed in A. (C) A dendrogram for graphs in B is constructed by hierarchical clustering of cell clusters, and a multilayer graph learning algorithm is applied using the tree structure. Each gene in each graph is then encoded into an embedding space, where genes with similar functions are embedded together.
GIANT integrates multimodality and multitissue data
The gene-embedding space produced by GIANT enables joint functional analyses across data modalities, tissues, and cell clusters. Therefore, the location of a gene in the space is expected to reflect the function the gene performs (which could be the same across several different cell types and tissues) instead of the specific data source that generates the value for that gene. For example, genes from different tissues can be closely embedded in space as long as they perform similar functions in these tissues. To assess the integration of various data sources, we visualized the entire embedding space and highlighted the locations of genes based on their respective data sources. Figure 2 presents the distribution of genes in the map based on their modalities (Fig. 2A), tissues (Fig. 2B), and cell clusters (Fig. 2C), respectively.
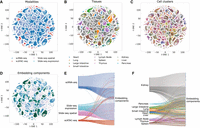
Distributions of data modalities, tissues, and cell clusters in the entire embedding space. (A) Distributions of data modalities in the embedding space; genes are colored by data modalities. (B) Distributions of tissues in the embedding space; genes are colored by tissues. (C) Distributions of cell clusters in the embedding space; genes are colored by cell clusters. (D) The embedding space is split into components; genes in different components are shown in different colors. (E) Cross-clustering between data modalities. (F) Cross-clustering between tissues. As can be seen, genes from several different tissues, and from multiple modalities, are clustered together, allowing for a joint analysis of this large data set.
As can be seen, the locations of genes are not determined by their data modalities or tissues, allowing us to use the embeddings to investigate shared functionalities across tissues and mechanisms being studied by each of the data modalities we used. This observation is confirmed by our quantitative evaluation. GIANT obtains a very low silhouette coefficient (0.016) for data modalities (Supplemental Note 1). We next compared GIANT's gene embedding with the embeddings of six cell-based data integration methods, which, similar to GIANT, can be used to integrate different modalities and tissues (Supplemental Note 2; Supplemental Figs. S1, S2). GIANT achieves a substantially lower silhouette coefficient of data modality compared with Harmony (Korsunsky et al. 2019), LIGER (Liu et al. 2020), Scanorama (Hie et al. 2019), scVI (Lopez et al. 2018), and GLUE (Cao and Gao 2022) and achieves a similar silhouette coefficient to that of Seurat (Supplemental Fig. S1H; Hao et al. 2021). In addition, for the scRNA-seq data used, which were obtained from five different platforms, we see very good integration by GIANT (silhouette coefficient −0.03) (Supplemental Fig. S3).
To further investigate which data sources tend to cocluster, we clustered the entire embedding space (Fig. 2D) and then looked at the data sources assigned to each of the resulting clusters (embedding components). Specifically, we applied the Leiden algorithm with a resolution of one (Traag et al. 2019) to cluster all genes in the embedded space into 172 components (Methods). We then determined the data modality or tissue for each gene in each component. Figure 2E shows the mapping from data modalities to components. Genes from Slide-seq expression and Slide-seq spatial graphs are always assigned to the same components, as these represent different measurements for the same gene. For the other modalities, we found that 14.5% (25 out of 172) of the components contain genes from at least two of the three modalities, scRNA-seq, scATAC-seq, and Slide-seq. To obtain baselines for demonstrating the integration of different modalities, we applied node2vec (Grover and Leskovec 2016) and Gene2vec (Supplemental Note 3; Du et al. 2019) on the same gene graphs used as input for GIANT and generated gene embeddings. We performed Leiden clustering on their embeddings, using a resolution parameter of one (same as we used for GIANT), to identify embedding components and assess how many contained genes from at least two modalities. In both node2vec and Gene2vec embeddings, the majority of components consisted of gene nodes from a single graph (Supplemental Fig. S4). Specifically, only 0.18% (one out of 554) of node2vec components and 0.91% (five out of 548) of Gene2vec components included genes from at least two of the three modalities. The results indicate that GIANT's embedding algorithm with the hierarchical alignment leads to better integration of data from different modalities compared with the baseline methods. Figure 2F shows the mapping from tissues to components. Genes from the heart (blue) and lung (orange), both closely related to the circulatory and the respiratory systems (Fessler 1997), frequently appear in the same components. Tissues from the immune system, including the lymph node (purple), thymus (pink), and spleen (brown) (Felten and Felten 1994), are also frequently coclustering. To quantify the clustering of functionally related tissues in our gene embeddings, we computed the tissue mean average precision (MAP) score (Cao and Gao 2022). In this analysis, the heart and lung were considered as one class and the lymph node, thymus, and spleen as another class, and each of the other tissues was treated as a separate class. MAP calculates for each gene the average precision within its K ordered nearest neighbors (Supplemental Note 4). We obtained a tissue average MAP score of 0.71 for GIANT's embeddings. The tissue average MAP scores derived from the other embeddings (Harmony, LIGER, Scanorama, scVI, Seurat, and GLUE) are much lower than that of GIANT (0.59 for Scanorama, which is the best among compared methods) (Supplemental Fig. S5). These results demonstrate that GIANT effectively integrated data across tissues and cell clusters and is able to group genes based on shared functions. However, gene nodes from graphs of different modalities (e.g., scATAC-seq, scRNA-seq) are not always merged in this data set. This is likely because of incomplete coverage of tissues for different modalities in the HuBMAP data set, leading to unmatched cell clusters across modalities. Additionally, biological variations in cell clusters collected in different modalities of the same tissues further contribute to this limitation (for details, see Discussion).
To further test the ability of GIANT on integrating data from different modalities, we collected single-cell mouse brain data from the Allen Brain Cell Atlas (Yao et al. 2023), which contains 2,341,350 cells from scRNA-seq and 3,938,808 cells from MERFISH spatial transcriptomics (Chen et al. 2015). Both modalities were derived from entire adult mouse brains, encompassing the same tissue regions and cell populations, serving as a good data set for testing the integration of different data modalities. In each modality, cells were grouped into 34 clusters, with each cell cluster annotated as a cell type. We built a gene coexpression graph for each scRNA-seq cell cluster and MERFISH cell cluster. We then ran GIANT to embed genes from the gene graphs (Supplemental Note 5). The embeddings demonstrated clear preservation of cell types, with genes from the same cell types clustered and genes from different modalities well mixed in the embedding space (Supplemental Figs. S6, S7). To compare GIANT with other methods, we again ran the cell embedding methods (Harmony, LIGER, scVI, Scanorama, Seurat, and GLUE) on this data set. We assessed cell-type preservation using cell-type average MAP scores (Supplemental Note 4) and modality mixing using the local inverse Simpson's index (LISI) (Supplemental Note 6; Korsunsky et al. 2019). LISI measures the diversity of neighboring nodes, with a value of two indicating perfect mixing for two modalities. GIANT achieves a cell-type average MAP score of 0.999, outperforming all other methods (Harmony: 0.913; LIGER: 0.864; scVI: 0.962; Scanorama: 0.935; Seurat: 0.542; and GLUE: 0.932). GIANT also obtains a LISI score of 1.91 for the mixing results, closer to the ideal value of two than those of competing methods (Harmony: 1.60; LIGER: 1.19; scVI: 1.38; Scanorama: 1.52; Seurat: 1.32; and GLUE: 1.63).
Genes closely embedded in the space show tissue-specific GO enrichment
We next tested the functional enrichment of nearby genes in the embedding space (Methods). Because of the large number cells of different tissues we analyzed, such analysis allowed us to identify functional groups spanning a large portion of the Gene Ontology (GO) categories in the embedding components. Specifically, this analysis identified 272,848 enriched biological process (BP) GO terms, 44,952 enriched cellular component (CC) GO terms, and 56,199 enriched molecular function (MF) GO terms (adjusted P ≤ 0.01) in the 1199 embedding components identified by the Leiden algorithm with a resolution of 50 (Supplemental Table S1).
We next analyzed the GO terms discovered in the embedding components associated with different tissues. Specifically, for each component, we looked at the top three tissues for genes assigned to that component. Next, we selected the top 5% enriched BP GO terms with the largest fold changes (ratio of the observed proportion of genes associated with a GO term in the embedding component to the expected proportion of genes associated with that GO term in the population) and adjusted P ≤ 1 × 10−5, and associated these GO terms with each of the top three tissues. To summarize the enriched GO terms for each tissue, we propagated these GO terms to higher levels along the GO hierarchies. The top GO categories enriched in each tissue are shown in Figure 3A, in which the size of each dot indicates the ratio of child terms of the noted GO category in all the enriched terms of the tissue, and the color of each dot indicates the fold change of the ratio over the average value of the ratios across tissues.
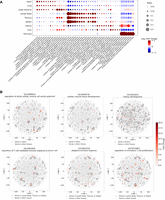
GO terms enriched in different embedding components. (A) The top categories of GO terms enriched in the embedding components associated with different tissues. The size of each dot indicates the ratio of child terms of the GO category in all the enriched terms of the tissue. The color of each dot indicates the fold change of the ratio over the average value of ratios in the column. (B) GO enrichment in different embedding components. Components with the GO term enriched (adjusted P < 0.01) are colored by the enrichment levels of the GO term, and the other components are shown in gray. Genes from tissues related to the GO term are highlighted by dark gray, and genes in the other tissues are colored by light gray.
We found that most of the top GO categories for each tissue (Fig. 3A, dark red dots) are strongly consistent with the known cellular functions of that tissue. For example, muscle-related GO categories (e.g., GO:0003012 muscle system process, GO:0006936 muscle contraction) are prominent for the heart, lung, and large intestine, which contain abundant cells of cardiac muscles, respiratory muscles, and smooth muscles (Severs 2000; Ratnovsky et al. 2008; Sanders et al. 2012). For immune tissues (lymph node, thymus, and spleen), immunity-related GO categories dominated the assignments, including “GO: 0002376 immune system process” and “GO:0006955 immune response.” Lipid metabolism-related GO categories (e.g., GO:0010876 lipid localization) are prominently discovered for the liver (Nguyen et al. 2008). In addition, metabolic process–related GO categories are significantly found for the pancreas, which could be explained by its key metabolic functions in the regulation of blood glucose levels (Röder et al. 2016). Examples of some tissue-specific GO terms are shown in Figure 3B. Each of them is enriched in different components in the embedding space (reddish regions), whereas they are more likely found in the components in which the related tissues reside (dark gray regions). In addition, we identified a list of GO terms enriched in tissue-specific embedding components, which are not directly related to the primary functions of the tissues but are likely associated with specific cell populations supporting the diverse activities of the tissues. These examples are listed in Supplemental Table S2. For example, “GO:0031344 regulation of cell projection organization” was found in a large intestine–specific embedding component. Although cell projection is commonly associated with neuronal cell types, this enrichment may relate to the formation of microvilli in absorptive cells of the large intestine (Walton et al. 2016). In another example, “GO:0019226 transmission of nerve impulse” was enriched in a kidney-specific embedding component. Although the kidney is not primarily a nerve-driven organ like muscle tissues or neurons, it does receive regulation from nervous systems that help control its functions (Neyra et al. 2021).
To confirm the GO findings, we further performed the same enrichment analysis using reactome pathways (Supplemental Note 7; Gillespie et al. 2022). Similar to what we observed for GO, pathways such as “R-HSA-1280218 adaptive immune system” in the lymph node, “R-HSA-1280215 cytokine signaling in immune system” in the thymus and spleen, and “R-HSA-390522 striated muscle contraction” in the heart were identified. In addition, we also observed “R-HSA-5663205 infectious disease” for both the thymus and lymph node (Supplemental Fig. S8).
We again compared GIANT with the six aforementioned cell-based embedding methods. The WGCNA module detection method (Langfelder and Horvath 2008) was applied in each cell cluster identified from each cell-based embedding (Supplemental Note 2). We found that compared with GIANT, there were much fewer unique GO terms enriched in the gene modules discovered from cell-based embeddings (Supplemental Fig. S1H). We also compared GIANT to a general method for clustering genes using all data. For this, we performed hierarchical clustering on the concatenated data matrix from different data modalities for each tissue (Supplemental Note 8). We found much fewer enriched unique GO terms in the identified gene clusters (2467 vs. 6972 of GIANT, P-value < 0.05). In addition, we observed fewer tissue-specific GO categories enriched in gene clusters associated with different tissues (Supplemental Fig. S9). Finally, we also compared GIANT with Limma (Ritchie et al. 2015), which is usually applied to correct for batch effects and so can be used for data integration. We performed the same cell clustering and gene clustering methods as used for GIANT to identify gene modules from the Limma-corrected data matrix (Supplemental Note 9). Again, there are much fewer enriched unique GO terms in the identified gene modules (3939 vs. 6972 of GIANT, P-value < 0.05). Additionally, there are fewer tissue-specific GO categories or GO categories not typically associated with tissue-specific functions found in gene modules associated with different tissues (Supplemental Fig. S10).
Testing GIANT integration on a developmental atlas data set
To further test the ability of GIANT to integrate data from different modalities, we used a human fetal cell atlas data set (Cao et al. 2020; Domcke et al. 2020). This data set contains single-cell data from 15 tissues and two data modalities, including the adrenal, cerebellum, cerebrum, eye, heart, intestine, kidney, liver, lung, muscle, pancreas, placenta, spleen, stomach, and thymus of scRNA-seq and scATAC-seq. Unlike the HuBMAP data, the fetal atlas provides cell-type annotations for cell clusters, allowing us to validate our embeddings at a more refined level. Results are presented in Figure 4, A and B (Supplemental Note 10). As the figure shows, GIANT is able to merge genes from different modalities and tissues. Specifically, several components in the embedding space are dominated by one cell type (Fig. 4C), although genes in these components are often from several tissues and both modalities. To quantify the conservation of cell-type information in our gene embeddings, we computed the cell-type average MAP score (Supplemental Note 4). We obtained a MAP score of 0.72 for GIANT's embeddings. For comparative analysis, we again applied the aforementioned cell-based embedding methods (i.e., Harmony, LIGER, Scanorama, scVI, Seurat, and GLUE) to this data set (Supplemental Figs. S11–S13). The cell-type average MAP scores derived from the embedding of these methods (0.39 of scVI, which is the best among compared methods) are much lower than that of GIANT (Supplemental Fig. S13G). Analysis using only scRNA-seq data from different tissues further supports our findings (Supplemental Note 10). GIANT achieved the highest cell-type MAP score of 0.73, outperforming all other methods (Harmony: 0.63; LIGER: 0.49; scVI: 0.65; Scanorama: 0.65; Seurat: 0.31, and GLUE: 0.62), demonstrating better preservation of cell-type information (Supplemental Fig. S14).
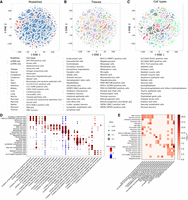
Application of GIANT on the human fetal atlas. (A) Distributions of data modalities in the embedding space. (B) Distribution of tissues in the embedding space. (C) Distribution of cell types in the embedding space. Each point in the plots represents a gene that is colored by its data source. (D) Top GO categories for different tissues. The size of each dot indicates the ratio of child terms of the GO category in all the enriched terms of the tissue. The color of each dot indicates the fold change of the ratio over the average value of ratios in the column. Shown cell types are those with at least 50 GO terms. (E) The enrichment level of different developmental GO terms in the embedding components associated with different cell types shown in D. Colors show the negative logarithm of the corrected P-values.
We also performed the same GO enrichment analysis in the embedding components, as done for Figure 3A, this time using cell types instead of tissues. Our analysis revealed a strong consistency between the enriched GO terms found within the embedding components of each cell type and the well-established cellular functions associated with those specific cell types (Fig. 4D; for the P-value for each GO term, see Supplemental Table S3). For example, signal transduction–related GO categories (e.g., GO:0007267 cell–cell signaling, GO:0009966 regulation of signal transduction) are mainly assigned to nerve cells, such as inhibitory interneurons (Roux and Buzsáki 2015), and enteric nervous system (ENS) neurons (Furness 2012), whereas immune-related GO categories (e.g., GO:0002682 regulation of immune system process, GO:0046649 lymphocyte activation) are mostly associated with immune cells, such as myeloid cells (Gupta et al. 2014), lymphoid cells (Blom and Spits 2006), and microglia (Graeber and Streit 2010). We also found many development-related GO terms enriched in embedding components for this fetal atlas data set. A closer examination revealed that the cell-type-specific developmental pathways are highly enriched in the embedding components associated with the corresponding cell types (Fig. 4E; for gene ratios and fold changes of these GO terms, see Supplemental Fig. S15), such as “neuron projection development” (GO:0031175) for inhibitory interneurons, “hematopoietic or lymphoid organ development” (GO:0048534) for lymphoid cells, and “erythrocyte development” (GO:0048821) for erythroblasts.
In addition, we found that 88.5% (1061 out of 1199) of the HuBMAP embedding gene components are enriched in the embedding components of the human fetal data set (Supplemental Fig. S16), indicating the ability of large-scale atlas data sets to reveal most of the significant biological processes.
To further explore the different gene–gene relationships profiled in the HuBMAP (adult human) and human fetal atlases, for each tissue that is common between the two atlases, we identified unique gene modules in the gene-embedding components of each atlas (Supplemental Note 11). Notably, we observed enrichment of genes associated with the function “regulation of cell differentiation (GO:0045595)” (P-value < 0.05) in fetal-specific gene modules across six out of seven tissues, compared with two tissues in adult-specific gene modules (Supplemental Fig. S17). In addition, genes associated with the function of “regulation of cell cycle (GO:0051726)” were also enriched in six out of seven tissues for fetal-specific gene modules but were only enriched in a single tissue for adult-specific gene modules. This suggests that, as expected, fetal cells are more proliferative compared with adult cells (Bunis et al. 2021). Conversely, GO terms “metabolic process (GO:0008152)” and “biosynthetic process (GO:0009058)” usually have higher enrichment levels in the adult-specific gene modules compared with fetal-specific gene modules across all tissues (Supplemental Fig. S17), suggesting varied metabolism of some substances at different developmental stages (Piquereau and Ventura-Clapier 2018). These findings imply distinct gene regulations depicted in the two atlases, which are captured by our gene embeddings.
Inferring gene functions from multimodality multitissue embeddings
The precise function of a specific protein (and the genes that code for it) may differ between tissues and cell types (Yeger-Lotem and Sharan 2015). Given the observations above that GIANT can group functionally similar genes together across data sources, we used the embeddings to infer novel gene functions for all the genes we embedded. We first clustered each gene's embeddings and then inferred the gene's functions in each cluster based on other coclustered genes with known functions in the graphs of this cluster (Methods).
We tested the coverage and precision of the functions that we can assign using GO. We found that 47.4% of all known GO functions for the genes are identified in at least one of their clusters, which means that our predictions cover roughly half of all known functions for these genes (recall). This number is much higher than the expected number given the number of GO assignments (compared with 6.9% expected for random assignments of 671,215 GO terms for the 1822 genes analyzed here). On the other hand, the ratio of known functions in our GO assignments for genes (precision) is 25.1%, which is also much higher compared with the ratio of known functions in the whole prediction space (1.5%) (Fig. 5A). Precision and recall of our predictions considering only child terms of other GO categories are shown in Figure 5A. We note that the data sets we used were healthy controls. Thus, they did not contain information about stress response, diseases, etc., which explains why several GO functions were not recovered for many genes. In addition, our gene function assignments rely on GO enrichment in neighboring genes within the embedding space. GO terms with very few genes do not reach the significance threshold to be called in such setups. For the precision, we considered the broadest predictions with a P-value threshold of 0.05, encompassing a substantial portion of novel predictions.
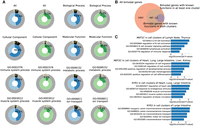
Inference of gene functions in cell clusters. (A) Blue circles show the ratios of known gene functions that are covered by our predictions (recall), and their outside circles indicate the ratios of our predictions in the whole prediction space (all the possible GO assignments). Green circles show the ratios of known gene functions in our predictions (precision), and their outside circles indicate the ratios of known functions in the whole prediction space. Titles indicate child GO terms of which categories are considered. Besides the whole GO space and three main GO branches, some representative GO categories in Figure 3A are also shown here. (B) The number of bimodal genes and the number of bimodal genes for which one or both functions are confirmed. (C) Inferred functions of two genes in different cell clusters.
Given this large coverage and precision, we next investigated genes that are assigned very different functions in at least two different clusters, which are termed as “bimodal genes” here. In particular, genes are selected as bimodal that met the following criteria: Among all the gene's clusters with at least five assigned GO terms, which are BP terms associated with no more than 200 genes to focus on specific GO terms that may show tissue specificity, at least two clusters do not have any common GO terms assigned. If a gene has more than two such clusters, the two with the most GO terms assigned are considered in this analysis. As a result, we identified 1063 bimodal genes, and 487 (45.8%) among them have known functions assigned in at least one cluster. For 121 (11.4%) of them, both clusters have functions that are already known even though they can be very different (Fig. 5B).
Figure 5C presents examples of bimodal genes with different known functions in different clusters. MEF2C is essential in B lymphopoiesis, as well as for B cell survival and proliferation in response to stimulation (Khiem et al. 2008; Wilker et al. 2008). On the other hand, it is known to be involved in vascular development and controls cardiac morphogenesis (Lin et al. 1997, 1998). Our method correctly assigns MEF2C the functions of B cell activation, lymphocyte activation, and regulation of immune response in the group of lymph node and thymus cell clusters, but it assigns this gene the functions of positive regulation of developmental process, as well as tube morphogenesis in a group of heart and lung cell clusters. MEF2C is also known for its important role in neuronal cell types (Li et al. 2008). Even though the HuBMAP data set does not contain brain tissue, which impacts the ability of the method to assign neurological functions to the gene, we did find a cluster of MEF2C that is associated with the function “GO:0010975 regulation of neuron projection development” in cell clusters of the large intestine. It was previously reported (Li et al. 2008) that MEF2C plays a role in the development of neuronal cell types in the ENS (located in the gut wall). However, further experimental validation is required to confirm this prediction. Similarly, RYR2 has been found to play an important role in triggering cardiac muscle contraction and to be related to cardiac arrhythmia (Bround et al. 2012), whereas it also mediates the flow of calcium ions across cell membranes in the large intestine (O'Donnell et al. 2019). Again, RYR2 is assigned the functions of regulation of heart rate, muscle contraction, and regulation of blood circulation in the group containing a heart cell cluster, whereas in the large intestine cell cluster, it is assigned to the ion transmembrane transport function. A complete list of predicted bimodal genes and their predicted functions can be found in Supplemental Table S4.
Gene embeddings enable the discovery of tissue-specific gene regulations
Identifying the regulatory programs of cells helps elucidate their diverse gene expression patterns. We further demonstrate the ability of our gene embeddings to discover gene regulations in cell clusters of different tissues. Similar to the previous subsection, we calculated the enrichment of genes regulated by the same transcription factor (i.e., TF regulon) in each embedding component (Methods). Specifically, this analysis identified 5815 enriched TF regulons (adjusted P ≤ 0.001) in the 1199 components. To compare the TFs identified in cells of different tissues, we associated discovered TFs with tissues using the top three tissues for each component as discussed above. TF overlaps among different subsets of tissues are shown in Figure 6A. Although large numbers of TFs (104 out of 435, 23.9%) were identified as significant for all the tissues, most TFs show specificity for different tissues. These observations are consistent with previous findings in the literature (Sonawane et al. 2017; Steuernagel et al. 2019). The full list of TFs identified in different tissues can be found in Supplemental Table S5.
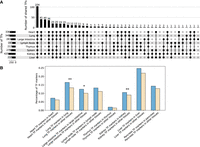
TFs discovered in different tissues. (A) TF intersections among different tissues. Bars on the left show the number of TFs identified in each tissue, and each bar on the top shows the number of TFs shared among tissues marked by the dots below it. Tissues with fewer than 20 TFs identified are omitted in this figure. The figure was drawn using UpSetR (Conway et al. 2017). (B) The percentage of tissue-specific TF markers in the corresponding tissue (blue) compared with their percentage in the other tissues (light yellow). Significant enrichment in blue groups: (*) P ≤ 0.05, (**) P ≤ 0.01; P-values are evaluated using one-sided Fisher's exact tests.
To further validate our discoveries, we examine whether TFs found in each tissue are enriched with TFs known to be specific to this tissue. For this, we used a prior list of tissue marker TFs (Xu et al. 2022). We find enrichment for tissue markers in all tissues and significant enrichment for three of the eight tissues (Fig. 6B).
Discussion
Recent efforts to generate atlas-scale single-cell data provide opportunities for joint analysis across tissues and modalities, which can enable new insights into cell functional plasticity, gene function, cell regulation, and disease. A key challenge for such analysis is how to effectively integrate data from different sources, such as tissues, modalities, and conditions, while enabling the discovery of system-level organizations and regulations.
We developed a new method that is the first to fully integrate atlas-level data from multiple tissues and modalities in a single unified embedding. For this, we use genes as the anchor points common to all data sets, modalities, and tissues. Our method first converts cell clusters in different tissues from different data modalities to gene graphs and then recursively embeds these graphs such that connected genes within a single graph are encouraged to have similar embeddings, whereas genes from graphs generated from similar cells are projected closely in the embedding space. Finally, a unified embedding space is produced in which the locations of genes reflect the functions they perform in the cells.
We applied our method to HuBMAP database with 10 different tissues, three different major modalities, and nine different single-cell platforms. As we have shown, despite the considerable heterogeneity of the input data, our method was able to successfully group genes based on their function and pathways. We further used the method to assign new functions to previously characterized genes and to explore tissue similarities and regulations. We compared GIANT with six cell-based embedding methods. Note that these cell-based methods are not expected to focus on genes, and these comparisons are mainly to emphasize the advantage of gene-based analysis.
An interesting finding is the number of genes that have at least two sets of functions (termed “bimodal genes”). Even with only 10 tissues, we identified more than 1000 genes with two or more diverse functions depending on the tissues. Although several studies have pointed out genes with multiple functions (Pritykin et al. 2015) mostly in different cell types or tissues (Khiem et al. 2008; Wilker et al. 2008), these prior findings were anecdotal and focused on very few genes. By analyzing much larger atlas data, we were able to determine that such bimodal genes are actually quite common. It is likely that the number of genes with multiple functions is actually much larger given the large number of tissues that were not included in our study. These findings indicate that even though the majority of genes have at least one associated function in large annotation databases (e.g., GO), there is still much work needed to fully characterize the functions of human genes.
For HuBMAP, we have mainly focused on tissue-level analysis as the first step for atlas-level integration. Although we used cell clustering (which likely represents cell types) as input, these clusters were not assigned to specific cell types, and so, our method can currently only provide information on tissue-level functions and regulations. However, as we showed with the fetal atlas data set, GIANT can be easily applied to integrate atlas data at the cell-type level as well.
Although GIANT relies on cell-based clustering for the initial processing (creating data set–specific graphs), it integrates data in the gene space rather than at the cell level. This provides several advantages as we discussed above and leads to better results as we showed. Although we used cell-type information for validation, the method does not assume such information is provided. It is therefore robust to different cell cluster assignments between modalities. The method performs clustering of cells within each modality, and so, the main requirement is that cells from the same type are similar in each of the different modalities, which is usually the underlying assumption of these technologies.
The rapid expansion of single-cell technologies has introduced various sequencing platforms that produce data with different qualities. For example, in contrast to transcript end sequencing technologies, full-length scRNA-seq covers the entire sequence of RNA molecules, enabling the detection of splice isoforms and genetic variants (Hagemann-Jensen et al. 2020; Hahaut et al. 2022). Applications of GIANT on such data, which offers richer information, could be tested in the future to determine if it yields novel biological insights.
GIANT constructs gene–TF graphs for scATAC-seq cell clusters, which connect genes to TFs who are known to bind to their promoter regions while the binding sites are open. This reduces the scATAC-seq data to the gene space, which, on the one hand, leads to information loss but, on the other, enables easier integration of data from different modalities.
GIANT successfully integrates data from three different modalities (the silhouette coefficient for data modality is 0.016, indicating very little impact of data modalities) (Supplemental Fig. S1H). This can also be observed when visualizing the gene embeddings of the kidney from both scRNA-seq and Slide-seq (Supplemental Fig. S18), in which Slide-seq data are distributed in the embedding among several different scRNA-seq gene clusters. However, gene nodes from individual modality graphs (scATAC-seq, scRNA-seq, etc.) are usually not merged because they are embedded together as part of their input graph. This is likely because graphs for different modalities have very different structures that capture gene functions from different perspectives (coexpression for scRNA-seq, spatial coexpression for Slide-seq, and c-regulation for scATAC-seq). In addition, for the HuBMAP data set, we do not have all modalities for all tissues. For example, the lymph node and thymus have only scRNA-seq data, but for the pancreas and small intestine, we only have scATAC-seq data. Slide-seq is only available for kidney data. Combined, this leads to a number of cell clusters from a modality that do not have a corresponding cluster in another modality, which limits the overall integration. Moreover, we observed that within each tissue, cell clusters between different modalities generally exhibit less overlap in their marker genes (determined by differential expression as described in Methods) compared with those from the same modality. Consistently, GO terms enriched in embedding components associated with cell clusters from different modalities show lower overlap compared with those from the same modality (Supplemental Fig. S19). The results suggest the existence of variations in cell states for data from different modalities even when they come from the same tissues. Still, the ability to integrate gene modules (or graphs in our representation) of GIANT is important and, as we show, can lead to relevant and novel biological findings. For example, for the gene function prediction task, among the 664 genes that appear in at least two of the three data modalities, 332 (50.0%) genes have predicted functions arising out of neighbors from more than one modality. Among the 1924 genes that appear in at least two tissues, 1606 (83.5%) genes have predicted functions arising out of neighbors from more than one tissue (Supplemental Fig. S20). We also note that GIANT can enforce a higher level of modality integration by increasing the regulation strength (λ); however, in such cases we observed lower-quality gene embeddings (Supplemental Fig. S21). Similarly, we found that cell-based embeddings, such as Seurat and GLUE, achieve higher levels of modality integration, but the resulting gene modules are of lower quality (Supplemental Fig. S1H).
GIANT uses a multistep approach to overcome batch effects. Specifically, rather than embedding the specific expression levels, which are batch sensitive, GIANT embeds the gene interaction graphs. These are more robust because they do not depend on specific values and combine information from multiple cells (Stuart et al. 2003). Next, GIANT projects genes from each gene graph into an embedding space, in which the positions of genes are meant to reflect their connections within the gene graph and therefore their functions. The embeddings are constrained by relationships between graphs from different data sets in the dendrogram, which is a regularizer that also reduces batch effects. This approach can be seen as a hierarchical alignment along the embedding.
There are a number of other methods that can also generate gene embeddings from single-cell data. These include GLUE (Cao and Gao 2022), DeepMAPS (Ma et al. 2023), SIMBA (Chen et al. 2024), and GSDensity (Liang et al. 2023). For example, GSDensity can associate genes with cells based on their distances within the same embedding space and therefore enables the analysis of pathway activities in cells. However, unlike GIANT, these methods only provide a single embedding for each gene, limiting their ability to capture the rewiring of gene networks under different conditions and cell types. In contrast, GIANT generates a unique embedding for each gene in each cell cluster, allowing users to study multiple functions of the same gene in different tissues or cell types, as demonstrated in our study.
GIANT generates gene embeddings for cell clusters from different tissues measured under various modalities within a unified space, providing a foundation for gene- or pathway-centric analyses in multimodal, multitissue, single-cell data. GIANT is not designed for cell embeddings. The embedding is encouraged to place gene nodes representing the same gene together. This cannot be directly applied to cells because there is no concept of the same cell in different data sets. Given this, GIANT cannot be applied to cell-level analysis. As discussed in this study, the GIANT embedding allows users to explore the diverse functional roles of genes across different conditions, cell types, and tissues. We demonstrated how GIANT embeddings captured developmental pathway differences in tissue development. Other potential usages are comparisons between healthy and disease tissues, using the GIANT embeddings generated for different cell types and cellular states, and multimodal comparisons between large-scale single-cell data sets across different species and groups.
Our embedding method is implemented in Python and is available along with the generated gene embeddings at GitHub (https://github.com/chenhcs/GIANT). We hope that as the analysis of atlas-level data becomes more common, our method would be able to be applied to additional atlases and data sets to integrate large-scale single-cell data.
Methods
Data sets
We integrated single-cell data from more than 60 individuals profiling 10 different tissues and three major data modalities from the HuBMAP consortium (HuBMAP Consortium 2019).
scRNA-seq data sets
scRNA-seq data sets were obtained for eight tissues: heart, kidney, large intestine, liver, lung, lymph node, spleen, and thymus. We obtained 14 data sets and a total of 30,241 cells for the heart, 15 data sets and 109,055 cells for the kidney, six data sets and 13,128 cells for the large intestine, five data sets and 7916 cells for the liver, nine data sets and 68,390 cells for the lung, four data sets and 40,449 cells for the lymph node, 10 data sets and 93,737 cells for the spleen, and three data sets and 10,838 cells for the thymus. These data were obtained from five platforms (Supplemental Table S6) and were uniformly processed. The uniform processing used the Salmon's Alevin method (Patro et al. 2017) for sequencing read mapping and abundance quantification, leading to a cell-by-gene count matrix for each data set.
scATAC-seq data sets
scATAC-seq data sets were obtained for eight tissues, including the heart, kidney, large intestine, liver, lung, pancreas, small intestine, and spleen. There are 14 data sets and in total 312,579 cells for the heart, 19 data sets and 272,526 cells for the kidney, six data sets and 8368 cells for the large intestine, two data sets and 50,748 cells for the liver, nine data sets and 205,048 cells for the lung, three data sets and 68,294 cells for the pancreas, three data sets and 7774 cells for the small intestine, and four data sets and 16,260 cells for the spleen. Again, data from three platforms (Supplemental Table S6) were combined using a uniform processing pipeline. For this, we used SnapTools (Fang et al. 2021) for read mapping and abundance quantification. SnapTools segments the genome into uniform-sized bins of 5000 bp, and a cell-by-bin matrix is created for each data set with each element indicating the count of sequencing fragments overlapping with a given bin in a certain cell.
Slide-seq data sets
Slide-seq (Rodriques et al. 2019), a spatial transcriptomics method, was used to profile kidney samples. There are in total 551,342 cells (DNA-barcoded beads) included in 19 Slide-seq data sets. Slide-seq generates short-read sequencing data that are equivalent to scRNA-seq data and were processed in the same way we processed all scRNA-seq data. In addition, the spatial location associated with each bead is provided.
The HuBMAP data from each modality (even from different platforms) were processed using the same pipeline, and the reads of different data sets were aligned to the same reference genome. It is highly recommended that other large-scale atlas analyses follow a similar procedure. Accession numbers for the HuBMAP data sets used in this study are listed in Supplemental Table S6. Data can be obtained from the HuBMAP data portal (https://portal.hubmapconsortium.org) via accession numbers.
The scRNA-seq data and the scATAC-seq data of the human fetal atlas were obtained from the Descartes interactive website (Supplemental Note 10; https://descartes.brotmanbaty.org/).
The scRNA-seq data and the MERFISH data of the Allen Brain Cell Atlas were obtained from the Allen Brain Map portal (Supplemental Note 5; https://portal.brain-map.org/atlases-and-data/bkp/abc-atlas).
Clustering cells with the HuBMAP modality-specific pipelines
For each tissue in each data modality, cells are merged by joining the data matrices of different data sets along the cell axis. The HuBMAP modality-specific data analysis pipelines, described below, are then used to cluster cells in each tissue of each data modality.
The HuBMAP scRNA-seq pipeline uses the SCANPY (Wolf et al. 2018) package to perform cell clustering. SCANPY first filters cells that express fewer than 200 genes and genes that are expressed in less than three cells. Each cell is then normalized by its total counts over all genes. The produced count matrix is log-transformed and scaled so that each gene has unit variance and zero mean, after which it is used as input to dimensionality reduction via PCA, computation of a k-nearest-neighbor (k-NN) graph in PCA space, and finally clustering with the Leiden algorithm (Traag et al. 2019). We further identify differentially expressed genes for each cell cluster against all the other clusters in the tissue using the Wilcoxon rank-sum test, allowing for estimating similarities between cell clusters.
The scATAC-seq processing pipeline uses the SnapATAC package (Fang et al. 2021) for data processing, which starts from each cell-by-bin count matrix. High-quality cells are identified for subsequent analysis based on three criteria: (1) total number of unique fragment count (>2000); (2) UMI count (>1000); and (3) mitochondrial read ratio (<50%). Bins are then filtered out if they overlap with the ENCODE blacklist (downloaded from https://mitra.stanford.edu/kundaje/akundaje/release/blacklists/), if they are in mitochondrial chromosomes, or if they belong to the top 5% invariant bins ranked by the coverage. From the filtered cell-by-bin matrix, we create a gene activity matrix. Gene coordinates are extracted and extended to include the 2000 bp upstream region (Hao et al. 2021). Gene activities are then computed based on the number of fragments that map to each of these regions, using the “calGmatFromMat” function in the SnapATAC package, and saved as a cell-by-gene matrix. Cell clusters are identified from the cell-by-gene matrix by using it as the input of the same scRNA-seq cell clustering workflow with the steps of normalization and rescaling, dimensionality reduction, k-NN graph computation, and clustering with the Leiden algorithm. After cell clustering, MACS2 (Feng et al. 2012) is used to call accessibility peaks over all cells for each cluster with at least 100 cells. Genes that are differentially active for each cell cluster against all the other clusters in the tissue are again calculated using the Wilcoxon rank-sum test from the gene activity matrix.
Because Slide-seq yields gene count matrices that are equivalent to those of scRNA-seq, the same cell clustering pipeline that was used for scRNA-seq is applied to Slide-seq data. The spatial information of cells is used to construct the gene spatial graphs as discussed below.
Constructing gene graphs for cell clusters
Gene graphs are constructed for cell clusters in all tissues and modalities. To focus on cell-type-specific genes, we filter genes based on their variance in the data sets. Specifically, the union of the 500 most variable genes from each tissue are selected, using the SCANPY package with the criteria defined in Seurat (Satija et al. 2015). The variable genes are identified from gene expression matrices for scRNA-seq and Slide-seq and from gene activity matrices for scATAC-seq. The union of these sets results in 7000 genes that are selected and used for building gene graphs and further analyses.
An unweighted undirected k-NN gene graph is computed for each scRNA-seq cell cluster from its normalized and log-transformed expression matrix. For each pair of genes, the Pearson's correlation of their expression values across cells in the data sets is computed. Each gene is then connected to the top 10 other genes with the strongest correlation.
A gene–transcription factor (TF) hypergraph is constructed for each scATAC-seq cell cluster by associating the peaks identified in each cell cluster with TFs and target genes. We identify peaks that contain TF motifs using the motifmatchr package, which provides information on 633 human TF binding motifs collected from the JASPAR 2020 (Fornes et al. 2020) database. A binary peaks-by-TF matrix is generated, in which an element “1” indicates that the TF's motifs are found in the region covered by the corresponding peak. A complimentary gene-by-peak matrix is generated, in which elements are set to “1” if the corresponding peaks are within the promoter regions of the corresponding genes. Here the promoter region is defined as the 500 bp upstream region of the transcription start site, which considers core and proximal promoters, as was previously done by Aibar et al. (2017). The dot product of the two aforementioned matrices results in a gene-by-TF matrix. We then construct a hypergraph by treating the gene-by-TF matrix as the incidence matrix, in which each column that corresponds to a TF is treated as a hyperedge, and genes with elements of “1” in this column are nodes connected by this hyperedge.
The expression matrix of Slide-seq is equivalent to that of scRNA-seq data. Therefore, for each Slide-seq cell cluster, we construct an unweighted undirected coexpression k-NN graph using the same method used for scRNA-seq data. In addition, we construct another spatial coexpression graph for each bead cluster that links coexpressed genes in neighboring beads. For this, we identify the neighbors of each bead using the Delaunay triangulation of the bead's spatial coordinates without using a distance threshold, which avoids the need to specify the parameters from users and is commonly used in other studies (Dries et al. 2021; Zhang et al. 2022). Then for each cluster, we compute the correlation of each pair of genes, in which one side is the expression of a gene in this cluster and another side is the mean expression of a gene in the neighboring clusters. Again, each gene is connected to the top 10 most coexpressed genes, and the graph is converted to a hypergraph in which each neighboring-cell gene is treated as a hyperedge.
Finally, we constructed 701 graphs with 1,318,103 nodes in total for 10 tissues and three data modalities.
Connecting gene graphs through a cell cluster dendrogram
We build a cell cluster dendrogram using hierarchical clustering (Pedregosa et al. 2011), in which cell clusters that are close together in the dendrogram are likely functionally similar. For this, we first construct
a distance matrix between cell clusters using the obtained top 50 differentially expressed or active genes of each cell cluster.
Each Slide-seq cell cluster has two copies, one for the expression graph and one for the spatial graph. Given two sets of
differentially expressed or active genes SA and SB of cell clusters A and cell cluster B, their distance is defined as
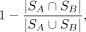 (1)
where
(1)
where 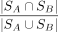 is the Jaccard index of the two gene sets. That is, if two cell clusters have totally different differentially expressed
or active gene sets, their distance will be one, but if they have the same gene sets, their distance will be zero. We then
build the dendrogram from the distance matrix using the agglomerative clustering algorithm with the criterion of minimizing
the average of distances when merging pairs of branches. The clustering process results in a dendrogram in which each leaf
is associated with a gene graph of a cell cluster.
is the Jaccard index of the two gene sets. That is, if two cell clusters have totally different differentially expressed
or active gene sets, their distance will be one, but if they have the same gene sets, their distance will be zero. We then
build the dendrogram from the distance matrix using the agglomerative clustering algorithm with the criterion of minimizing
the average of distances when merging pairs of branches. The clustering process results in a dendrogram in which each leaf
is associated with a gene graph of a cell cluster.
Learning gene embeddings from gene graphs and the cell cluster dendrogram
To identify genes with similar functions or genes participating in the same pathway, we aim to embed each gene in each gene graph into a d-dimensional feature space in an unsupervised fashion. For this, we use the multilayer graph learning objective proposed by Zitnik and Leskovec (2017), which relies on connections between genes within a single graph and combines this with inter-graph relationships between genes in different graphs. Formally, let V be a given set of N genes {v1, v2, …, vN} and Gi = (Vi, Ei) be one of the K gene graphs in the data set (i = 1, 2, …, K), where Ei denotes the edges between nodes Vi ⊆ V within the graph. The dendrogram is a full binary tree T defined over a set of 2K − 1 elements, where each of the K leaf elements is a gene graph. We define π(i) as the parent of the element i in the tree and, reversely, c(i) as the children of the element i in the tree. The problem is thus to learn mapping functions f1, f2, …, fK, each attached with one graph, such that function fi maps nodes in Vi to feature representations in ℝd. fi is equivalent to a matrix of |Vi| × d parameters.
We started from the assumption that genes with similar neighborhoods in each single graph perform similar functions and therefore
should be embedded closely together. Particularly, for the coexpression graph, in many cases there is a strong relationship
between coexpression and functions. The idea has been widely used in the literature to study gene functions using scRNA-seq
data (McKenzie et al. 2018; Cha et al. 2023). GIANT does not just look for coexpression in a single cell or tissue but rather focuses on coexpression across several
tissues and modalities, which is more likely to result from real functional implications. The following objective function
is used to achieve this goal:
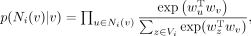 (2)
where Ni(v) are the neighbors of node v in graph Gi, and wv = fi(v), which is the embedding of node v in this graph. The conditional likelihood p(Ni(v)|v) is equivalent to the product of a series of softmax functions; maximization of p(Ni(v)|v) therefore tries to maximize the classification probabilities of v’s neighbors based on learned node embeddings. With that, the objective for each graph i is set to
(2)
where Ni(v) are the neighbors of node v in graph Gi, and wv = fi(v), which is the embedding of node v in this graph. The conditional likelihood p(Ni(v)|v) is equivalent to the product of a series of softmax functions; maximization of p(Ni(v)|v) therefore tries to maximize the classification probabilities of v’s neighbors based on learned node embeddings. With that, the objective for each graph i is set to
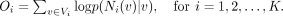 (3)
(3)
We assume that nearby cell clusters in the dendrogram are functionally similar, which implies that the same gene should perform
similar functions in these cell clusters, and therefore, its embeddings in the corresponding graphs should be similar. For
this, we introduce a mapping function fi (where i = K + 1, …, 2K − 1) for each internal (i.e., nonleaf) element, similar to those of leaf elements, which map nodes to feature representations
in ℝd. Recursively, the node set in an internal element is the union of the nodes in its child elements, 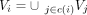 (where i = K + 1, …, 2K − 1). The following regularization term is then introduced for nodes in each element i of T:
(where i = K + 1, …, 2K − 1). The following regularization term is then introduced for nodes in each element i of T:
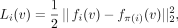 (4)
where node v in the child elements is encouraged to share similar embeddings with the node v in the parent element under the Euclidean norm. We obtain the regularization for an element by summing over the terms of
all its nodes:
(4)
where node v in the child elements is encouraged to share similar embeddings with the node v in the parent element under the Euclidean norm. We obtain the regularization for an element by summing over the terms of
all its nodes: 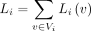 . For an internal element i, its mapping function fi in the optimization iteration t can be solved by the following closed-form solution given the mapping functions of all the other elements in T computed from the previous iteration t − 1, which minimizes the regularization terms involving fi:
. For an internal element i, its mapping function fi in the optimization iteration t can be solved by the following closed-form solution given the mapping functions of all the other elements in T computed from the previous iteration t − 1, which minimizes the regularization terms involving fi:
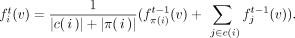 (5)
(5)
With the objective function and regularization term defined, the model tries to solve the following optimization problem to
find the best mapping functions f1, f2, …, fK of leaf elements:
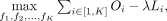 (6)
where λ is the hyperparameter of the regularization strength. To optimize this function, an iterative learning process is
performed, in which leaf mapping functions and internal mapping functions are updated alternately. The leaf mapping functions
f1, f2, …, fK are updated by performing one epoch of stochastic gradient descent that optimizes Equation 6, whereas the internal mapping functions fK+1, fK+2, …, f2K−1 are updated using Equation 5. At each iteration t, the optimization of a mapping function depends on the results of its parent or child elements from the previous iteration,
t − 1.
(6)
where λ is the hyperparameter of the regularization strength. To optimize this function, an iterative learning process is
performed, in which leaf mapping functions and internal mapping functions are updated alternately. The leaf mapping functions
f1, f2, …, fK are updated by performing one epoch of stochastic gradient descent that optimizes Equation 6, whereas the internal mapping functions fK+1, fK+2, …, f2K−1 are updated using Equation 5. At each iteration t, the optimization of a mapping function depends on the results of its parent or child elements from the previous iteration,
t − 1.
In practice, we set the dimension hyperparameter d as 128 and the regularization strength λ as 1.0, which makes the model achieve the best performance. When using a λ in the range between 1.0 to 10.0, the model performs better by merging information between gene graphs instead of focusing on single gene graphs (λ in the range of 0.0 to 0.1). In addition, we observed that the results stay about the same across a range of choices of hyperparameter values for d and λ (Supplemental Fig. S22). The neighborhood N(v) for each node in each graph is constructed using Node2vec's random walk algorithm (Grover and Leskovec 2016). We implemented distributed training that applies gradients on each node parallelly based on the Gensim package (Rehurek and Sojka 2010) written in Cython. With this, the algorithm is scalable to hundreds of graphs and millions of nodes. The runtime and memory usage for running GIANT on both the HuBMAP and human fetal data sets, along with the comparison to other methods, are reported in Supplemental Table S7. The runtime and memory usage on querying the nearest neighbors from the GIANT embedding are reported in Supplemental Table S8.
Splitting embedding space into components and identifying enriched GOs and TF regulons in them
The learned mapping functions map each gene in each cell cluster to a point in the embedding space. To identify overlapping data sources and discover functions and pathways enriched in different regions of the space, we split the embedding space into embedding components by clustering gene points in it. To do the clustering, we generate another graph in which each point in the space (gene) is connected to its 30 nearest neighbors. The Leiden algorithm (Traag et al. 2019) with resolutions of one (for identifying overlapping data sources) or 50 (for discovering enriched functions) is then applied to cluster the genes. GO enrichment is performed for genes in each component against all the genes clustered using the GOATOOLS package (Klopfenstein et al. 2018), in which a gene from a cell cluster is considered as an object, and all the genes from all the cell clusters form the population. P-values from randomization experiments follow a uniform distribution, indicating the validity of this statistical test (Supplemental Fig. S23). GO annotations with the evidence code “IEA,” namely, electronic annotation without manual review, are ignored in the analysis to avoid circular reasoning. GO annotations are propagated through the GO hierarchies with “is_a” and “part_of” relationships. False-discovery rate (FDR) correction with Benjamini–Hochberg is applied to adjust the enrichment P-values.
We also download manually curated TF–target regulatory relationships from the TRUST v2 (Han et al. 2018) database. Enrichment of TF regulons (genes regulated by the same TF) in each component is computed using the same method described above.
Inferring multicellular functions of genes
To identify new functions for genes, we relied on genes with known functions that are coembedded with them. Specifically, for one gene, we calculate the Euclidean distance between each pair of the gene's embeddings of different graphs and then grouped all its graphs into clusters using agglomerative clustering, in which two clusters will stop being merged if the distance between them is above 1.5. Then, for each graph in a cluster, the gene's 50 nearest neighbors in the graph are identified based on the Euclidean distance between gene embeddings. A “common neighbor” set for the gene in this cluster is finally selected based on two criteria: (1) the neighbor gene must appear in the nearest neighbors of at least half of the graphs in the cluster and (2) the neighbor gene is in the 50 most frequent neighbors based on the number of its appearance in nearest neighbors. Next, GO enrichment analysis is performed using the common neighbors. GO terms that are enriched in the neighbors (P < 0.05) are assigned to the gene as predicted functions. In this analysis, we only predict functions of genes expressed in at least 30% of the cells in each cell cluster. Using “common neighbors” for performing this analysis as opposed to analyzing individual cell clusters increases robustness. By first performing the clustering of gene graphs and then the identifying common neighbors and enriched functions, we reduce the amount of false positives.
Software availability
The source code of GIANT and the gene embeddings generated from GIANT for both the HuBMAP data set and the human fetal atlas data set are available at GitHub (https://github.com/ chenhcs/GIANT), at Zenodo (https://doi.org/10.5281/zenodo.10515930), and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The work in this article was partially supported by National Institutes of Health (NIH) grants OT2OD026682, 1U54AG075931, and 1U24CA268108 to Z.B.-J.
Author contributions: H.C., N.D.N., and Z.B.-J. conceptualized the study. H.C., N.D.N., and Z.B.-J. designed the algorithm and methodology. M.R. and H.C. performed data preprocessing. H.C. and N.D.N. developed the software. H.C. and Z.B.-J. performed the analysis. H.C., N.D.N., and Z.B.-J. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279631.124.
-
Freely available online through the Genome Research Open Access option.
- Received May 28, 2024.
- Accepted February 3, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








