
Abstract
Since the advent of long-read sequencing, achieving longer read lengths has been a key goal for many users. Ultra-long-read sets (N50 ≥ 100 kb) produced from Oxford Nanopore sequencers have improved genome assemblies in recent years. However, despite progress in extraction protocols and library preparation methods, ultra-long sequencing remains challenging for many sample types. Here, we compare various methods and introduce the FindingNemo protocol that: (1) optimizes ultra-high-molecular-weight (UHMW) DNA extraction and library cleanup by using glass beads and hexamminecobalt(III) chloride (CoHex), (2) can deliver high ultra-long sequencing yield of >20 Gb of reads from a single MinION flow cell or >100 Gb from PromethION devices (R9.4–R10.4 pore variants), and (3) is scalable to using fewer input cells or lower DNA amounts, with extraction to sequencing possible in a single working day. By comparison, we demonstrate that this protocol surpasses previous methods by enabling precise determination of input DNA quantity and quality through cell counting, sample dilution, and homogenization techniques.
The first ultra-long (UL) sequencing protocols on nanopore sequencing instruments resulted in highly contiguous human genome assemblies (Jain et al. 2018a; Quick 2018). Later, some methods improved on these assemblies through increased yields (Miga et al. 2020; Logsdon et al. 2021). Oxford Nanopore Technologies (ONT) subsequently launched UL sequencing kits, starting with ULK001 on R9.4 flow cells and more recently with ULK114 on R10.4.1 flow cells. The ULK001 protocol utilized extraction and cleanup steps using silicon-coated discs from Circulomics Inc. (https://www.pacb.com/wp-content/uploads/Guide-overview-Nanobind-CBB-kit.pdf), which was subsequently replaced with the ONT star-shaped matrix for library precipitation in the ULK114 kit. These UL kits introduced a range of protocol improvements both in library preparation and sequencing performance to maximize read length and generated reads with an N50 > 100 kb from both the MinION and PromethION flow cells.
In our experience, we have found that the most critical step in obtaining high-throughput, high-occupancy UL sequencing libraries is the DNA fragmentation step, followed by the final cleanup step prior to loading the library. The UL protocol uses a transposase enzyme complex (transposome) to fragment DNA. During fragmentation, the DNA should be homogeneous and accessible to this fast-acting transposome. Two methods are effective in reducing the number of transposase cuts to increase read length: either using a high concentration of high-molecular-weight (HMW) DNA as shown by Jain et al. in sequencing the human genome (Jain et al. 2018a) or diluting the transposase relative to DNA (Jain et al. 2018b). UL libraries are difficult to clean up using conventional magnetic solid-phase reversible immobilization (SPRI) approaches as the DNA can be hard to elute and so sheared during this; SPRI beads are mostly used for polymerase chain reaction (PCR) cleanup where the fragment length is much shorter (Rodrigue et al. 2010; Stortchevoi et al. 2020). In addition, the library is a complex of motor protein and DNA, and so alcohol-based precipitation and washes are not compatible. In our search for suitable alternative approaches, we found that hexamine cobalt (III) (CoHex) cations stabilize and condense DNA and reasoned that they may enable alcohol-free precipitation of nanopore sequencing libraries (Allers and Lichten 2000; Kankia et al. 2001).
Early versions of the ONT UL protocol required extraction and library cleanup starting from 6 million human cells or an equivalent input amount of DNA (Methods; https://www.pacb.com/wp-content/uploads/Guide-overview-Nanobind-CBB-kit.pdf). The complete extraction and library preparation protocol takes between 2 and 3 days in the laboratory. However, different sample types often require tailored extractions to provide optimal DNA for sequencing, so we sought to use a wide range of sample extraction protocols to obtain UL reads on ONT platforms. We also explored reducing both sample input requirements and the total time for library preparation for sequencing scalability.
Here, we describe the FindingNemo protocol (named after the characteristic orange color of the CoHex buffer) for the generation of high-occupancy UL reads on nanopore platforms. This protocol can generate equivalent or more throughputs to disc-based methods and may have additional advantages in tissues and nonhuman cell material. The protocol can also be tuned to enable extraction from as few as 1 million human cell equivalents or 5 µg of human ultra-HMW (UHMW) DNA as input and enables extraction to sequencing in 1 working day. The cleanup method can be used to generate UL libraries from DNA extracted with phenol–chloroform, sodium dodecyl sulphate (SDS) lysis, and cetyltrimethylammonium bromide (CTAB) approaches as well as commercial kits such as the NEB Monarch HMW gDNA kits and those from Circulomics (now Pacific Biosciences). ONT released similar protocols using a spermine-based precipitation buffer (PPT) instead of CoHex and no DNA-binding substrate (ONT, ULK001) and then incorporating a star (ONT, ULK114) after our initial protocol release on protocols.io (Cahyani et al. 2021). We compare all these methods here.
Results
The FindingNemo protocol compiles and optimizes available UL sequencing protocols
Previously developed protocols
Both the standard and UL sequencing workflows consist of three main steps: DNA extraction, library preparation, and library cleanup prior to flow cell loading (Fig. 1A). UL sequencing necessitates minimal DNA shearing throughout the library preparation process and during the extraction methods to maintain UHMW DNA. Early experiments pioneering UL sequencing used the transposase-based rapid kit (ONT, e.g., RAD004) to obtain reads with an N50 > 100 kb (Jain et al. 2018a). This method eliminated the need for DNA shearing before library preparation. However, the yield from rapid runs was typically low, which might be a consequence of the protocol itself. The lack of purification or cleanup of rapid libraries prior to flow cell loading likely contributed to reduced yields compared to ligation-based sequencing approaches. To better understand the complex interplay of DNA quality, library preparation, read length, and yield, we tested a range of UL protocols aimed at maximizing both read length and yield (Fig. 1B).
Summary of the FindingNemo toolkit. (A) Similarities and differences between the standard and UL protocols that center on the way UHMW DNA is handled. Created with BioRender (https://www.biorender.com). (B) The route of FindingNemo toolkit consists of five main steps, with options at the extraction, library preparation, and cleanup steps. Options with asterisks (*) are our current laboratory workflow for the UL protocol.
The goal in obtaining UL reads with the rapid kit is to ensure that each DNA molecule is cut only once by a transposase complex, maximizing read length. Initially, Quick (2018) achieved this by saturating the transposase reaction with UHMW DNA (Table 1A). An alternative approach taken by Logsdon et al. (2021) was to reduce the amount of transposase for a fixed amount of DNA (Table 1B). However, the sequencing output of this method, when applied to the CHM13 cell line (Logsdon et al. 2021), was lower overall compared to the publicly available nanopore data for GM24385 and GM24631 ligation-based libraries (Supplemental Fig. S1A). We hypothesized that this was most likely due to suboptimal pore occupancy (i.e., the number of available pores sequencing at any time), resulting from the reduced number of adapted DNA ends available for sequencing because of the lower amount of transposase. In testing the Quick protocol, we observed two different read length distributions (see Supplemental Fig. S1B,C). While both had N50s of ∼90 kb, one was dominated by shorter reads. This suggests that the DNA was not cut uniformly. Moreover, although these protocols generated UL reads, they suffered from high variability in output N50, occupancy, and yield (Supplemental Fig. S1B,C). Typically, as N50 increases, yield and occupancy drop (Supplemental Table S1A,B).
Compilation of UL library preparation protocols, optimized in human GM12878 cells
| Previous protocols | New protocols | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Quick's UL, Quick 2018 | Logsdon's UL, Logsdon 2020 | Rocky Mountain Tyson 2019 | Krazy StarFish | ULK001 | RAD004-Nemo | ULK001-Nemo | ULK114 | ULK114-Nemo | |
| A | B | C | D | E | F | G | H | I | |
| Input cell numbera | 50 million | 20–70 million | >2 million | >2 million | 6 million | 1–6 million | 1–6 million | 6 million | 6 million |
| Input DNA amount | 15 µg | 2–3 µg | >7.5 µg | >7.5 µg | 40 µg | 5–40 µg | 5–40 µg | 40 µg | 40 µg |
| ONT library preparation kit | RAD004 | RAD004 | LSK109 | RAD004 | ULK001 | RAD004 | ULK001 | ULK114 | ULK114 |
| Fragmentation volume | Small (15 µL) | Small (18 µL) | Medium (60–100 µL) | Medium (2 × 100 µL) | Large (1 mL) | Large (0.5–1 mL) | Large (0.5–1 mL) | Large (1 mL) | Large (1–1.2 mL) |
| Library cleanup protocol | No | No | No | Partial using filter paper | Nanobind (circulomics) | Nemo | Nemo | No | Nemo |
| Processing time (excluding incubations) | 15 min | 2.5 h | 18–24 h | 30 min | ∼2.5 h | ∼2.5 h | ∼2.5 h | ∼2.5 h | ∼2.5 h |
| Input library per MinION flow cell | 10–15 µg | 2–3 µg | 1–3 µg | 2–4 µg | 5–6 µg | 1–5 µg | 1–5 µg | NA | NA |
| No. of loads per library (MinION) | 1 | 1 | 1–3 | 3 | 6 | 1–6 | 1–6 | NA | NA |
| Expected yield per MinION flow cell | ∼3 Gb | 1–2 Gb | 10–>20 Gb (depends on N50) | 6–8 Gb (3 loads) | >20 Gb (3 loads) | >15 Gb (3 loads) | ∼20 Gb (3 loads) | NA | NA |
| Input library per PromethION flow cell | NA | NA | NA | NA | 10–13 µg | 10–13 µg | 10–13 µg | 10–13 µg | 10–13 µg |
| No. of loads per library (PromethION) | NA | NA | NA | NA | 3 | 1–3 | 1–3 | 3 | 3–4 |
| Expected yield per PromethION flow cell (Gb) | NA | NA | NA | NA | NA | NA | 24 Gb (1 load) | ∼85 Gb | up to >100 Gb |
| UL read length (>100 kb) | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes |
[i] (NA) Not available.
[ii] aCell number was dependent on the extraction protocol used; here, it is listed as the number of cells required by the original protocol. If cell number is unknown, use the DNA amount.
The Rocky Mountain protocol (Table 1C; Tyson 2019) was developed to use the ligation kit (ONT, e.g., LSK109) to address the yield issues experienced with the rapid kit. This protocol utilized varying concentrations of salts and polyethylene glycol (PEG) to precipitate longer reads after the library preparation step. The ligation kit requires light shearing of the input DNA, resulting in non-UL N50s albeit with much improved yields (Tyson 2019). Therefore, this protocol was further modified to use the rapid kit followed by precipitation of the fragmented DNA using a filter paper disc or star as a form of SPRI matrix (Krazy StarFish [KSF]) (Table 1D). The KSF protocol worked consistently in producing UL N50s, but occupancies and yields were still suboptimal, perhaps a consequence of having DNA cleanup prior to sequencing adapter ligation, consequently producing many free adapters (Methods; Supplemental Fig. S1D,E; Supplemental Table S1C,D).
New and developing protocols
ONT released a rapid kit for UL sequencing, SQK-ULK001 (hereafter ULK001), in which the transposase reaction is performed in a large volume (Table 1E), keeping the DNA concentration at ∼50 ng/µL. We hypothesized that larger reaction volumes facilitated even diffusion and mixing of DNA and transposase complexes in a more homogeneous state in solution, a feature that we included in our Nemo protocols (Table 1F,G). Efficient transposase reactions result in consistent UL sequencing output as shown by the high rates of occupancy (Fig. 2; Supplemental Figs. S1F–H, S2; Supplemental Table S1). Occupancy as a parameter is specifically defined by the percentage of sequencing pores (i.e., pores in “adapter” and “strand” states) compared to all pores in available states (i.e., “adapter,” “strand,” and “pore” states).
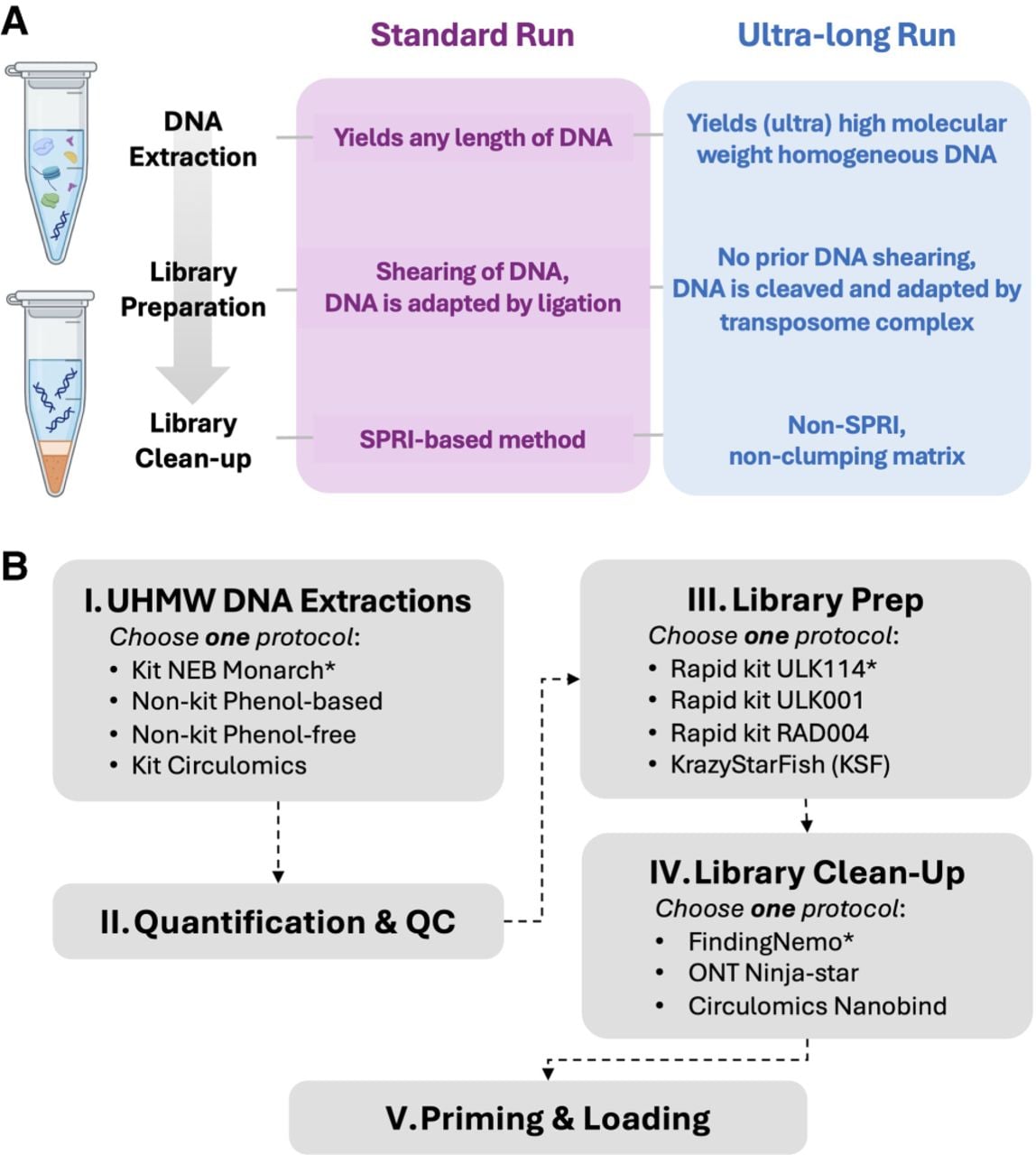
Validation of the FindingNemo protocol. Sequencing outputs between rapid kit-based library preparation protocols (ULK001-Nemo and RAD004-Nemo protocol) show comparable performance when compared to the control ULK001 protocol. (A) Workflow of the library preparation protocols, (B) total yields subcategorized by read lengths, (C) time lapse of yields, (D) read length distributions (dashed black vertical lines denote N50s), and (E) time lapse of relative pore occupancies. Each library was loaded three times (i.e., total DNA equivalent to 3 million GM12878 cells) on a MinION R.9.4.1 flow cell following a nuclease flush protocol. All data are after 65 h of sequencing on the GridION platform.
We next tested the performance of RAD004 and ULK001 rapid kit-based protocols, modifying parameters such as the number of input cells, the RAD004 dilution buffer, and the cleanup steps while maintaining the large volume ratio, and compared these to the control ULK001 protocol (Fig. 2A). Regardless of cleanup method (Nanobind disc or glass beads), ULK001-based protocol outputs were comparable in terms of N50s and occupancies (Fig. 2B–E). Meanwhile, the output of the library prepared with the RAD004 protocol showed slightly lower performance in general (Fig. 2B–E; Supplemental Table S1G).
The total yield of an UL sequencing is not a direct measure of the library quality as yield is also flow cell dependent. The control ULK001 library produced the highest total yield (Fig. 2B,C) likely because of the highest pore count of the flow cell (Supplemental Fig. S3A; Supplemental Table S1E). Normalizing yields between flow cells by taking a ratio of yield to the number of pores utilized during a period of data collection (i.e., yield per pore) allows comparison of run performance. When normalized, the yields per pore of the two ULK001 libraries were comparable (Supplemental Table S1E,F). In all three libraries, the distribution of read lengths against read quality was similar (Supplemental Fig. S3B), as well as sequencing speed (Supplemental Fig. S3C). These similarities imply that either of the rapid kit-based protocols could be used to successfully produce UL sequencing outputs. Considering all sequencing output parameters, the ULK001 was the optimal protocol for UL sequencing compared to the RAD004 (Supplemental Table S2).
The FindingNemo protocol efficiently purifies and accurately quantifies UHMW DNA and UL libraries
DNA precipitation and recovery: maintaining quality and homogeneity
Prior to the launch of the ULK001 kit, we developed a protocol to clean the library after adapter ligation using DNA precipitation chemistry compatible with the motor protein–DNA complex. The filter paper from the KSF protocol was replaced with glass beads, adapting the glass bead's effectiveness to act as a DNA-binding substrate as in the Monarch extraction kit protocol (NEB, T3050). We also surveyed compounds with well-studied DNA precipitating (Deng and Bloomfield 1999; Ouameur and Tajmir-Riahi 2004) and selected spermine and CoHex for testing DNA precipitation and recovery profiles using glass beads (Supplemental Fig. S4A). Optimum final concentrations of the compounds were tested empirically based on previous studies (Pelta et al. 1996).
The ULK001 protocol is carried out in a dilute condition of ∼50 ng/µL UHMW DNA. We tested the impact of DNA concentration (10, 20, and 50 ng/µL) on precipitation and recovery rate. CoHex is the best precipitant for both recovery and homogenization of UHMW DNA at a concentration range of 20 ng/µL to <50 ng/µL (Supplemental Fig. S4B–D). The optimal DNA recovery of ∼75% was obtained using CoHex at a DNA concentration of 20 ng/µL. Spermine shows a significantly higher recovery rate at 50 ng/µL DNA but show greater heterogeneity and sample-to-sample variation (Supplemental Fig. S4B–D).
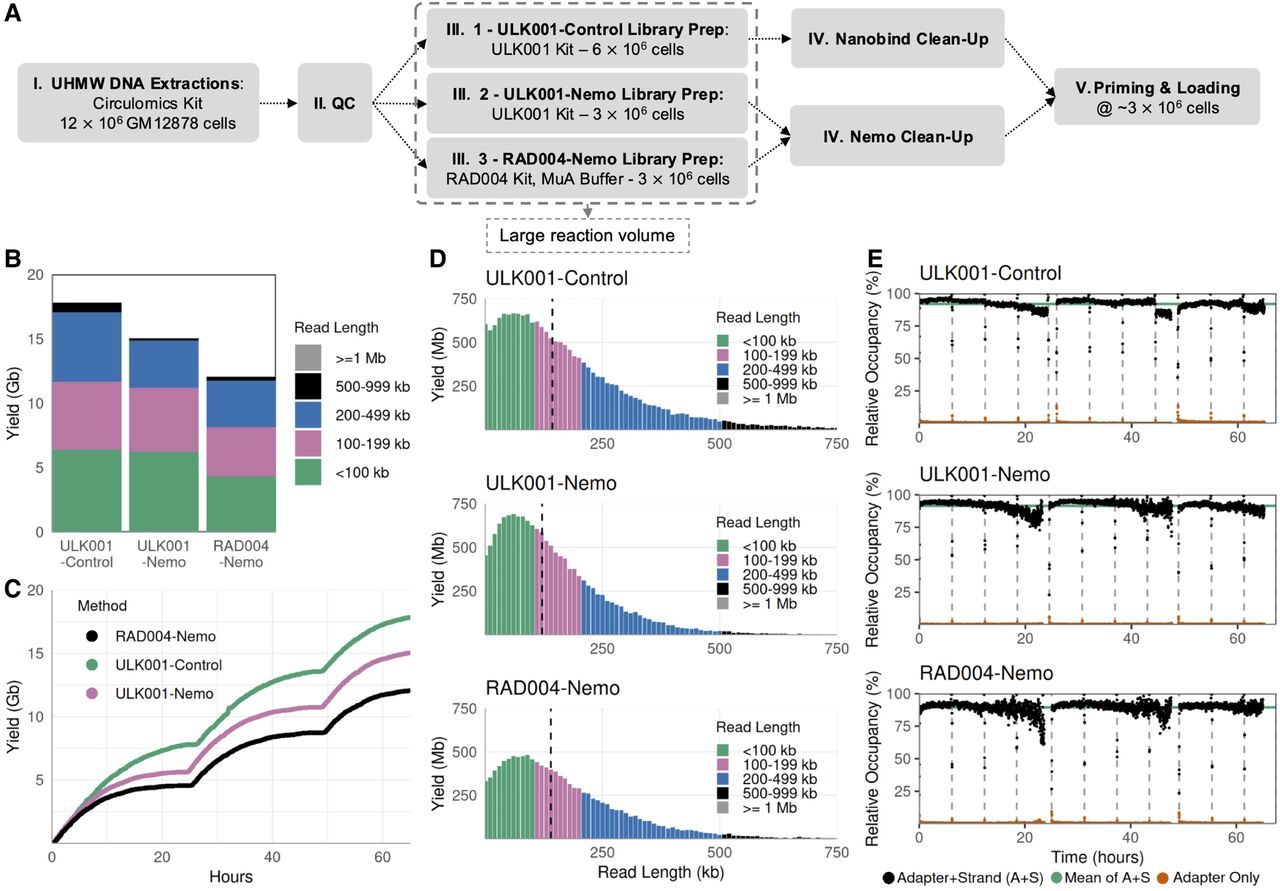
The early ULK001 kit version used a spermine-based precipitation buffer (PPT buffer). We tested this alongside our FindingNemo and spermine cleanup protocols, using a DNA input of 40 ng/µL, and split the adapted library into three cleanup preparations. These runs yielded similar UL sequencing metrics (Fig. 3A). Albeit with a slightly longer N50, ONT's PPT protocol showed the lowest yield per pore, presumably from the lack of any washing step after the library precipitation that might cause the library to have a higher rate of blocking than the others (Fig. 3B).
Choosing the best precipitating agent that ensures fast homogenization. (A) Read length distribution of libraries precipitated with different buffers; dashed black vertical lines denote N50s. (B) Sequencing metrics after 6 h of run (excluding the first 10 min). All libraries were extracted from GM12878 cells using Monarch direct lysis protocol with an input DNA concentration of ∼40 ng/µL split into three cleanup preparations, loaded on MinION R.9.4.1 flow cells and sequenced on GridION platform.
Homogeneity is measured by calculating the coefficient of variation percentage (%CV) of the DNA concentration. We take at least three concentration measurements from the top, middle, and bottom parts of the solution using a NanoDrop spectrophotometer device. The smaller the %CV, the more homogeneous and vice versa. Recovered DNA became more homogeneous after being in solution for a longer period, except when precipitated using CoHex at 10 ng/µL DNA concentration (Supplemental Fig. S4E). Previous studies showed that a charge neutralization of 88%–90% is required for DNA condensation (Deng and Bloomfield 1999). We hypothesize that at 10 ng/µL DNA, excess CoHex cations affected the water hydration at the DNA–cations interface (Deng and Bloomfield 1999) resulting in a more condensed, difficult-to-dissolve DNA as observed during pipetting. Moreover, NanoDrop is more sensitive when measuring absorbance of the differentially condensed DNA in the solution, which explains the larger variation in DNA concentration compared to Qubit measurements (Supplemental Fig. S4C,D). In contrast, spermine-precipitated DNA shows the least homogeneity in solution (Supplemental Fig. S4E). Spermine is also less stable than CoHex; as a solution, it is readily oxidized at room temperature (Sigma-Aldrich). CoHex was chosen to precipitate DNA at a concentration range of 20–40 ng/µL UHMW DNA.
The importance of DNA homogeneity and optimum concentration during UL library preparation was shown in the use of a concentrated and nonhomogeneous UHMW DNA in sequencing, i.e., more than 60 ng/µL DNA in reaction (Supplemental Fig. S5). The sequencing output of this library typifies the output of an undercut library where N50 reaches above 100 kb, but accumulated yield and occupancy are low, as well as a rapid decrease of sequencing pore number (Supplemental Fig. S5D). In essence, ensuring the homogeneity of extracted UHMW DNA and setting the reaction concentration below 50 ng/µL provide the optimum conditions for UL sequencing. Longer elution of the library (e.g., overnight) at room temperature with gentle rotation may also help untangle long DNA molecules, facilitating further homogenization and eventually improving UL sequencing output.
Quantification of UHMW DNA
One of the complications in UL sequencing is in accurately quantitating the UHMW DNA. There is often large variation between the two most used quantification methods: the spectrophotometric- and fluorometric-based methods, represented by NanoDrop and Qubit 3, respectively (Supplemental Fig. S4F). The viscous nature of UHMW DNA also makes it difficult to withdraw a homogeneous small volume to be measured, even when using a positive displacement pipette. Therefore, we followed a novel Qubit concentration measurement of UHMW DNA to complement the standard NanoDrop method. This method uses Jurkat genomic DNA as the baseline standard for UHMW DNA and a glass bead to homogenize samples before each measurement (‘Giron’ Koetsier and Cantor 2021). We modified it by combining small volumes from three to four different parts of the DNA solution to average out concentration differences within the solution (Cahyani et al. 2021). An accurate concentration measurement requires an ideal ratio of NanoDrop to Qubit DNA values between 1 and 1.5 (Simbolo et al. 2013).
As expected, sheared DNA homogenized faster and better than UHMW DNA as shown by the smaller %CV of the sample a few hours after elution (Supplemental Fig. S6A). This fits with the fact that shorter DNA molecules will diffuse faster than longer ones. Longer DNA molecules may also have more complex structure and behavior in solution (Robertson and Smith 2007). The %CV of both types of samples were all below 50% indicating that the FindingNemo method could homogenize DNA solution relatively quickly (Supplemental Fig. S6A). It also filtered out contaminants in the input DNA as shown by the increase of the NanoDrop absorbance values after purification (Supplemental Fig. S6B; Koetsier and Cantor 2019). Last, the FindingNemo protocol did not interfere with the DNA quality as shown by their size distribution on the pulsed-field electrophoresis gel before and after DNA cleanup (Supplemental Fig. S6C).
The FindingNemo protocol versus undercut UL libraries
Combining the FindingNemo approach with either the Quick's or KSF protocols improved sequencing yield but did not significantly improve occupancy or N50 (Supplemental Table S3). We hypothesize that the transposase reaction upstream of the library cleanup is a more critical step in producing optimal UL sequencing outputs. Suboptimal transposase fragmentation may result in long DNA molecules without adapters in the library, because of an insufficient number of transposome complexes compared to DNA molecules, a situation we refer to as an “undercut.”
An undercut library may also occur if the transposome complex-to-DNA ratio is balanced, but the DNA solution is not homogeneous. Viscous, nonhomogeneous UHMW DNA is difficult to dilute and can have varying DNA densities at different points in the solution. Consequently, the cutting frequency of the transposome complex will be higher where DNA is more accessible and will be lower where long DNA molecules remain in a compacted, less-accessible mass. This scenario counterintuitively results in a library with a short N50 despite the HMW nature of the input DNA (Supplemental Fig. S8—0 rpm sample).
It is also important to consider the interaction between UHMW DNA molecules and the sequencing pores, especially just before a mux scan occurs. During this period, only pores with actively threading (sequencing) DNA molecules will continue sequencing; pores without DNA are inactivated. This process terminates at a fixed time, which is 10 min by default. After this period, all pores are inactivated. Molecules still traversing a pore at that point will be ejected, but UHMW DNA increases the risk of stalling or blocking nanopores. At a sequencing speed of 400 bases per sec, any molecule longer than 240 kb is at risk of blocking a pore and negatively impacting yield. Therefore, in the FindingNemo protocol, we modified this script parameter (wind_down) from 600 to 1800 sec (Cahyani et al. 2024).
The FindingNemo protocol in UHMW DNA extraction
Extractions without kits
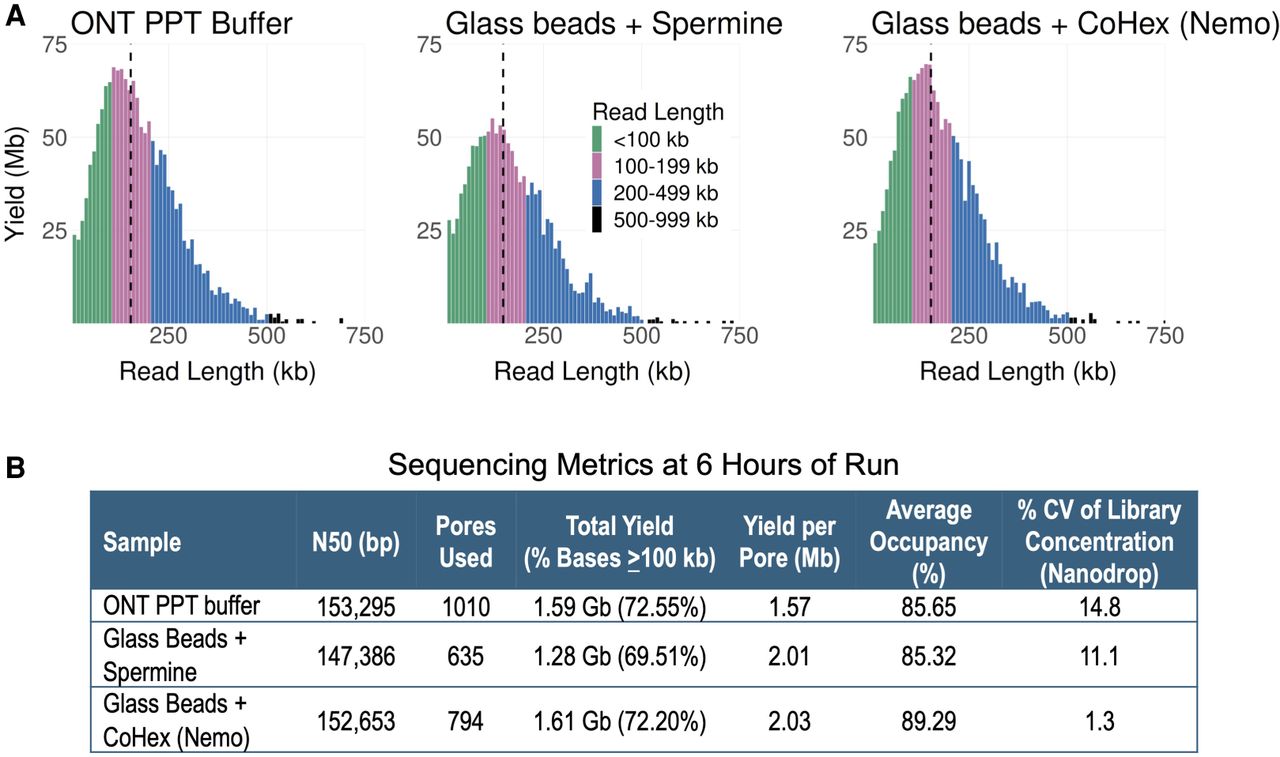
We next compared the effect of DNA extraction methods on UL sequencing. The original approaches by Quick and Logsdon et al. utilized phenol–chloroform isolation and alcohol-based precipitation with some differences (Table 2A,B). We succeeded in scaling down these protocols in combination with glass beads to use less cell input and shorten hands-on time to at least half (Table 2C). To reduce the toxicity from phenol use, we also developed phenol-free glass beads–based extraction protocols utilizing either SDS, CTAB, or CTAB with CoHex as the lysis buffer (Table 2D,E). These extraction protocols produced (U)HMW DNA, characterized by the distinctly narrow vertical lines/smears and DNA in the wells of the pulsed-field gel electrophoresis results (Supplemental Fig. S7).
Compilation of UHMW DNA extraction methods for UL sequencing, optimized in human GM12878 cells
| Without kits (phenol-based) | Without kits (phenol-free) | Kit-based | |||||
|---|---|---|---|---|---|---|---|
| Quick's phenol protocol Quick 2018 | Logsdon's phenol protocol Logsdon et al. 2021 | Phenol + glass beads | CTAB or SDS Lysis + glass beads | CTAB–CoHex Lysis + glass beads | NEB Monarch Cell & Blood Kit | Circulomics Nanobind CBB Big DNA Kit | |
| A | B | C | D | E | F | G | |
| Standard cell input | 50 million | 20–70 million | 1–5 million | 1–5 million | 1–5 million | 0.5–5 million | 6 million |
| Phenol–chloroform | Yes | Yes | Yes | None | None | None | None |
| Lysis buffer | SDS | SDS | SDS | CTAB or SDS | CTAB and CoHex | Proprietary | Proprietary |
| SPRI substrate | None | None | 3 mm glass beads | 3 mm glass beads | 3 mm glass beads | 4 mm glass beads | Nanobind disc |
| Precipitation reagents | Ethanol, ammonium acetate | Ethanol, ammonium acetate | Ethanol, ammonium acetate | Isopropanol, ammonium acetate | CoHex | Isopropanol, proprietary binding buffer | Isopropanol, proprietary binding buffer |
| DNA extraction time | 4 h | 4 h + overnight | 2 h | 1 h | 1 h | ∼30 min | 2 h |
| DNA resuspension time | 2 days | 2 days | min. overnight | min. 2 h | min. overnight | min. 1 h (standard overnight) | overnight |
We also successfully produced UL runs using these DNA samples with varying degrees of occupancies and yields (Fig. 4A–J). Only the extraction protocol using a combination of CTAB and CoHex in the lysis buffer could not produce UL N50 (Fig. 4F). The inclusion of CoHex during lysis was to test whether stable DNA toroid aggregates could be induced (Deng and Bloomfield 1999; Ouameur and Tajmir-Riahi 2004), so that DNA length could be kept intact during extraction and eventually improve sequencing output. The CoHex precipitated DNA was then washed with ethanol, and it rendered DNA elution from the beads more difficult. Ethanol is required in the wash buffer as it rinses salt, contaminants, and other impurities from the cell extract while simultaneously affecting DNA condensation (Arscott et al. 1995; Oda et al. 2016). More optimization is needed to obtain a consistent rate of elution and recovery in the CTAB–CoHex lysis protocol. Nevertheless, the option of using one of two oppositely charged surfactants as lysis agents, i.e., cationic CTAB (Arseneau et al. 2017) and anionic SDS (Xia et al. 2019), provides flexibility in extracting UHMW DNA from diverse sample types and context.
Combinatorial effects of extraction and library cleanup methods, either with or without the use of kits, on the UL sequencing outputs. (A–I) Read length distributions of the libraries: Graph titles denote the used extraction kit/method and the cleanup protocol; dashed black vertical lines denote N50s. (J) Sequencing metrics after 6 h of run (excluding the first 10 min). DNA was extracted from GM12878 cells. Each library was loaded on a MinION R9.4.1 flow cell and run on the GridION platform.
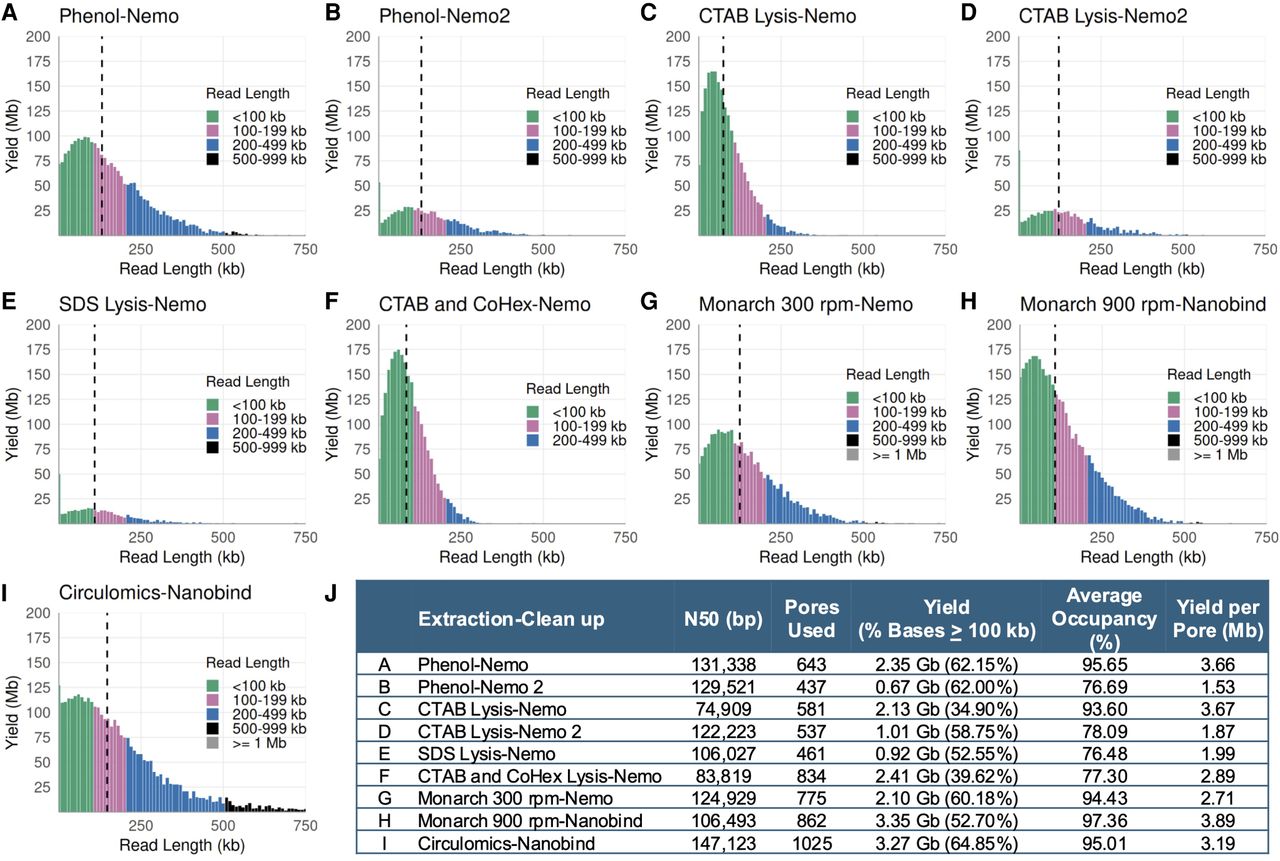
Extractions using kits
We tested a range of (U)HMW DNA extraction kits during our method development and found that Nanobind CBB Big DNA (Circulomics) and Monarch (NEB) were best suited for our purposes (Table 2F–G). The Nanobind CBB Big DNA Circulomics kit could be used to extract HMW or UHMW DNA, depending on the chosen protocol. DNA extracted with the UHMW protocol showed higher size distribution on a pulsed-field electrophoresis gel compared to the HMW protocol (Supplemental Fig. S7A vs. S7B). Libraries prepared from UHMW DNA samples extracted using the Circulomics kit produced UL reads and good sequencing metrics (Fig. 4I,J).
The Monarch kit protocol initially involved a two-step DNA extraction: first extracting nuclei from cells and then isolating DNA from the nuclei (Nuclei Prep). It was later refined to a “Direct Lysis” method, which bypassed the “Nuclei Prep” step by lysing cells and nuclei simultaneously to release DNA. This method yielded high-quality UHMW DNA from GM12878 cells, as shown in pulsed-field gel samples (Supplemental Fig. S7A,B). Libraries prepared from these DNA samples indicated that the Direct Lysis approach performed slightly better than the Nuclei Prep method (Supplemental Fig. S8).
We also found that the shaking speed during lysis in the Monarch protocol influenced DNA homogeneity, which affected sequencing N50 and occupancy (Fig. 4G–H; Supplemental Fig. S8; Supplemental Table S4). Lower lysis speeds yielded longer DNA fragments by keeping DNA more intact. However, no shaking during lysis could result in a nonhomogeneous UHMW DNA solution, negatively affecting the transposase reaction during library preparation (Supplemental Fig. S8—0 rpm). This resulted in a library with non-UL read N50, the lowest occupancy, and yield compared to other libraries (Supplemental Fig. S8). The best UL output was achieved by preparing libraries from DNA lysed at a medium speed of 600–900 rpm (Fig. 4G–H; Supplemental Fig. S8; Supplemental Table S4).
Sequencing parameters and performance of the FindingNemo protocol
Maximum yield is obtained at a read N50 between 90 and 110 kb
On a MinION, yields in excess of 3 Mb per pore after 6 h of sequencing could be obtained at 93% occupancy, reinforcing that high occupancy is essential for high yield (Fig. 4J). However, UL libraries with many short fragments had reduced occupancy as short reads pass quickly through the pores, lowering total yields (Fig. 4B,D,E,J). This underscores the importance of homogeneously cut UHMW DNA in UL library preparation. Additionally, targeting an N50 between 90 and 110 kb is ideal for optimal UL sequencing output, as N50 is inversely proportional to yield (Fig. 4J; Supplemental Table S4)
Scalable library loading amount with the FindingNemo protocol
We also tested the impact of the amount of input DNA and library loaded per flow cell. When the input amount of UHMW DNA was <5 µg, sequencing output parameters were markedly reduced (Supplemental Table S4, Monarch-01 vs. Monarch-05). To produce the optimum N50, occupancy, and yield, DNA equivalent to at least 1 million GM12878 cells (or at least 5 µg) should be used for library preparation, and a minimum of 2 µg of this library was loaded onto a MinION flow cell. At least 2 million cells and 4 µg of library should be used on a PromethION flow cell (Supplemental Table S4, Monarch-01 vs. Monarch-02).
FindingNemo in 1 day: from cells to UL sequencing in 1 working day
The relatively fast homogenization rate of the FindingNemo cleanup method, combined with the fast DNA extraction when using the Monarch kit, can support the preparation of UL libraries from fresh or frozen cells to sequencing in just a working day (Supplemental Fig. S9A; Cahyani 2021). We can obtain UL N50s using this protocol; however, yields are markedly reduced (Supplemental Fig. S9B–D), likely as a result of the heterogeneity of DNA samples. Yield reductions were exacerbated by the low occupancy level and the abundance of short reads and would be interpreted as undercut libraries (Supplemental Fig. S9B–D, Runs 2 and 3). Moreover, the FindingNemo cleanup protocol might not significantly remove these highly abundant short reads because of their high concentration in the libraries.
Using the FindingNemo protocol in diverse sample types, chemistries, and platforms
The FindingNemo protocol improved N50, occupancy, and yield with ULK114 kit
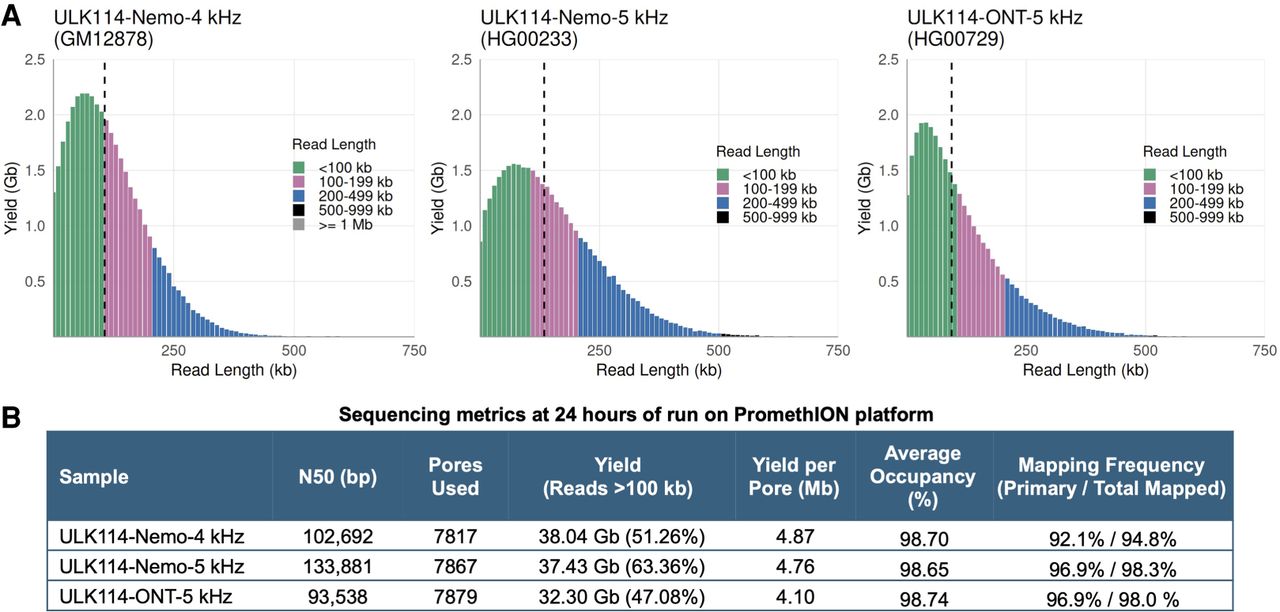
In 2022, ONT released a new pore and chemistry running on the R10.4.1 flow cells and a new UL kit, ULK114. This included new sequencing scripts and a sample rate shift from 4 to 5 kHz. We tested the ULK114 kit according to the manufacturer's instructions and with the FindingNemo protocol on three human cell lines (Fig. 5). Libraries prepared with the FindingNemo protocol showed longer N50s, optimal yield, and occupancy (Fig. 5A,B). This protocol consistently produced higher yields, regardless of kit chemistry and sequencing programs. Additionally, the 5 kHz script increased the overall mapping rate of reads compared to the 4 kHz program (Fig. 5B).
UL N50s were obtained using the new ULK114 kit from three cell lines. (A) Read length distribution of GM12878 library run with the earlier ULK114 script version (left panel), while the other two libraries (middle and right panels) were run with the most current 5 kHz version of the script. Vertical dashed black lines indicate N50s. (B) Run metrics of the three libraries showed that the Nemo protocol performed better than ONT's.
Ratio of transposase to DNA
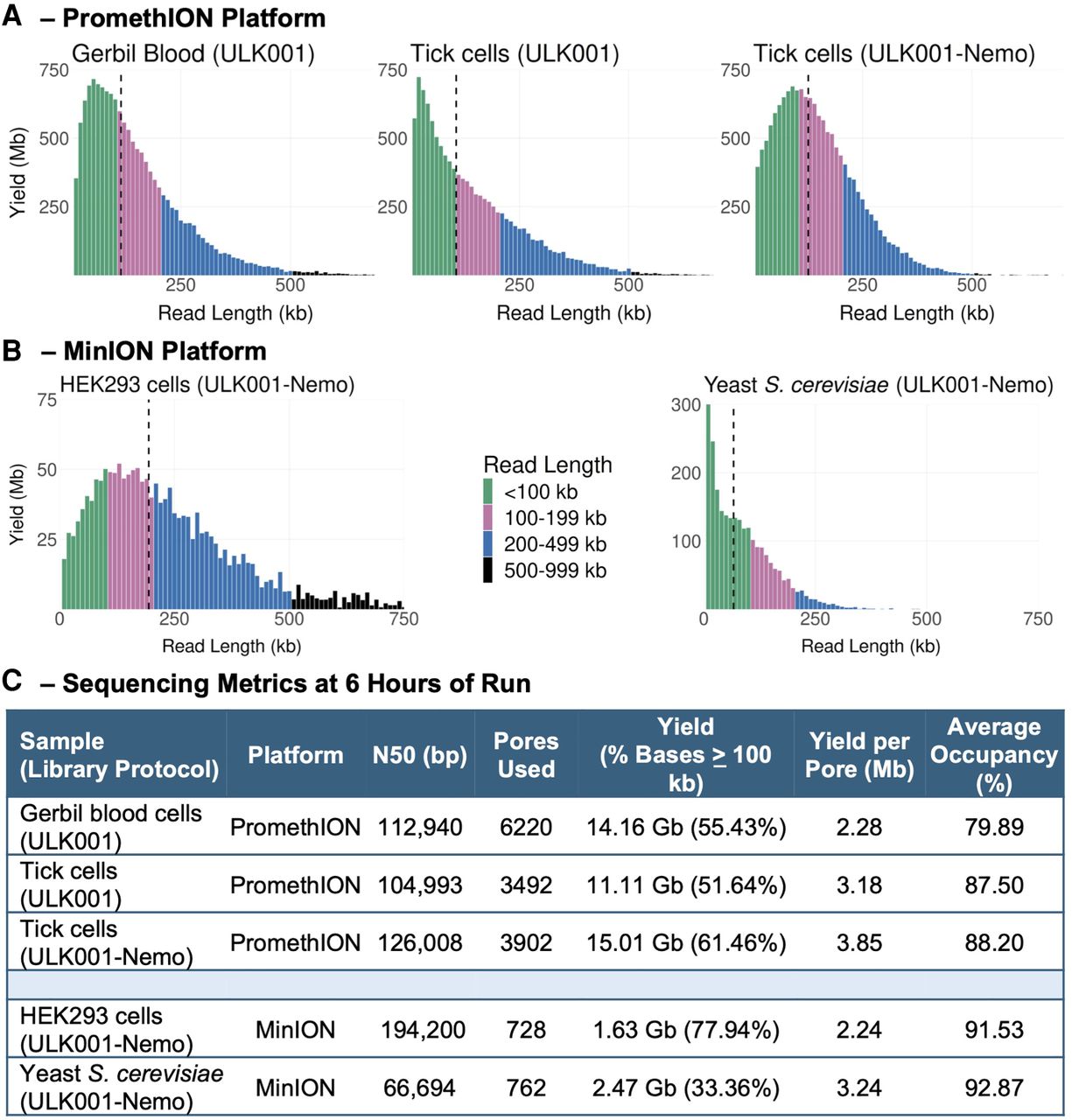
The ratio of transposase to DNA is crucial for optimizing UL sequencing. The ULK001 protocol specifies 1 µL of transposase fragmentation mix (FRA) per 1 million human cells, corresponding to 5–6 µg of UHMW DNA for a 3.2 Gb genome (Table 3). We used this ratio for all GM12878 (Figs. 2Figure 3.Figure 4.–5) and HEK293 (Fig. 6) runs. Additionally, we tested different FRA to DNA ratios in Mongolian gerbil, tick Amblyomma variegatum, and yeast Saccharomyces cerevisiae (Table 3). These ratios produced UL sequencing outputs for the gerbil and tick samples (Fig. 6A,C). Although the yeast run did not yield UL read N50s (Fig. 6B,C), it showed a significant increase in read length compared to existing nanopore-sequenced data (Giordano et al. 2017; Salazar et al. 2017).
UL sequencing output and metrics of non-GM12878 cells and different sequencing platforms: gerbil blood, tick A. variegatum, yeast S. cerevisiae, and human HEK293 cells using either the ULK001 or the ULK001-Nemo protocol (as labeled). (A) Read length distributions of libraries run on PromethION R9.4.1 flow cells; dashed black vertical lines denote N50s. (B) Read length distribution of the HEK293 and yeast S. cerevisiae libraries run on MinION R9.4.1 flow cells. (C) Sequencing metrics of the libraries. Each metric was shown after 6 h of run (excluding the first 10 min).
Ratio of FRA to DNA in different genomes
| Species | Genome size | Cell input number | DNA equivalent (μg) | FRA volume (μL) |
|---|---|---|---|---|
| Human GM12878 | 3.2 Gb | 1 × 106 | 5–6 | 1 |
| Human HEK293 | 3.2 Gb | 1 × 106 | 6–8a | 1 |
| Mongolian Gerbil | ∼2.7 Gb | Unknown | 10 | 1.5 |
| Tick A. variegatum | ∼6.0 Gb | 1 × 106 | 11–13 | 2.5 |
| Yeast S. cerevisiae (n) | 12 Mb | 2 × 108 | 4–5 | 6 |
[i] aExtracted DNA yield is likely to be more than normal diploid human cell line because of its hypotriploid nature (Lin 2014).
Genome ploidy
Genome ploidy anomalies must be considered when preparing UL libraries. For instance, about 4.2% of HEK293 cells are hypotriploid (Synthego HEK293). This means the DNA mass extracted from the same number of cells is higher than from the GM12878 cell line, altering the FRA to DNA ratio (Table 3) and potentially affecting sequencing output by increasing read length and decreasing yield. Sequencing HEK293 DNA with the Monarch kit confirmed this, producing N50s over 190 kb but with lower yields (Fig. 6B,C). To maximize yield and N50, it is better to use the FRA volume to DNA mass ratio rather than the absolute cell number when genome ploidy information is available.
Sample types: to count or to weigh?
The UL protocol requires the same number of human cells per microliter of FRA, whether from a cell line or nucleated blood. However, counting cells in blood samples can be impractical, as seen in the gerbil UL run (Table 3). In such cases, the FRA to DNA mass ratio can be used, provided the DNA is extracted to maintain native chromosome length and properly quantified (Methods). Using this ratio, UL libraries were produced from gerbil blood following the original ULK001 protocol (Fig. 6B,C).
The FindingNemo protocol works on different nanopore platforms
We ran pairwise sequencing of identical libraries on both the MinION and PromethION platforms, showing that PromethION produced more yield per pore (Supplemental Table S5). With nearly six times as many pores as a MinION, PromethION is expected to generate more data. The best PromethION results had an N50 of 126 kb and a yield of 3.85 Mb per pore at 88% occupancy (Fig. 6C—tick cells ULK001-Nemo). On the MinION platform, the best results ranged from 106 to 147 kb N50, with yields of 3.19–3.89 Mb per pore and at least 95% occupancy (Fig. 4J). These metrics were all obtained after 6 h of sequencing.
Relative performance of extraction kits in tick samples
We compared two extraction kits, Circulomics and Monarch, for UL sequencing of a tick cell line with a ∼6 Gb genome (Fig. 6A,C). Although flow cell occupancies were similar, the Monarch-extracted DNA had longer N50s, higher yields, and a greater proportion of UL reads (Fig. 6A right panel, C). The Circulomics library had more short reads, likely because of undercutting (Fig. 6C, middle panel). The Circulomics-extracted DNA concentration was ∼50 ng/µL, while Monarch-extracted DNA concentration was ∼26 ng/µL, reflecting differences in sample homogeneity. Moreover, Monarch extractions yielded DNA with a longer size distribution, as shown by pulsed-field electrophoresis gel (Supplemental Fig. S10). The Monarch protocol is also more flexible, allowing modifications to target different read length distributions by adjusting lysis speed (Methods).
Discussion
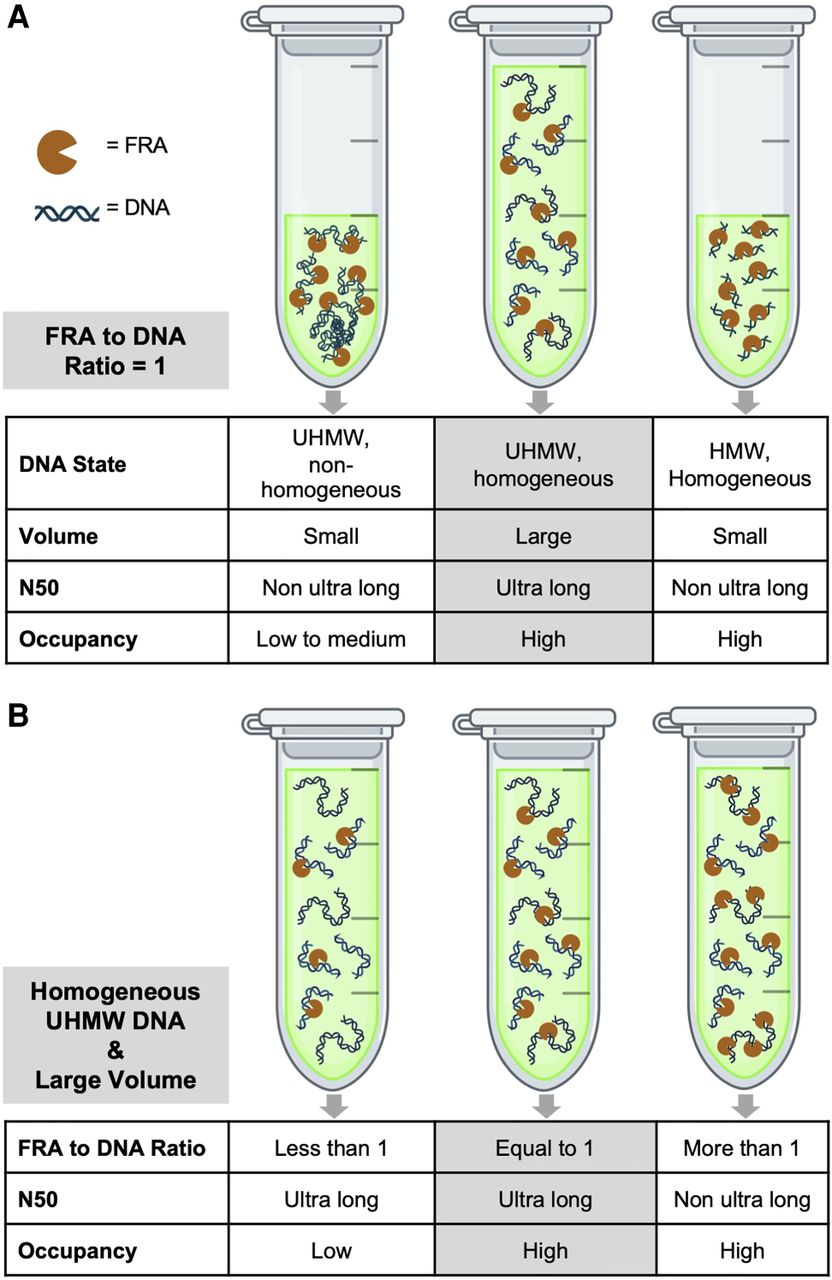
High-quality, uniformly long DNA is essential for UL reads on Oxford Nanopore sequencers. Our results support the hypothesis that to obtain this length uniformity, transposase reactions should occur in a dilute and homogeneous DNA solution. This can be achieved in a large reaction volume and by thorough mixing to obtain a “properly cut” library in contrast to “undercut” as explained before or “overcut,” which is a higher ratio of FRA to DNA molecules (Fig. 7A,B). Additionally, nonadapted DNA molecules may “crowd” the space around the pores, thereby decreasing the diffusion rate of the adapted DNA molecules to be tethered to the pores. The adapted long DNA molecules themselves may inactivate or block the pores during sequencing because of their lengths.
Optimal conditions for ultra-long sequencing. To achieve optimal ultra-long (UL) sequencing output, the FRA-to-DNA molecule ratio should be 1:1 in a homogeneous solution, verified through accurate cell counting and/or DNA mass measurement. (A) Hypothetical cutting frequency under three conditions of reaction volume and DNA length when the FRA-to-DNA ratio is 1:1. (B) Hypothetical cutting frequency under three conditions of FRA-to-DNA ratio and DNA length when the reaction volume is large. This figure was created with BioRender (https://www.biorender.com).
With these challenges, control of DNA quality and quantity is of primary importance. Proper quantification of the number of input cells or tissue weight has direct consequence to controlling DNA yield and to some extent, length distribution. Extended elution and homogenization are also beneficial; the longer that an UHMW DNA sample is allowed to equilibrate, the more homogeneous it becomes. This reduces the benefit of our FindingNemo in 1 day protocol where both maximal read lengths and yields are important. However, as we show, a rapid UL library preparation is possible, albeit with lower total yield. Further optimization is required, especially at the homogenization of DNA postextraction, to make this protocol more robust.
Chromosome length distribution of a genome may limit the maximum read N50 that can be obtained, as was the case with the yeast S. cerevisiae UL library. Prior knowledge of the genome size and chromosome lengths combined with empirical tests may be required to find the optimum ratio of transposase to DNA amount and produce maximum sequencing N50 and yield. This is a challenging part of UL sequencing as it needs fine tuning and adjustments depending on the sample types used.
We anticipate that the FindingNemo protocol will be generally applicable to tissue samples, could be readily multiplexed, and is likely compatible with automation. Finally, open-source development and sharing of protocols is crucial to enable the widest access to cost-effective high-throughput UL sequencing and troubleshooting. We established the LongRead Club on protocols.io to house these protocols and promote reproducibility (https://www.protocols.io/workspaces/long-read-club/about). The FindingNemo protocol described here is a valuable addition to the repertoire of UL nanopore sequencing methods available.
Methods
Genomic DNA extraction from cells
Cell sources
GM12878 cells were grown by Darren Crowley (University of Nottingham) and additionally purchased from Coriell Institute for Medical Research, Camden, NJ, USA. Yeast S. cerevisiae cells were given by Stephen Gray (University of Nottingham). Tick A. variegatum (AVL/CTVM17) cells were obtained from Alistair Darby's group (University of Liverpool). The HG00233 and HG04054 cells were obtained from Danny Miller's laboratory (University of Washington, Seattle, WA, USA). Last, HEK293 cells were obtained from New England Biolabs (NEB) in Ipswich, MA, USA.
Kit-based extraction
Kits used in the extraction of UHMW DNA were Monarch HMW DNA Extraction Kit for Cells & Blood (NEB T3050) and Nanobind CBB Big DNA Kit (Circulomics SKU NB-900-001-01) plus the auxiliary Nanobind UL Library Prep Kit (Circulomics SKU NB-900-601-01) according to the manufacturer's protocols with modifications as previously described for the Monarch protocol (Cahyani et al. 2021). The Monarch nuclei prep approach followed the original protocol (NEB T3050), while the direct lysis approach combined the prep and lysis buffers into one step (NEB Direct Lysis). Lysis speed at 600 up to 900 rpm resulted in an optimum UL sequencing output and a minimum of overnight incubation of DNA in solution helped homogenization.
Phenol–chloroform extraction with glass beads
This is a scaled-down version of Quick's original protocol (Quick 2018), and the modifications are as previously described (Cahyani et al. 2021). In summary, pellet of 5 million cells was washed with phosphate-buffered saline (PBS) (Fisher Scientific 15453819) and lysed with an SDS buffer (0.5% SDS, 100 mM NaCl, 25 mM EDTA pH 8.0, and 10 mM Tris-HCl pH 8.0) in the presence of RNase A (Qiagen 19101, 20 µg/mL) at 37°C for 5 min. Proteinase K (Qiagen 19131, 200 µg/mL) was added and incubated at 56°C for 15 min. Cell lysate was split into two 5PRIME phase-lock gels (Quanta-Bio 2302820). BioUltra TE-saturated phenol (Merck 77607) was added at 1:1 volume ratio. Sample was homogenized by vertical rotation at 20–30 rpm for 10 min and then centrifuged at 4000g for 10 min. The aqueous phase was transferred into new phase-lock gel tubes. DNA purification was repeated with a second round of phenol:chloroform:isoamyl alcohol = 25:24:1 volume ratio. In the third round, only chloroform:isoamyl alcohol = 24:1 was used. After the last centrifugation, the aqueous phase from the two tubes was combined in a 5 or 15 mL tube and added with 5 M ammonium acetate (Sigma-Aldrich A-7330) (0.4 volume), three glass beads (3 mm diameter), and absolute ethanol (2.5 volume). The tube was inverted by hand 20–30 times to precipitate DNA onto the glass beads (or placed in a tube rotator at 10 rpm for 3 min). The supernatant was removed, and bound DNA was washed twice with 60%–70% ethanol. Beads with DNA were poured into a bead retainer and quickly spun to remove excess ethanol (or absorbed with a filter paper or tissue). DNA was quickly eluted from beads by pouring them into a 2 mL LoBind (Eppendorf 0030108051) tube containing 10 mM Tris-HCl pH 9.0 and incubating at 37°C for 30 min with regular wide-bore pipette mixing. Incubation was continued overnight at room temperature. Afterward, using a bead retainer, DNA was spun down at maximum speed for 1 min.
Phenol-free, home-brew extractions
This protocol is adapted from the Monarch HMW DNA Extraction Kit for Cells & Blood as described previously (Cahyani et al. 2021). In summary, a pellet of 1–3 million cells was washed with PBS and lysed in either SDS or CTAB buffer with 200 µg Proteinase K and incubated at 56°C for 15 min (preferably with shaking at 600–700 rpm). The SDS buffer composition was the same as used in the phenol-based extraction described in the previous section. The CTAB buffer compositions were 2% CTAB, 1.4 M NaCl, 25 mM EDTA pH 8.0, and 10 mM Tris-HCl pH 8.0. After lysis, 100 µg RNase A was added and incubated at 37°C for 5 min. To precipitate DNA, 5 M ammonium acetate (0.4 volume), two to three glass beads (depending on cell number used), and isopropanol (0.9 volume) were added. Sample was mixed on a rotator at 9 rpm for 5 min (or inverted by hand 20–30 times). Liquid was removed by pipetting, and bound DNA was washed twice with 60%–70% ethanol. Beads with DNA were poured into a bead retainer and quickly spun to remove excess ethanol (or absorbed with a filter paper or tissue). DNA was quickly eluted with 10 mM Tris-HCl pH 8.0 in a 2 mL tube and incubated at 37°C for 30 min with regular wide-bore pipette mixing. Incubation was continued overnight at room temperature. Afterward, using a bead retainer, DNA was spun down at a maximum speed for 1 min.
CTAB–CoHex extraction
This protocol is similar to the home-brew CTAB method as described above with the following modifications. The CTAB–CoHex buffer compositions used were 2% CTAB, 20 mM CoHex, 1.4 M NaCl, 25 mM EDTA pH 8.0, and 10 mM Tris-HCl pH 8.0. To precipitate DNA, sample was diluted with 10 mM Tris-HCl pH 8.0, so CoHex concentration became ∼5 mM and NaCl was ∼400 mM, i.e., the ratio of CTAB–CoHex lysis buffer to Tris-HCl was 1:2.5. Then, —two to three glass beads (depending on cell number used) were added. Sample was mixed on a rotator at 9 rpm for 5 min (or inverted by hand 20–30 times). Liquid was removed by pipetting, and bound DNA was washed twice with an ethanol buffer (50% ethanol and 50% of 1 mM CoHex). Beads with DNA were poured into a bead retainer and quickly spun to remove excess ethanol (or absorbed with a filter paper or tissue). DNA was quickly eluted with 10 mM Tris-HCl pH 8.0 in a 2 mL tube and incubated at 37°C for 30 min with regular wide-bore pipette mixing. Incubation was continued overnight at room temperature. Afterward, using a bead retainer, DNA was spun down at a maximum speed for 1 min.
Yeast extraction using CTAB and glass beads
Yeast pellet (∼50 µL volume or equivalent to 200 million cells) was washed twice in cold PBS and resuspended in 480 µL SpheroBuffer (1 M sorbitol, 100 mM Na2HPO4, and 100 mM EDTA) (Cahyani et al. 2024). Lyticase enzyme (5 U/µL) was added (20 μL). Sample was incubated at 30°C for 30 min, and spheroplast formation was checked from time to time. Sample was lysed by adding 480 µL of 2× CTAB lysis buffer (4% CTAB, 2 M NaCl, 50 mM EDTA pH 8.0, and 20 mM Tris-HCl pH 9.0), mixed well with wide-bore pipet tip as the buffer is viscous, added with 400 µg Proteinase K (20 mg/mL), and incubated at 56°C for 15 min while shaking at 700 rpm. To remove RNA, 400 µg of RNase A (100 mg/mL) was added followed by incubation at 56°C for 10 min without shaking. Sample was cooled off at room temperature and added with 400 µL of 5 M ammonium acetate and 100 µL of 5 M NaCl (Thermo Fisher Scientific AM9759) and mixed by a vertical rotator for 1 min at 9 rpm. Sample was then centrifuged for 7 min at 16,000g, and the lysate supernatant was removed to a new 2 mL tube. Three glass beads and 0.5 mL isopropanol were added, and the sample was mixed on a vertical rotating mixer at 9 rpm for 5 min. Between 0.5 and 0.7 mL liquid was removed and replaced with the same amount of isopropanol and was rotated for an additional 3 min. When DNA had bound tightly around the beads, the liquid was discarded, and bound DNA was washed twice with 1 mL of 70% ethanol. Beads with DNA were poured into a bead retainer and quickly spun to remove excess ethanol (or absorbed with a filter paper or tissue). DNA was quickly eluted with elution buffer (EB) (ONT) or 10 mM Tris-HCl pH 9.0 in a 2 mL tube and incubated at 37°C for 30 min with regular wide-bore pipette mixing. Incubation was continued overnight at room temperature. Afterward, using a bead retainer, DNA was separated from the beads by centrifugation at a maximum speed for 1 min.
Quantification of UHMW DNA
Two nucleic acid quantification methods, fluorometric-based Qubit (Thermo Fisher Scientific, Waltham, MA, USA) and spectrophotometric-based NanoDrop (Thermo Fisher Scientific), were used in parallel to assess both the quantity and purity of the extracted DNA. The quantification follows the published protocol (‘Giron’ Koetsier and Cantor 2021) with some modification as previously described (Cahyani et al. 2021). In brief, DNA is sampled from four different points in the tube: top, upper-middle, lower-middle, and bottom part of the solution. Then, a glass bead was added, and sample was vortexed at full speed for 1 min for concentration measurements. For Qubit measurement, Jurkat genomic DNA (Thermo Fisher Scientific SD1111) was used as a concentration standard instead of the lambda DNA provided by the manufacturer (‘Giron’ Koetsier and Cantor 2021). To measure DNA homogeneity in solution, %CV of concentration was calculated by directly measuring concentrations of three to four different points of the solution using NanoDrop.
Quality check of UHMW DNA using PFGE
To assess DNA quality and read length distribution, 200–300 ng extracted DNA was run on 0.75% gold agarose gel (SeaKem 50150). Pulsed-field gel electrophoresis (PFGE) was carried out using Pippin Pulse device (Sage Science) at a preset mode of “5–430 kb” for 16 h. Gel was stained with RedSafe DNA stain (ChemBio 21141) and visualized with a Bio-Rad GelDoc imaging system.
Rapid kit-based UL library preparation
UL kit Nemo
The ONT UL kits, i.e., ULK001 and ULK114, are based on the cleaving and tagging of the DNA by the transposome complexes to insert transposase adapters, which then are ligated to the sequencing adapters (ONT ULK001). Our modifications of the ULK001 method were described in previously published protocol (Cahyani et al. 2021). To summarize, DNA extracted from 1 to 6 million cells was diluted to a final concentration of 20–40 ng/µL (in the final reaction volume). All mixing was done with wide-bore pipette tips. Meanwhile on ice, the FRA dilution buffer (FDB) was well mixed with FRA (1 µL per 1 million human cells DNA) and then combined to the DNA solution and quickly incubated at 23°C for 5–10 min (depending on the reaction volume). Enzyme was inactivated at 70°C for 5 min and cooled down to room temperature. Rapid adapter-F (RAP-F) was added (0.83 µL per 1 µL FRA used), and the sample was incubated at room temperature for at least 30 min.
The ULK114 modifications are similar to ULK001 and are described in the newly published and updated protocol also on protocols.io (Cahyani et al. 2024). In summary, library preparation to run on a PromethION flow cell needs extracted DNA from 6 million cells that was diluted to a final concentration of 20–40 ng/µL (in the final reaction volume). On ice, the dilution buffer (FDB) was well mixed with FRA (6 µL per 6 million human cells DNA) and then combined to the DNA solution and quickly incubated at 23°C for 10 min with 9 rpm rotation. Enzyme was inactivated at 75°C for 10 min and cooled down to room temperature. Sequencing adapter (RA) was added (5 µL per 6 µL FRA), and the sample was rotated at 9 rpm for 30 min.
RAD004-Nemo
The RAD004-Nemo protocol followed the same route as the ULK001-Nemo protocol described previously (Cahyani et al. 2021). However, the transposase and adapter used were the ones from the RAD004 kit (i.e., FRA and RAP, respectively). The dilution buffer used to replace FDB was the 4× MuA buffer (100 mM Tris-HCl pH 8.0, 40 mM MgCl2, 440 mM NaCl, 0.2% Triton X-100, and 40% glycerol).
Krazy StarFish
The KSF protocol is an alternative UL sequencing that uses a filter paper instead of glass beads. It is a cheap, quick-and-dirty method to produce UL sequencing based on the RAD004 kit. This method was described previously (Cahyani et al. 2021) and summarized as follows. An amount of ∼7.5 µg DNA in 75 µL volume was mixed well with 25 µL 4× MuA buffer. Meanwhile in another tube on ice, 25 µL 4× MuA buffer was diluted with 74 µL water and added with 1 µL FRA (or up to 1.2 µL FRA if DNA was quite viscous). These two solutions were mixed well on ice, aliquoted into two PCR tubes, and treated at 30°C for 1 min, 80°C for 1 min, and then cooled to room temperature. Reactions were pooled into a single tube, and 12 µL 5 M NaCl was added and mixed gently. A KSF filter (made from Whatman filter paper no. 3 with a star hole puncher) was submerged in the solution, and 142 µL isopropanol added and mixed by inversion of 20–30 times allowing UHMW DNA to collect and condense onto the filter. Liquid was removed, and filter paper was washed twice with 60%–70% ethanol. Excess ethanol was removed by pipetting, and paper was air-dried for 2 min. After the paper was moved to a clean tube, the library was eluted by adding 120 µL of elution buffer (10 mM Tris-HCl pH 8.0). Before sequencing, 37.5 µL library was added with 0.5 µL RAP and incubated for at least 30 min at room temperature.
Nemo cleanup: UHMW DNA library purification with glass beads and CoHex
The Nemo protocol is at the heart of this study and was previously described as an alcohol-free library cleanup method (Cahyani et al. 2021, 2024). Typically, for DNA equivalent to 1 million cells or more, three glass beads were used. In brief, glass beads were added to the library to be cleaned up in a 1:1 volume ratio of 10 mM CoHex (2×), i.e., final CoHex concentration is 5 mM. Sample was rotated at 9 rpm for 3 min or 20–25 times if using hand inversion. Liquid was removed, and beads were washed twice with polyethylene glycol wash (PEGW) buffer (10% PEG-8000 and 0.5 M NaCl) for 3 min each. Excess buffer was removed with a fine pipette tip after a quick spin of the tube. Alternatively, the beads with DNA could be poured into a bead retainer (home made or using the NEB-T3004), and the buffer was absorbed by tapping on a filter paper/tissue. Beads should not be let dry, were quickly eluted with 10 mM Tris-HCl pH 9.0 or EB (ONT), and were incubated at 37°C for 30 min with regular wide-bore pipette mixing. Incubation was continued overnight (preferable) at room temperature. Afterward, using a bead retainer on a new tube, DNA was spun down at a maximum speed for 1 min.
Flow cell priming and library loading
All flow cells were primed according to the ULK001 and ULK114 protocols. Library was loaded onto a primed FLO-MIN106 or FLO-PRO002 (R9.4.1), or FLO-MIN114 or FLO-PRO114 (R10.4.1) flow cells for sequencing on a GridION or PromethION, respectively, using the accompanying MinKNOW version software. Loaded UL library was let to tether on the flow cell for at least 30 min before run was started using the ONT UL script with mux scan of every 6 h for R9.4.1 and the default 1.5 h for R10.4.1.
FindingNemo in 1 day
To run UL sequencing within the same day as the DNA extraction, FindingNemo in 1 day protocol was developed as previously described (Cahyani 2021). In brief, cells were extracted using the Monarch kit and lysed at 700–800 rpm. Eluted DNA was incubated at 37°C and regularly mixed for at least 2 h. After quantification, we showed that DNA was sufficiently homogeneous (%CV < 100%), and the library was prepared following the ULK001-Nemo protocol. Eluted library was further incubated at 37°C for at least 30 min with regular pipette mixing, quantified, and loaded on a primed flow cell.
Nanoplot data analysis and plotting
Violin plots of sequencing speed and distribution graphs comparing read lengths and read quality were created using the summary file data processed by the NanoPlot tool (De Coster et al. 2018).
R data analyses and plotting
Read length histograms, occupancy, yield, and other sequencing metrics were analyzed from the summary, mux, and duty time (or in newer filename version, pore activity) output files using in-house R (R Core Team 2024) scripts available on GitHub (https://github.com/cinswasti/FindingNemo) and as Supplemental Code.
Sequence sources and alignment
For human work, the reference genome used is the GRCh38 (hg38) (NCBI RefSeq assembly GCF_000001405.40). Sequence data were aligned to this reference genome using minimap2 (Li 2021). Nanopore data of GM24385 and GM24631 are publicly available on Amazon Web Services (AWS) registry and can be accessed using the command line “aws s3 ls -–no-sign-request s3://ont-open-data.”
Data access
The FASTQ data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB76809. The FindingNemo protocol can be found as Supplemental Protocol and at the protocols.io website (https://dx.doi.org/10.17504/protocols.io.5jyl8p38rg2w/v1).
Competing interest statement
M.L., J.T, J.Q., and N.L. were members of the MinION early access program and have received free flow cells and sequencing reagents in the past. All have received reimbursement for travel and accommodation to speak at events organized by ONT.
Acknowledgments
We thank Giron Koetsier (NEB), Simon Mayes (ONT), and Kelvin Liu (Circulomics) for lending their expertise and/or advanced product trials. We thank Darren Crowley and Luke Simpson for supplying the GM12878 cells, Stephen Gray for the yeast cells, Danny Miller and Miranda Galey (University of Washington) for the Human Genome (HG) cells, Lesley Bell-Sakyi and Alistair Darby (University of Liverpool) for the tick A. variegatum cells, Giron Koetsier (NEB) for the HEK293 cells, and John Mulley (Bangor University) for the use of the gerbil's sequencing metrics. John Tyson is grateful to Terrance Snutch. This work was supported by the Wellcome Trust (grant number 204843/Z/16/Z, I.C., N.H., N.L., and M.L.).
Author contributions: N.L., M.L., J.T., and J.Q. conceived the project. I.C. led the work with contributions from J.Q., J.T., N.H., and C.M. I.C. drafted the manuscript with input from all the authors. All the authors reviewed, revised, and approved the final version of the manuscript.
Notes
[4] Supplementary material [Supplemental material is available for this article.]
[5] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279943.124.
[6] Freely available online through the Genome Research Open Access option.
References
- ↵Allers T, Lichten M. 2000. A method for preparing genomic DNA that restrains branch migration of Holliday junctions. Nucleic Acids Res 28: 6. 10.1093/nar/28.2.e6
- ↵Arscott PG, Ma C, Wenner JR, Bloomfield VA. 1995. DNA condensation by cobalt hexaammine(III) in alcohol–water mixtures: dielectric constant and other solvent effects. Biopolymers 36: 345–364. 10.1002/bip.360360309
- ↵Arseneau J-R, Steeves R, Laflamme M. 2017. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol Ecol Resour 17: 686–693. 10.1111/1755-0998.12616
- ↵Cahyani I. 2021. FindingNemo in OneDay: ultra-long ONT library preparation from cell to flowcell in one day. https://www.protocols.io/view/findingnemo-in-oneday-ultra-long-ont-library-prepa-14egnzqzyg5d/v1 [accessed November 4, 2024].
- ↵Cahyani I, Tyson J, Holmes N, Quick J, Loman N, Loose M. 2021. FindingNemo: a toolkit of CoHex- and glass bead-based protocols for ultra-long sequencing on ONT platforms. protocols.io. https://www.protocols.io/view/findingnemo-a-toolkit-of-cohex-and-glass-bead-base-dm6gpwkn1lzp/v1 [accessed March 11, 2024].
- ↵Cahyani I, Tyson J, Holmes N, Quick J, Loman N, Loose M. 2024. FindingNemo (v.kit14): a toolkit for DNA extraction, library preparation and purification for ultra long nanopore sequencing. protocols.io. https://www.protocols.io/view/findingnemo-v-kit14-a-toolkit-for-dna-extraction-l-5jyl8p38rg2w/v1/metadata [accessed April 17, 2024].
- ↵De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. Nanopack: visualizing and processing long-read sequencing data. Bioinformatics 34: 2666–2669. 10.1093/bioinformatics/bty149
- ↵Deng H, Bloomfield VA. 1999. Structural effects of cobalt-amine compounds on DNA condensation. Biophys J 77: 1556–1561. 10.1016/S0006-3495(99)77003-7
- ↵Giordano F, Aigrain L, Quail MA, Coupland P, Bonfield JK, Davies RM, Tischler G, Jackson DK, Keane TM, Li J, 2017. De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Sci Rep 7: 3935. 10.1038/s41598-017-03996-z
- ↵‘Giron’ Koetsier PA, Cantor EJ. 2021. A simple approach for effective shearing and reliable concentration measurement of ultra-high-molecular-weight DNA. BioTechniques 71: 439–444. 10.2144/btn-2021-0051
- ↵Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, 2018a. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060
- ↵Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, Paten B, Haussler D, Willard HF, Akeson M, Miga KH. 2018b. Linear assembly of a human centromere on the Y chromosome. Nat Biotechnol 36: 321–323. 10.1038/nbt.4109
- ↵Kankia BI, Buckin V, Bloomfield VA. 2001. Hexamminecobalt(III)-induced condensation of calf thymus DNA: circular dichroism and hydration measurements. Nucleic Acids Res 29: 2795–2801. 10.1093/nar/29.13.2795
- ↵Koetsier G, Cantor E. 2019. A practical guide to analyzing nucleic acid concentration and purity with microvolume spectrophotometers. New England Biolabs 1: 1–8. https://www.neb.com/-/media/catalog/application-notes/mvs_analysis_of_na_concentration_and_purity.pdf?rev=be7c8e19f4d34e558527496ea51623dc.
- ↵Li H. 2021. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37: 4572–4574. 10.1093/bioinformatics/btab705
- ↵Lin YC, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, Reumers J, Moisse M, Plaisance S, Drmanac R, 2014. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun 5: 4767. 10.1038/ncomms5767
- ↵Logsdon G. 2020. HMW gDNA purification and ONT ultra-long-read data generation v1. https://www.protocols.io/view/hmw-gdna-purification-and-ont-ultra-long-read-data-bchhit36 [accessed December 13, 2024].
- ↵Logsdon GA, Vollger MR, Hsieh PH, Mao Y, Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC, Rhie A, 2021. The structure, function and evolution of a complete human chromosome 8. Nature 593: 101–107. 10.1038/s41586-021-03420-7
- ↵Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA, 2020. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585: 79–84. 10.1038/s41586-020-2547-7
- ↵Oda Y, Sadakane K, Yoshikawa Y, Imanaka T, Takiguchi K, Hayashi M, Kenmotsu T, Yoshikawa K. 2016. Highly concentrated ethanol solutions: good solvents for DNA as revealed by single-molecule observation. Chemphyschem 17: 471–473. 10.1002/cphc.201500988
- ↵Ouameur AA, Tajmir-Riahi H-A. 2004. Structural analysis of DNA interactions with biogenic polyamines and cobalt(III)hexamine studied by Fourier transform infrared and capillary electrophoresis. J Biol Chem 279: 42041–42054. 10.1074/jbc.M406053200
- ↵Pelta J, Livolant F, Sikorav J-L. 1996. DNA aggregation induced by polyamines and cobalthexamine. J Biol Chem 271: 5656–5662. 10.1074/jbc.271.10.5656
- ↵Quick J. 2018. Ultra-long read sequencing protocol for RAD004. protocols.io 1–16. 10.17504/protocols.io.mrvc566
- ↵R Core Team. 2024. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Robertson RM, Smith DE. 2007. Strong effects of molecular topology on diffusion of entangled DNA molecules. Proc Natl Acad Sci 104: 4824–4827. 10.1073/pnas.0700137104
- ↵Rodrigue S, Materna AC, Timberlake SC, Blackburn MC, Malmstrom RR, Alm EJ, Chisholm SW. 2010. Unlocking short read sequencing for metagenomics. PLoS One 5: e11840. 10.1371/journal.pone.0011840
- ↵Salazar AN, Gorter de Vries AR, van den Broek M, Wijsman M, de la Torre Cortés P, Brickwedde A, Brouwers N, Daran J-MG, Abeel T. 2017. Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113-7D. FEMS Yeast Res 17: fox074. 10.1093/femsyr/fox074
- ↵Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT, Scarpa A. 2013. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One 8: e62692. 10.1371/journal.pone.0062692
- ↵Stortchevoi A, Kamelamela N, Levine SS. 2020. SPRI beads-based size selection in the range of 2-10kb. J Biomol Tech 31: 7–10. 10.7171/jbt.20-3101-002
- ↵Synthego HEK293. HEK293 cells: background, applications, protocols, and more. synthego.com. https://www.synthego.com/hek293 [accessed March 12, 2024].
- ↵Tyson J. 2019. Rocky Mountain adventures in genomic DNA sample preparation, ligation protocol optimisation/simplification and ultra long read generation v1. https://www.protocols.io/view/rocky-mountain-adventures-in-genomic-dna-sample-pr-7euhjew [accessed November 4, 2024].
- ↵Xia Y, Chen F, Du Y, Liu C, Bu G, Xin Y, Liu B. 2019. A modified SDS-based DNA extraction method from raw soybean. Biosci Rep 39: BSR20182271. 10.1042/BSR20182271