
Integrating short-read and long-read single-cell RNA sequencing for comprehensive transcriptome profiling in mouse retina
Abstract
The vast majority of protein-coding genes in the human genome produce multiple mRNA isoforms through alternative splicing, significantly enhancing the complexity of the transcriptome and proteome. To establish an efficient method for characterizing transcript isoforms within tissue samples, we conducted a systematic comparison between single-cell long-read and conventional short-read RNA sequencing techniques. The transcriptome of approximately 30,000 mouse retina cells was profiled using 1.54 billion Illumina short reads and 1.40 billion Oxford Nanopore Technologies long reads. Consequently, we identify 44,325 transcript isoforms, with a notable 38% previously uncharacterized and 17% expressed exclusively in distinct cellular subclasses. We observe that long-read sequencing not only matches the gene expression and cell-type annotation performance of short-read sequencing but also excel in the precise identification of transcript isoforms. While transcript isoforms are often shared across various cell types, their relative abundance shows considerable cell type–specific variation. The data generated from our study significantly enhance the existing repertoire of transcript isoforms, thereby establishing a resource for future research into the mechanisms and implications of alternative splicing within retinal biology and its links to related diseases.
The mouse retina is a complex neuronal tissue composed of over 130 unique neuronal cell types that are categorized into seven major cell classes (Masland 2012; Grünert and Martin 2020; Yan et al. 2020). Each cell type differs in its morphology, function, location, and transcriptomic profile (Jeon et al. 1998; Grünert and Martin 2020). Alternative splicing of pre-mRNA is a crucial mechanism that enhances the diversity of transcriptome and proteome (Wang et al. 2015). It regulates gene expression by producing multiple mRNA variants from a single gene, allowing for diverse protein production. This process enables cells to adapt their function during development and in response to environmental changes, underscoring its pivotal role in the complexity of multicellular organisms. Similar to other neural tissues, the retina exhibits a notable enrichment of tissue-specific splicing events (Liu and Zack 2013; Aísa-Marín et al. 2021). Previous research endeavors have unveiled various aspects of retina-specific splicing, including the identification of retina-specific exons, transcript isoforms, and splicing regulators (Murphy et al. 2016; Ciampi et al. 2022). A comprehensive understanding of the expressed transcript isoforms, coupled with insights into retina cell type–specific splicing events as well as the expression patterns of transcript isoforms at the single-cell level is indispensable for understanding the underlying mechanisms of splicing and gene regulation. Furthermore, a complete catalog of splicing isoforms in individual cell-type contexts could guide the accurate prediction of the effect of genetic variants in disorders related to the retina (Aísa-Marín et al. 2021).
Single-cell RNA sequencing (scRNA-seq) has widely been used to characterize cell-specific transcriptomic differences in various neuronal tissues (Tian et al. 2021). Currently, scRNA-seq data primarily consist of short-read sequencing technologies because of their high accuracy, efficiency, and low cost (Amarasinghe et al. 2020). However, short-read RNA technology is limited to sequencing either the 5′ or 3′ end of the transcript, limiting the ability to quantify RNA transcript isoforms (Byrne et al. 2019). Long-read sequencing is an ideal technology to effectively identify alternative splicing and sequence heterogeneity in mRNA transcripts (Kovaka et al. 2019). Indeed, recent studies of long-read-based scRNA-seq technology have shown that distinct mRNA transcript isoforms have been observed among different cell types (Tian et al. 2021). Unfortunately, to date, RNA transcript isoforms in the mouse retina have not been systematically annotated and quantified in a cell type–specific context.
In this study, we adapted protocols from the 10x Genomics and implemented a modified pipeline (Fig. 1A; Methods) to conduct Illumina short- and Oxford Nanopore Technologies (ONT) long-read sequencing and data analysis at the single-cell level. By profiling the transcriptome of single cells from mouse retinas with short and long reads, we aimed to primarily assess the performance of long-read sequencing at the gene expression level and conduct an unbiased characterization of the isoform catalog at the single-cell level in the mouse retina, which will be the first extensive examination of isoforms in mouse retina, offering valuable insights into their diversity and abundance. Additionally, we aimed to identify novel isoforms and those specific to particular cell classes, thereby enhancing our understanding of isoform heterogeneity and its implications for cell function. Another goal was to explore the differential usage and expression of isoforms across cell classes and to determine whether incorporating splicing information can enhance cell subclustering and provide deeper insights into cell diversity. Lastly, our study also included specific isoform investigations using the extensive data set, which will further our exploration, uncover additional discoveries, and advance our understanding of biological mechanisms.

Overview of the experimental design and basic summary statistics between Illumina short-read (SR) and ONT long-read (LR) scRNA-seq. (A) Summary of the study design, with an overview of the Illumina SR and ONT LR scRNA-seq data processing pipeline. (B) Summary of cell count in each major retina cell class and subclass. (C) Uniform Manifold Approximation and Projection (UMAP) visualization of cells in each major retina cell class for SR (left upper) and LR (left lower) and dot plots for viewing the expression level of known major cell class markers between SR (right upper) and LR (right lower) data. (D) Scatter plot for single-cell gene expression levels calculated by two approaches in each major retina cell class. (E) Concordance of major cell class annotation results between SR and LR. Only cells detected in both approaches were shown here. (F) Concordance of BC type annotation results between SR and LR approaches. Only cells detected in both approaches were shown here. (G) UMAP visualization of cells in each BC type for SR (left upper) and LR (left lower) and dot plots for viewing the expression level of known BC type markers between SR (right upper) and LR (right lower) data.
Results
Single-cell RNA-seq with short- and long-read technologies
To benchmark and assess the performance of short-read and long-read single-cell sequencing technologies comprehensively, we performed the two technologies on cells isolated from four mouse retina samples. These included two biological replicates from wild-type retinas and two samples enriched in amacrine cell (AC) and bipolar cell (BC), leading to a more comprehensive coverage of rare cell types. A thorough comparison of short-read and long-read sequencing methods was conducted, as detailed in Supplemental Table S1.
In total, the transcriptomes of over 30,000 single cells from four mouse retinas were profiled, generating 1.54 billion Illumina short reads and 1.40 billion ONT long reads. The sequencing achieved high read coverage, with an average of over 45,000 reads per cell, and exhibited comparable depth between the short- and long-read data sets. The median read length for ONT was ∼1000 bp, corresponding to the average size of full-length transcripts. The average base call quality score was ∼12.5, corresponding to an estimated error rate of ∼5.6% (Supplemental Fig. S1).
Cell clustering and annotation were initially conducted on the short-read scRNA-seq data set (Supplemental Data). Following the analysis pipeline previously described (Li et al. 2024), a total of 29,191 cells that passed quality filters were integrated and clustered, forming six major groups (Fig. 1B,C). Cell clusters were annotated using established cell class–specific marker genes (Supplemental Fig. S2; Li et al. 2024). Six major cell classes in the retina were identified, including 13,525 rod cells, 8863 BCs, 4571 ACs, 1218 cone cells, 869 Müller glial cells (MGs), and 145 retinal ganglion cells (RGCs). As expected, the two biological replicates of wild-type retina displayed a similar distribution of cell-type proportions, with rod cells being the most abundant (Supplemental Table S2). In contrast, the samples enriched for ACs and BCs showed a significant increase in the proportion of the corresponding cell classes (Supplemental Table S2). Given the known heterogeneity within AC, BC, and RGC classes, based on their distinct transcriptomic, morphological, and functional properties, notable cell heterogeneity was observed, particularly in AC and BC clusters. To attain higher resolution, ACs were further clustered into three subclasses: GABAergic, Glycinergic, and non-GABAergic non-Glycinergic (nGnG), while BCs were further clustered into 14 cell types (Fig. 1B).
To compare the performance of long-read to short-read sequencing, we conducted cell clustering and annotation using long-read data generated from the same set of samples. As depicted in Figure 1A, long reads that passed quality filtering were demultiplexed based on cell barcodes (CBs) identified by short-read sequencing (details in Methods), resulting in ∼518 million long reads across the four samples (Supplemental Table S3). Upon mapping, cell clustering was performed, and the resulting clusters were annotated using known marker genes, following the same pipeline as the short-read data set. Consistently, six major cell classes were identified with cell proportions similar to the short-read approach (Fig. 1C,D; Supplemental Fig. S2). The consistency of cell class annotations between the short-read and long-read data sets was analyzed by comparing individual cell annotations (Fig. 1E). Over 98.0% of cell class assignments (28,606 out of 29,191 cells) were consistent, with BCs showing an agreement of 99.8%. This high level of concordance was also evident at higher cell cluster resolutions. For instance, in BC cell types, a concordance of 97.4% (8629 out of 8863 cells) was observed at the individual cell-type level (Fig. 1F). The long-read data identified an additional BC type, BC4, that was missed in the short-read data (Fig. 1G).
We further assessed the correlation of gene expression by comparing the transcriptomics across all cell classes from both data sets. As illustrated in Supplemental Figure S3, a strong positive correlation in gene expression was observed, with an overall Pearson's r value of 0.87 (P-value < 2.2 × 10−16). This strong correlation was consistent across all cell classes, with Pearson's r values ranging from 0.84 to 0.90 (Fig. 1D, P-value < 2.2 × 10−16). Therefore, our results indicated that when sequenced at similar depths, both short-read and long-read data sets exhibited comparable sensitivity and high concordance in cell identification, clustering, and annotation.
Mouse retina isoform catalog and unique splicing profiles across cell classes
One of the key advantages of long read is the improved ability of detecting transcript isoforms. To assess the robustness of the long-read sequencing approach in discovering splicing isoforms of different cell classes in mouse retina, we performed a detailed isoform analysis including identification, classification, and quantification on the long-read data. Under a stringent cutoff and filtering criteria (see Method), as shown in Figure 2A, using annotated transcripts in GENCODE (vM25) as the reference, a total of 44,325 transcript isoforms were identified. Most transcript isoforms (38,247, 86.2%) belonged to protein-coding genes. Sixty percent of isoforms match with known isoforms, including 19,821 isoforms matching a reference transcript at all splice junctions (full splice match, FSM) and 7517 isoforms partially matching to consecutive splice junctions of the reference transcripts (incomplete splice match, ISM). The remaining 40% represents novel isoforms, including 15,894 isoforms of known genes with novel splice junctions of known splicing sites (novel in catalog, NIC), 38 isoforms of known genes with novel donor and acceptor sites (novel not in catalog, NNC), and 1055 isoforms matching the concatenation of two or more separate genes (fusion). It was interesting to note that novel isoforms (NNC, NIC, and fusion) tended to be expressed at lower levels (Fig. 2B), which could provide a partial explanation for why these isoforms remained undetected in previous studies. Therefore, single-cell long-read sequencing greatly increased the number of isoforms detected.
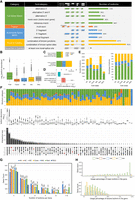
Overview of single-cell isoform catalog in the mouse retina. (A) Classification of isoforms according to their splice sites when compared to reference annotations and the number of isoforms in each category detected by the entire data set. (B) Box plot showing the isoform abundance in different categories specified in (A). The adjusted P-value shows the group differences. (C) Number and classification of isoforms detected in both single-cell and bulk RNA-seq data compared to isoforms uniquely detected using single-cell data. (D) Isoform abundance of isoforms detected in both single-cell and bulk RNA-seq data compared to isoforms uniquely detected using single-cell data. (E) Summary of number (left) and percentage (right) of isoforms in different categories for each cell class, colored by categories specified in A. (F) UpSet plot showing overlap of isoforms in cell classes, where number and percentage of isoforms shared by different cell classes were indicated in the top bar charts, colored by categories specified in (A), and the isoform abundance in each group were showed in the box plot. (G) Bar plot of the number of distinct isoforms expressed per gene. Genes with more than five distinct transcripts were merged. (H) Histogram showing the differential transcript usage in the gene of the two most abundant isoforms of each gene, grouped and colored by categories specified in A.
To validate the isoform detection, we sequenced RNA from another wild-type mouse retina sample aged P50 at the bulk level, obtaining ∼37 million ONT long reads and conducted isoform identification and classification (Supplemental Fig. S4; Methods). Consequently, 13,030 (30%) out of 44,325 transcript isoforms detected by the single-cell data set were also identified in the bulk data. The majority (9227) of these isoforms were classified as FSM, while those uniquely detected by single-cell data included a higher portion of novel isoforms (Fig. 2C). The low level of ISM detected in bulk data suggested that significant ISMs were likely artifacts. However, the substantial amount of NIC confirmed by bulk (3317 out of 15,894) indicated that the identification of much of the NIC was accurate. Additionally, 10 out of 38 NNCs in the single-cell data set were validated by bulk data. In terms of abundance, isoforms uniquely identified in the single-cell data set had lower expression levels (Fig. 2D, adjusted P-value < 2.2 × 10−16) compared to those validated in bulk RNA-seq data, consistent with the high coverage of long reads in single-cell data. Additionally, we examined the transcripts identified exclusively using bulk RNA-seq data (15,304) and found that a significant portion of those (6834, 45%) were filtered out under the stringent abundance criteria applied to single-cell data. This indicated over 70% (19,864 out of 28,334) of isoforms detected using bulk RNA-seq data were captured with the scRNA-seq data set, even though the data sets were generated from different mouse retina samples.
Subsequently, we systemically examined the identified 44,325 transcript isoforms in each cell class. The number of novel isoforms identified across different cell classes varied significantly, with over 13,000 novel isoforms found in rod cells followed by BC and AC (Fig. 2E), correlating with the number of cells from each cell class profiled in our data set (Fig. 1B). Despite the difference in raw isoform numbers, overall similar distribution of different isoform categories across cell classes was observed with known isoforms accounting ∼65% (Fig. 2E). In contrast, the proportion of different isoform categories varied significantly depending on their expression pattern (Fig. 2F). For example, out of the transcript isoforms that were expressed across all cell classes, ∼20% of these isoforms were newly discovered. In contrast, for isoforms that showed a more restricted expression pattern, the proportion of novel isoforms was generally higher. It was worth noting that although the vast majority of isoforms were expressed in at least three cell types, 16.7% of isoforms were expressed exclusively in one cell class, including 2945, 914, 108, 206, 77, and 3133 isoforms identified for AC, BC, cone, MG, RGC, and rod, respectively.
Consistent with previous studies (Tian et al. 2021), an average of four isoforms, ranging from 2 to 28, were observed for the majority of genes (68%) (Fig. 2G). Similar distribution was observed across all cell classes (Fig. 2G). Subsequently, we classified the structural categories (Supplemental Table S4) and compared the expression levels (Fig. 2H) of the two most abundant isoforms for each gene. Approximately 34% of genes exhibited a novel isoform among their top two isoforms. Moreover, for half of the genes (6336 out of 14,490) expressed in the retina, the major isoform constituted over 90% of the total gene expression, which indicated these genes had a dominant isoform.
Several features of known and novel isoforms were compared, including the number of exons (Fig. 3A), differential transcript usage (DTU) within genes (Fig. 3B), and the length of the open reading frame (ORF) (Fig. 3C). The results revealed that while the distribution of exon numbers and ORF lengths between known and novel isoforms exhibited similar patterns, their usage within genes exhibited significant differences. Most of the known isoforms accounted for over 90% of the total gene expression, whereas the novel isoforms showed lower expression levels. We then extended our comparison to examine the differential usage within genes of known and novel isoforms across different cell classes (Fig. 3D), and they exhibited consistent trends. This pattern also held true for isoforms specific to cell classes (Fig. 3E).
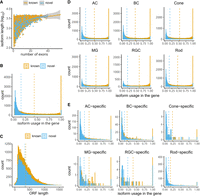
Comparison of novel and known isoforms in general and across different cell classes. (A) Scatter plot showing isoform length and exon numbers for known and novel isoforms. (B) Histogram showing the distribution of known and novel isoforms on expression level in the gene. (C) Histogram showing the distribution of known and novel isoforms on ORF length. (D) Histogram showing the distribution of known and novel isoforms expressed in different cell classes on expression level in the gene. (E) Histogram showing the distribution of cell class–specific known and novel isoforms on expression level in the gene.
Most genes display varied isoform usage among cell classes/subclasses/types
Since the vast majority of genes expressed multiple isoforms, it was interesting to examine whether genes demonstrated DTU among the cell classes, subclasses, or types illustrated in Figure 1B. Considering the high dropout rate in single-cell data, DTU analysis was performed by merging cells from the same cluster into a pseudo-bulk sample to calculate the proportion of isoform usage across different cell classes. Consistent with the observation where only a small portion of isoforms showed cell class–specific expression, most if not all isoforms of a given gene tended to be expressed across all cell classes. However, the proportion of different isoforms varied significantly among cell classes. For example, Pcbp4 is a gene predicted to facilitate mRNA 3′-UTR-binding activity. It acts upstream of or within the negative regulation of the DNA damage response and mRNA splicing via the spliceosome. Although all isoforms of Pcbp4 were expressed in all cell classes, varied usage among different cell classes was observed (Fig. 4A,B). As shown in Figure 4B, the transcript (colored in yellow) that was predominantly expressed in ACs was lowly expressed in rods, exhibiting significant difference (P-value = 1.06 × 10−88, Fig. 4C). Conversely, a transcript (colored in dark blue) was significantly more prevalent in rods than in BCs (P-value = 3.86 × 10−98, Fig. 4C). Another example is gene Prkcz, which belongs to the PKC family of serine/threonine kinases, and plays roles in diverse cellular activities like proliferation, differentiation, and secretion. Similarly, for the two isoforms of Prkcz, the isoform comprising 15 exons was predominantly expressed in ACs, BCs, and RGCs, while the isoform with 18 exons accounted for over 90% expression in cones, MGs, and rods (Fig. 4D).
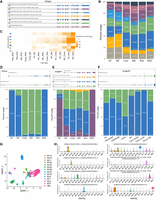
Statistics of differential usage of transcript isoforms across cell classes and subclasses. (A) Top 7 most abundant transcript isoforms of gene Pcbp4 detected. The transcription orientations for all the isoforms are from left to right. Differentially spliced sites were outlined in black. (B) Percent usage of each Pcbp4 isoform, with colors corresponding to isoforms in A. All other less abundant isoforms not plotted in A were merged and represented as gray bars. (C) Heatmap showing the significance of differential isoform usage between two major cell classes (x-axis) using Fisher's exact tests. For each isoform (y-axis, colored by isoform specified in A), the greater the disparity in isoform usage between the first and second cell classes, the lower the P-value. (D) Isoform plots and usage bar charts of Prkcz showing different isoform expression patterns across major retina cell classes. The transcription orientations for all the isoforms are from left to right. (E) Isoform plots and usage bar charts of Impdh1 showing different isoform expression patterns across major retina cell classes. The transcription orientations for all the isoforms are from left to right. The 17 bp exon was outlined in black. (F) Isoform plots and usage bar charts of Snap25 showing different isoform expression patterns across AC and BC subclasses. The transcription orientations for all the isoforms are from left to right. (G) UMAP visualization of 8863 BCs clustered using isoform as features and colored by BC subclass annotations from SR scRNA-seq. (H) Violin plot showing selected isoforms differentially expressed across BC subclasses.
Another example is Impdh1, which has associations with inherited human retinal disorders like Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP). We identified several novel isoforms of Impdh1, which include a novel 17 bp exon previously unreported in the Reference Sequence (RefSeq) transcripts (Fig. 4E, upper). The inclusion of the 17 bp exon resulted in a reading frameshift and ORF elongation (37 amino acids) with an alternative stop codon. The transcripts incorporating this exon demonstrated significant expression in BCs, cones, MGs, and rods (Fig. 4E, lower). The primary isoform of Impdh1 expressed in mouse photoreceptors was not the canonical Impdh1, suggesting that changes in the canonical protein's function were probably not the primary cause of retinal degeneration. Additionally, this exon exhibited a high degree of conservation in humans, and we identified several single nucleotide variants located within 10 bp upstream or downstream from the novel exon in our in-house RP patient cohort, as indicated in Supplemental Table S5. These variants were not present in the general populations in the Genome Aggregation Database (gnomAD) (Karczewski et al. 2020). It would be intriguing to explore its correlation with human diseases in subsequent studies.
Subsequently, we analyzed the pattern of isoform usage within the subclasses of AC and BC. Mirroring what we observed in major cell classes, it appeared that most isoforms were present across all subclasses, though the distribution of these isoforms shifted between subclasses. For example, as shown in Figure 4F, one isoform of Snap25 (colored in blue) was mainly found in GABAergic and Glycinergic subclasses, while another one (colored in green) constituted over half of the expression in nGnG ACs. Furthermore, the expression ratios of these two isoforms varied between cone BCs and rod BCs.
To assess transcript isoform usage in individual cell types, we further counted the isoform usage for each individual BC, resulting in a cell-by-isoform matrix. We then undertook cell clustering with isoforms serving as distinguishing features (Fig. 4G). While no additional cell clusters emerged compared to gene expression-based annotations, we observed differential expression patterns of isoforms among BC types and identified several isoforms that were predominantly expressed in specific BC types (Fig. 4H). Notably, some of these were novel isoforms.
In summary, our study revealed that genes exhibited diverse isoform usage patterns across major retina cell classes and subclasses. Additionally, we have identified distinct expression patterns of isoforms across BC types, as well as isoforms that are primarily expressed in specific cell types.
Gene fusion isoforms in mouse retina
A surprising finding was the identification of a substantial number (1055) of potential fusion transcripts, 114 of them were validated using the above-mentioned bulk RNA-seq data. These fusion transcripts were unlikely due to experimental artifacts as supported by the following evidence: (1) The fusions were substantiated by a minimum of 5 UMIs from long reads that spanned a fusion breakpoint (details in Methods), therefore, indicating these were independent events and unlikely to be artifact. (2) All detected fusions occurred within the same chromosome, with no indication of interchromosomal fusions, indicating it was unlikely due to template switch error during polymerase chain reaction (PCR). (3) Notably, the genes involved in each fusion were found on the same strand, further suggesting that these fusion transcripts were biologically relevant events.
Notably, 903 out of 1055 gene fusions occur in coding regions. We then extracted topologically associating domains (TADs) identified from an adult mouse retina bulk Hi-C data set (NCBI Gene Expression Omnibus [https://www.ncbi.nlm.nih.gov/geo/] accession number GSE135465) in a previous study (Norrie et al. 2019). As a result, 831 out of 1055 gene fusions occur within these TADs, 114 partially overlap with TADs, and 110 (10.43%) are located outside TAD regions (Supplemental Table S6).
Most of these fusions were characterized by the combination of two genes, though instances of three-gene fusions were observed (Fig. 5A,C). Subsequently, we assessed the proximity of the fused genes. Some exhibited adjacent fusion, while others skipped over nearby genes to fuse with more distant counterparts (Fig. 5B,C). When examining the exon count and abundance of fusions, categorized by gene proximity, it became evident that adjacent fusions contained a greater number of exons compared to the nonadjacent counterparts (Fig. 5D, adjusted P-value = 1.8 × 10−7), and variations in abundance were noted between the two groups (Fig. 5E, adjusted P-value = 0.0073).

Gene fusions in mouse retina. (A) Example visual of fusions (in green) aligned to the RefSeq reference (in blue) formed by two or three known genes. (B) Example visual of fusions (in green) aligned to the RefSeq reference (in blue) formed by adjacent or distant known genes. (C) Bar chart illustrating the count of fusions, categorized by the number of fused genes and their adjacency to one another. (D) Box plot showing the exon number of fusions categorized by the adjacency of the fused genes. (E) Box plot showing isoform abundance of fusions categorized by the adjacency of the fused genes. (F) Histogram illustrating the distribution of fusion expression ratios compared to the combined expression of fused genes, with log transformation applied. (G) Bubble chart depicting the 13 molecular functions of Gene Ontology enrichment analysis ranked by fold enrichment, utilizing gene set always in fusions. (H) Example visuals of fusions (in green) aligned to the RefSeq reference (in blue). The top illustration depicted a gene (Ifit3) that can fuse with multiple other genes (Ifit2 and Ifit3b). The lower visualization highlighted the alternative splicing within the fusion (Phf6-Hprt). (I) UpSet plot showing the intersection of fusions in major retina cell classes, where number of fusions shared by different cell classes were indicated in the right bar charts.
We then compared the abundance of the fusions against the expression of all isoforms present in the implicated genes. The proportions between fusion utilization and other isoform usage showed a broad distribution (Fig. 5F). Ninety genes were exclusively expressed in fusions. Functional clustering of these genes unveiled common pathways (Fig. 5G), notably those associated with immunity, such as the T cell receptor binding (GO:0042608), which was logical considering the need for adaptability in immune processes.
In addition, upon detailed examination of the detected gene fusions, we also observed that: (1) Certain genes could partner with multiple other genes, resulting in different fusions (Fig. 5H, upper). (2) Some fusions underwent alternative splicing events (Fig. 5H, lower), which would be interesting for drug target-related studies. We further evaluated the overlap of fusions across various cell classes (Fig. 5I) and pinpointed certain fusions unique to each cell class, tallying 164 in rods, 107 in ACs, and 27 in BCs.
Down-sampling analysis and sequencing saturation
To evaluate the sensitivity of sequencing depth on isoform detection and determine if our sequencing reached saturation, we conducted a simulation by randomly sampling 1%, 10%, and 50% of the data set and performed the analysis with the same pipeline. As expected, the number of isoforms detected positively correlates with the number of sequencing reads used in the analysis (Fig. 6A). However, the increasing rate varied among different structural categories. For example, the number of FSMs detected increased by 41.3% from 10% to 50% of the full data set and only increased slightly by 10.2% from 50% to the full data set (Fig. 6A; Supplemental Fig. S5A), indicating they were approaching saturation and canonical isoforms can be detected by a moderate number of reads. Conversely, the quantity of novel isoforms, such as NICs, kept escalating. There was a 111.3% increase in their numbers when comparing 10% of the full data set to 50%, and a further 26.4% increase was observed when expanding from 50% of the data set to the entire data set, indicating that it has not been saturated even with the full data set, probably due to their low expression level (Fig. 6A; Supplemental Fig. S5A).
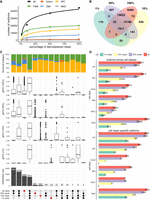
Down-sampling analysis. (A) Classification of isoforms according to their splice sites when compared to reference annotations and number of isoforms in each category detected using 100%, 50%, 10%, and 1% of demultiplexed LRs. (B) Venn diagram showing the intersection of the isoform sets detected using all, 50%, 10%, and 1% of demultiplexed LRs. (C) UpSet plot showing the intersection of isoforms detected using 100%, 50%, 10%, and 1% of demultiplexed LRs, where the number and percentage of isoforms shared by different cell classes are indicated in the bottom and top bar charts, colored by categories specified in A. The box plot shows the DTU in the gene before filtering with 2% cutoff for the isoforms in each group. (D) The bar plot shows the number of all isoforms (above) and cell class–specific isoforms (bottom) in each major retina cell class detected using 100%, 50%, 10%, and 1% of demultiplexed LRs.
We further examined the overlap of isoforms detected using 1%, 10%, 50%, and all long reads. Some isoforms were identified in data sets with fewer sequencing reads but were absent when more reads were used (Fig. 6B). For instance, 1075 isoforms were detected using 50% of the long reads, but they were not present in the comprehensive isoform set derived from all long reads. This discrepancy arose because we excluded isoforms that had a usage of <2% of the total usage for their respective genes, in order to mitigate background noise (Fig. 6C). Thus, when we down sampled the reads, the isoforms with low abundance could be kept by chance if their proportion becomes higher than 2%.
As we increased the number of reads used for isoform identification, the number of isoforms detected in each cell class followed a similar upward trend (Fig. 6D, upper) and the fraction of FSMs within the identified isoform set saw a downtick (Supplemental Fig. S5C–E). The count of identified isoforms in the six retina cell classes experienced a notable rise, ranging from 30% to 60%, when comparing the data from 10% of the full data set to 50%. Subsequently, when expanding from 50% of the data set to the complete data set, a further increase ranging from 11% to 18% was observed. This pattern suggested that isoform detection within each cell class approached a state of saturation. In addition, upon examining the overlap of isoforms across cell classes for each data set, we observed that 21.75% (8012 out of 36,832), 26.58% (5555 out of 20,899), and 27.32% (2208 out of 8082) isoforms from sets determined using 50%, 10%, and 1% of long reads, respectively, were expressed across all cell classes. This compared to 20.65% (9153 out of 44,325) in the complete data set, suggesting a smaller number of reads more tend to identify the common isoforms across cell classes. Additionally, the identification of cell class–specific isoforms has not yet reached saturation, as evidenced by a significant 56.0% increase in isoform number when the data set was expanded from 50% to its full extent (Fig. 6D, lower).
Discussion
Short-read scRNA-seq, now as a standard method, is highly effective in profiling gene expression to identify cell types and trajectories (Hwang et al. 2018), however, there is a limitation in the length of the sequenced reads. Short-read scRNA-seq can be divided into two main approaches: transcript counting methods (e.g., 10x Genomics) that sequence transcript ends, and whole transcript methods (e.g., Smart-seq2) (Picelli et al. 2013) that cover entire RNA sequences. Transcript counting methods can profile many cells but have limited capacity for splicing or isoform information, whereas whole transcript methods provide more details on exon alternative splicing but offer lower throughput. Previous short-read scRNA-seq studies employing whole transcript methods have revealed significant variations in isoform expression among individual cells (Shalek et al. 2013; Marinov et al. 2014). However, these analyses predominantly concentrated on alterations in specific exons or splice junctions, a limitation imposed by the nature of short-read sequencing. The reconstruction of isoforms remained suboptimal for sequences longer than 1 kb, with only ∼40% of molecules assignable to a specific isoform (Hagemann-Jensen et al. 2020). This technological limitation leaves a gap in understanding the full extent of alternative splicing and the diversity of isoform expression both within and across single cells and underscores the need for improved methodologies in accurately capturing and characterizing longer isoforms.
Long-read sequencing technology aims to resolve this issue entirely; however, its adoption in single-cell research is currently hindered by the lack of established protocols and data analysis pipelines (De Paoli-Iseppi et al. 2021). In our study, we developed a workflow designed to streamline the analysis aspect of high-throughput long-read RNA sequencing data, enabling the identification of transcript isoforms at the single-cell level. Our workflow modified the 10x Genomics scRNA-seq protocol (Gupta et al. 2018) in order to comprehensively resolve the full-length transcriptome of the mouse retina, and this approach can be applied to other cDNA libraries. In comparing the long-read and short-read data sets, we discovered highly comparable results in both gene expression and cell annotation. Our analysis revealed a 98.0% consistency in cell class categorization between long-read and short-read data sets, and a closely aligned 97.4% agreement at the BC-type level. In short, the long-read sequencing approach demonstrated its reliability in detecting single-cell transcriptomes based on gene expressions, making it plausible to use long-read sequencing exclusively for scRNA-seq in the future (Shiau et al. 2023).
The integration of long-read sequencing with single-cell sequencing techniques holds the promise of filling the existing gaps in isoform information. Using this pipeline, we conducted the first comprehensive characterization of full-length transcription isoforms within individual cells of the mouse retina. Over 16,000 novel transcripts were identified at lower expression levels compared to previously annotated ones. Although some of these transcript isoforms have already been identified in previous studies (Murphy et al. 2016; Norrie et al. 2019; Ling et al. 2020; Ray et al. 2020) using various methods, reference annotations such as GENCODE and RefSeq have not kept pace with the increasing amount of sequencing data available. Specifically, capture-based methods, which were designed to target and enrich particular isoforms, thereby uncovered a greater range of isoform diversity within selected genes (Ray et al. 2020). In contrast, our approach provided an unbiased detection method for isoforms across all genes, though its sensitivity was limited by the depth of sequence and abundance of the transcript. As a result, our study may not capture the full breadth of isoform diversity found in studies using more targeted techniques. For instance, in our isoform catalog, we identified 67 out of the 4116 isoforms across 30 genes from Ray et al. (2020) (Supplemental Table S7), which was likely due to the lower read abundance for these genes in our data set (Supplemental Table S8) compared to target enrichment method. However, these 67 isoforms appeared to be more abundant than other isoforms within their respective genes, suggesting our approach was able to capture the more abundant isoforms (Supplemental Fig. S6A). Increasing sensitivity without target enrichment would have required greater sequencing depth. Additionally, our bioinformatic pipeline excluded certain isoforms due to strict abundance filtering, as well as noncanonical splice isoforms because of the reference annotation used. For instance, unique Crb1 isoforms like Crb1-b, reported by Ray et al. (2020), were filtered out and not included in our final isoform catalog (Supplemental Fig. S6B). We then reconducted isoform identification using Ray et al. (2020)’s GTF as a reference annotation, rather than GENCODE, subsequently revealed more isoforms (Supplemental Fig. S6B). Therefore, we expect novel transcripts will be further discovered with a combination of increasing of sequencing depth and optimizing the analysis pipeline, such as including de novo transcript assembly.
In validating the identified isoforms using long-read bulk RNA-seq data, we found that a substantial portion (30%) of these isoforms could be confirmed, including both known and novel ones, despite the more than 10-fold difference in read depth for isoform detection between the two data sets. This underscored the reliability of long-read scRNA-seq for detecting transcript isoforms. The comparison between bulk and single-cell data revealed distinct advantages and limitations in isoform detection. Bulk RNA-seq data provided broader coverage, leading to the validation of numerous isoforms identified in the single-cell data set. However, single-cell data uniquely captured isoforms that were less abundant, which might be overlooked in bulk RNA-seq due to averaging effects across heterogeneous cell populations. Our analysis highlighted that while bulk RNA-seq can validate the presence of many isoforms, scRNA-seq is crucial for uncovering isoforms present in minor cell populations or those with low expression levels. Additionally, considering that many isoforms detected using bulk data (over 70%) could be detected in single-cell data when the abundance filtering criteria were relaxed, we could predict that a significantly larger number of true novel isoforms could be identified by loosing the criteria for single-cell analysis. Limitations in the current studies, such as sequencing depth, chemistry artifacts, stringent cutoffs, and the capabilities of software tools, suggest that some isoforms remain undetected. Addressing these limitations could lead to the identification of additional isoforms, enhancing our understanding of transcriptomic complexity. Future improvements in sequencing technologies and computational methods are essential for overcoming these challenges and providing a more comprehensive isoform landscape.
Additionally, we pinpointed 7383 isoforms specific to cell classes, many of which were novel and exhibited low expression levels. The identification of cell class–specific isoforms holds potential applications in various fields, such as immunotherapy, where cell surface proteins play a pivotal role. Our analysis of the mouse retina showed the distribution of novel transcripts was consistent across different retinal cell classes and unveiled a common pattern where all major cell classes in the tissue expressed a combination of diverse isoforms rather than a single canonical isoform. We frequently observed intricate splicing variations between the two most abundant isoforms of a gene. Furthermore, many genes displayed varying patterns of isoform usage among different cell classes and subclasses. Another noteworthy finding was that retinal cells could express different isoforms, even when their overall gene expression levels did not significantly differ from each other. This implied that identifying cell class–specific genes based solely on gene expression levels was not sufficient to characterize the vast transcriptional diversity. By examining the transcriptomic profiles of individual cells, researchers can gain a deeper understanding of how different isoforms are used in specific cellular contexts, shedding light on the intricacies of gene regulation and function within heterogeneous cell populations.
In addition to what we have demonstrated in this study, this data set has a wide range of applications across various genomic research areas. One such application is the exploration of single-cell gene fusions, which can provide valuable information about aberrant gene interactions and potential drivers of diseases. By leveraging long-read scRNA-seq data, we were able to identify a total of 1055 intrachromosomal gene fusions within the mouse retina. We analyzed the fusions within the 3D genome structure using TAD from the bulk Hi-C data of mouse retina and our results showed that over 10% of gene fusions were outside the TAD boundaries. This suggests potential new avenues of research for understanding gene fusions occurring beyond TAD constraints. It is plausible that these fusions may be associated with cell type–specific TADs not captured by bulk Hi-C data. Currently, however, no publicly available mouse retina cell type–specific 3D genome architecture, such as single-cell Hi-C, exists for further investigation, though we are keen to monitor future developments and revisit this question. In addition, these identified gene fusions exhibited intriguing characteristics. For instance, certain genes had the capability to partner with multiple other genes, resulting in diverse fusion events. Furthermore, some of these fusions underwent alternative splicing events, further diversifying the transcriptomic landscape. What is particularly noteworthy is that certain genes can fuse with others that are not immediately adjacent to them on the same chromosome, highlighting the complexity and flexibility of gene fusion events in the context of scRNA sequencing data. These findings may have implications in various fields, including cancer research, where gene fusions and differential isoform expression can have significant clinical relevance. At the meantime, fusion detection is still a more complex bioinformatics procedure compared to other transcript categories. We also examined the percentage of length of the fusion transcript mapped to each of the fused genes. As shown in Supplemental Figure S7, the length ratios between the two fused genes were widely distributed and not well balanced. To further exclude the possible artifacts due to the mapper, we also performed fusion detection using an alternative read mapping tool STARlong (Dobin et al. 2013). With stringent filtering and a lower mapping rate of 20.68%, 538 out of 1055 fusions were detected, indicating most of the fusions were likely to be true. The bioinformatics algorithms require improvement for fusion detection, and subsequent functional tests are necessary to better understand this phenomenon.
In conclusion, the long-read scRNA-seq approach was proved to be highly effective in identifying both known and novel isoforms. Our study stands as the first unbiased characterization of full-length transcription isoforms in single cells within the mouse retina. Our analytical approaches shed light to the transcriptome profiling and isoform discovery at the single-cell level, which can be applied to human samples, propelling isoform-focused research of aging and disease context.
Methods
Sampling and animal procedures
All mice were handled in accordance with the policies on the treatment of laboratory animals by the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine, and the studies were conducted in adherence to the Animal Models of Retinal Development and Diseases protocol approved by Biomedical Research and Assurance Information Network. Stock C57BL/6J mice were purchased from the Jackson Laboratories, Bar Harbor, ME (stock number: 000664). The C57BL/6J mice were maintained on a 12 h light–dark cycle at 23°C, standard mouse LabDiet 5V5R (Purina), and water was provided ad libitum throughout the study. The mice were managed and housed by the Baylor College of Medicine Center for Comparative Medicine. Adult mice were euthanized using CO2 gas and isoflurane asphyxiation for 5 min, followed by cervical dislocation.
Single-cell cDNA library preparation and Illumina short-read sequencing
Samples and scRNA-seq cDNA and library generation were described previously (Li et al. 2024). Briefly, scRNA-seq cDNA and library were generated using 10x Genomics Chromium Single cell 3′ Reagents Kits v2 and v3, following the manufacturer's instructions. The short-read sequencing library was sequenced on an Illumina NovaSeq 6000 sequencer (151 bases + 151 bases).
Nanopore long-read single-cell library preparation and sequencing
Nanopore library was generated from scRNA-seq cDNA following the Nanopore Single-cell transcriptomics with cDNA prepared using 10x Genomics protocol (version Jan2022) using the SQK-PCS111 Ligation Sequencing Kit; 35–55 fmol of the library was sequenced on PromethION FLO-PRO002 R9.4.1 flow cells. The following options were used: 72 h of run time, and real-time basecalling with high-accuracy mode.
Nanopore long-read bulk RNA-seq library preparation and sequencing
Total RNA was extracted from male C57BL/6J mouse retina using the Direct-zol RNA Microprep Kits (Zymo Research). The sequencing libraries were then generated using the ONT cDNA-PCR Sequencing Kit V14 (SQK-PCS114). And 35–55 fmol of the library was sequenced on PromethION FLO-PRO002 R10.4 flow cells using the following procedure: 72 h of run time, and real-time basecalling with super high-accuracy mode. The read quality control plots in Supplemental Figure S4 were generated using pycoQC v2.5.2 (Leger and Leonardi 2019).
Illumina short-read scRNA-seq data analysis
All four samples’ FASTQ data were processed using Cell Ranger (7.0.1) to create a gene count matrix for each. Contamination from background transcripts in the preserved true cells was mitigated using SoupX (Young and Behjati 2020). Subsequently, DoubletFinder (McGinnis et al. 2019) was employed to estimate and remove potential doublets, particularly focusing on those with a high proportion of simulated artificial doublets. The gene count matrices were then fed into the standard Seurat (Hao et al. 2021) pipeline, with SCTransform v2 (Choudhary and Satija 2022) undertaking the normalization process. The samples were clustered at a resolution of 0.6. Annotations for the retina cell class were added manually using well-known retina marker genes. The subclass annotation for AC and BC was obtained through the utilization of “single-cell ANnotation using Variational Inference” (scANVI) (Gayoso et al. 2022), using an in-house meta reference described in Li et al. (2024). Data integration from all four data sets was carried out using Seurat. Figure 1C and F illustrate the results of the clustering and cell annotation.
Oxford Nanopore long-read scRNA-seq data preprocessing
Basecalling was carried out on the raw ONT FAST5 data using Guppy (version 6.1.5). The Nanopore FASTQ reads were inspected for adapter sequences and poly(A/T) sequences of 15 nt or longer. We identified poly(A) (or T) sequences with a minimum of 75% adenine content within 170 nt from both read ends, generating stranded (forward) reads via Single Cell Long Read (SiCeLoRe) (Lebrigand et al. 2020) (https://github.com/ucagenomix/sicelore) utilizing the NanoporeReadScanner.jar module. The refined FASTQ data were then used to align the genome using minimap2 (Li 2018) (v2.24, https://github.com/lh3/minimap2) with the parameter “-ax splice -uf ‐‐MD ‐‐sam-hit-only ‐‐junc-bed”, referencing the GRCm38/mm10 genome, and annotated with GENCODE vM25. Using SiCeLoRe's Nanopore_BC_UMI_finder.jar module, we identified the location of the barcode sequence for each alignment by searching the flanking sequence to the CB. The CBs and gene correlation discovered from the Illumina short-read data served as a reference to find and trim in the long reads, with the dynamic edit distance adjusting the maximal edit distance according to the complexity of the search set. The sequences found after the CB were treated as UMIs (unique molecular identifiers) and were trimmed accordingly. Based on SiCeLoRe, similar to the barcode edit distance, the UMI edit distance was dynamically adjusted based on the complexity of the search set. After CB/UMI assignment and correction, the processed BAM was used for further analysis.
Detection and quantification of isoforms using ONT long-read scRNA-seq data
We used Flair (Tang et al. 2020) (https://flair.readthedocs.io/en/latest/index.html) to summarize the alignment for each read by grouping reads with similar splice junctions (<5 bp) to get a raw isoform annotation. The final nanopore-specific reference isoform assembly is made by aligning raw reads to the first-pass assembly transcript sequence using minimap2 to identify isoforms with high confidence. “Stringent” mode was used to make sure all supporting reads were full-length (80% coverage and spanning 25 bp of the first and last exons).
flair correct -q LR.bed -g mm10.fasta -f GENCODE.v25.mm10.annotation.gtf -o LR_corrected.bed
flair collapse -q LR_corrected.bed -g mm10.fasta -f GENCODE.v25.mm10.annotation.gtf -m /path/to/minimap2 -sam /path/to/samtools -o LR -r demultiplexed_reads.fq ‐‐stringent
Pseudo-bulk samples were produced by merging isoform counts at the level of cell class, cell subclass, and individual cells. Cell class groupings originated from the Seurat clustering depicted in Figure 1C, while AC/BC subclass groupings were derived from scANVI clustering presented in Figure 1G. The “flair quantify” function in Flair was used to create a UMI count matrix for each tier, in which rows correspond to chosen transcripts and columns represent groups (cell classes/subclasses).
flair quantify -r reads_manifest.tsv -i LR.isoforms.fa -m /path/to/minimap2 -sam /path/to/samtools -o LR.flair.quantify ‐‐isoform_bed LR.isoforms.bed ‐‐sample_id_only ‐‐generate_map ‐‐tpm ‐‐stringent
Isoform classification and ranking for Oxford Nanopore long-read scRNA-seq data
SQANTI3 (Tardaguila et al. 2018) (version 5.1.2, https://github.com/ConesaLab/SQANTI3) was used to compare the transcripts identified to the reference with rules mode and default parameters. The isoform classification was extracted from the SQANTI3 result and plotted in Figure 2A. Results from comparisons across different cell classes were plotted using the R (R Core Team 2023) package ComplexUpset (Krassowski et al. 2022). We ranked transcript abundance for each gene that had multiple isoforms and obtained the alternative splicing events from the most expressed isoform and the second most expressed isoform.
Isoform filtering for Oxford Nanopore long-read scRNA-seq data
We implemented several filters to hone the isoforms according to specific parameters. The first parameter was intrapriming events: isoforms showing 12 or more adenines at the genomic level within the 20 bp downstream from the transcription terminating site (TTS) were filtered out. RT-Switching was the next parameter: we removed isoforms that may have been affected by reverse transcription errors, resulting in the formation of new, noncanonical splice junctions. Isoforms linked to degraded RNA, as indicated by the retention of intronic sequences, were also considered for exclusion. Gene abundance and isoform abundance were also taken into account: we eliminated genes with a UMI count of 10 or less, and isoforms with a UMI count of 5 or less. Moreover, we excluded isoforms with a UMI count of <2% of the total UMI count for the respective gene to account for background noise. The SQANTI3 was used to annotate and filter based on the first three parameters, while the last three were applied manually after isoform quantification. These filtering steps were used to refine the isoform data set, ensuring a more precise and reliable set of isoforms for additional analysis.
Isoform detection, classification, and filtering using Nanopore long-read bulk RNA-seq data
Once again, we used Flair for isoform identification. Unlike in scRNA-seq data analysis, we used the “flair align” module to perform long-read alignment to the reference genome (mm10), which was also built using minimap2. The command and parameters used are detailed below:
flair align -g mm10.fasta -r ./Ms_bulk/Ms_bulk.fastq.gz -o ./Ms_bulk/flair_align ‐‐threads 36
Next, splice junction correction and read collapse were conducted using “flair correct” and “flair collapse,” respectively, with the same commands as the ONT long-read scRNA-seq data procedure. Then we ran SQANTI3 on the detected isoforms and compared with the reference transcript in GENCODE v25 under rules mode for structural category classification.
Lastly, we conducted isoform filtering with the following criteria, similar to scRNA-seq data, except the ones regarding abundance: (1) intrapriming events; (2) RT-Switching; and (3) intron retention.
Isoform comparison between long-read scRNA-seq and bulk RNA-seq data sets
The software gffcompare (Pertea and Pertea 2020) (version 0.12.6) was used with the command “gffcompare -i input -o out -A -K” to compare transcript isoforms in the GTF files generated using ONT long-read single-cell and bulk RNA-seq data after isoform filtering. The output file “out.tracking” contained IDs for transcripts that overlapped between these two data sets and those exclusively identified in each one. With the transcript IDs, we tracked back to the isoform annotation and quantification results to compare features like abundance, and structural category in each group.
Differential isoform usage analysis across cell classes/subclasses/types
Utilizing the matrix detailing isoform expression by cell class/subclass, the transcript structures and the percentage usage visualizations in Figure 4 were constructed via the “plot_isoform_usage” function in Flair. The significance of differential isoform utilization across cell classes was determined using the “diff_iso_usage” function in Flair, which is based on Fisher's exact tests. After scaling the matrix of cell-by-isoform expression, the outcomes were incorporated into UMAP visualizations, as depicted in Figure 4 and facilitated by Seurat.
Down-sampling analysis
The down-sampling data sets were achieved by randomly subsampling the demultiplexed long reads (1%, 10%, 50%) using seqtk (version: 1.3-r115-dirty, https://github.com/lh3/seqtk) and rerunning the pipeline with the same parameters and filtering. The gffcompare (Pertea and Pertea 2020) (0.12.6) program was used with the command “gffcompare -i input -o out -r GENCODE.vM25.annotation.gff3 -R” to compare transcript isoforms annotations obtained from the three down-sampling data sets and the complete isoform catalog. Results from these comparisons were plotted using R packages VennDiagram (Fig. 6B; Chen and Boutros 2011) and ComplexUpset (Fig. 6C).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE255520. The analyzed transcript isoform data generated in this study have been submitted to the UCSC Genome Browser (https://genome.ucsc.edu/s/wmeng1018/SC_transcript_isoform_mouseRetina). The code and scripts used in this study are available at GitHub (https://github.com/RCHENLAB/LR_scRNA-seq_manuscript) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Rui Chen Lab at the Baylor College of Medicine and was funded by the Chan Zuckerberg Initiative (CZF2019-002425). We acknowledge support to the Gavin Herbert Eye Institute at the University of California, Irvine from an unrestricted grant from Research to Prevent Blindness and from National Institutes of Health core grant P30 EY034070. We thank eyeGENE for providing patient samples collected at the National Eye Institute. We acknowledge Dr. Salma Ferdous for her contribution to improving the proofreading and English language in our manuscript.
Author contributions: M.W., R.C., and Y.L. conceived the study design. Y.L. collected mouse samples and performed experiments and sequencing. M.W. developed the pipeline, conducted data analysis, and wrote the manuscript while J.W. and R.C. provided feedback. S.H.O. provided helpful comments and discussion. Y.C. performed the experiment on long-read bulk RNA-seq. R.C. planned and supervised the research and wrote the manuscript. All authors provided critical feedback and read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279167.124.
- Received February 20, 2024.
- Accepted November 21, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








