
Multisite long-read sequencing reveals the early contributions of somatic structural variations to HBV-related hepatocellular carcinoma tumorigenesis
- Tianfu Zeng1,3,
- Haotian Liao2,3,
- Lin Xia1,3,
- Siyao You1,
- Yanqun Huang1,
- Jiaxun Zhang1,
- Yahui Liu1,
- Xuyan Liu1 and
- Dan Xie1
- 1Laboratory of Omics Technology and Bioinformatics, Frontiers Science Center for Disease-related Molecular Network, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China;
- 2Division of Liver Surgery, Department of General Surgery and Laboratory of Liver Surgery, and State Key Laboratory of Biotherapy and Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China
-
↵3 These authors equally contributed to this work.
Abstract
Somatic structural variations (SVs) represent a critical category of genomic mutations in hepatocellular carcinoma (HCC). However, the accurate identification of somatic SVs using short-read high-throughput sequencing is challenging. Here, we applied long-read nanopore sequencing and multisite sampling in a cohort of 42 samples from five patients. We found that adjacent nontumor tissue is not entirely normal, as significant somatic SV alterations were detected in these nontumor genomes. The adjacent nontumor tissue is highly similar to tumor tissue in terms of somatic SVs but differs in somatic single-nucleotide variants and copy number variations. The types of SVs in adjacent nontumor and tumor tissue are markedly different, with somatic insertions and deletions identified as early genomic events associated with HCC. Notably, hepatitis B virus (HBV) DNA integration frequently results in the generation of somatic SVs, particularly inducing interchromosomal translocations (TRAs). Although HBV DNA integration into the liver genome occurs randomly, multisite shared HBV-induced SVs are early driving events in the pathogenesis of HCC. Long-read RNA sequencing reveals that some HBV-induced SVs impact cancer-associated genes, with TRAs being capable of inducing the formation of fusion genes. These findings enhance our understanding of somatic SVs in HCC and their role in early tumorigenesis.
As the most prevalent form of primary liver cancer, hepatocellular carcinoma (HCC), which mostly forms after years of chronic liver disease, against a background of severe liver scarring and typically cirrhosis, has been ranked as one of the leading causes of cancer-related death worldwide (Villanueva 2019; Müller et al. 2020). More than half of HCC cases globally occur in China, where chronic hepatitis B virus (HBV) infection is the major etiological factor, accounting for over 60% of cases (Llovet et al. 2021; Bray et al. 2024; Chen et al. 2024a). Somatic DNA alterations are known as pivotal drivers of HCC tumorigenesis and progression. Mutations in the TERT promoter are the most prevalent (∼60%) genetic alterations in HCC (Schulze et al. 2016), and the TERT promoter is a recurrent insertion site of HBV DNA. Other recurrently mutated genes primarily harbor somatic single-nucleotide variants (SNVs) within their coding regions, including TP53 (∼30%), CTNNB1 (∼30%), and ARID1A (∼10%), which affect the cell cycle, WNT signaling, or chromatin remodeling (Khemlina et al. 2017). Nevertheless, these most prevalent mutations remain undruggable at present (Zucman-Rossi et al. 2015), and there are no mutations available in clinical practice to predict therapeutic response, highlighting the incomplete understanding of the mutational landscape of HCC.
Structural variants (SVs) are large genomic alterations (>50 bp), distinct from small variants like SNVs and short insertions and deletions (indels), as they often arise from different mechanisms (Abyzov et al. 2015). SVs are generally defined as insertions (INSs), deletions (DELs), duplications (DUPs), inversions (INVs), and translocations (TRAs), and are known to play an important role in cancer pathogenesis (George et al. 2015; Waddell et al. 2015). Over the past decade, utilizing short-read high-throughput sequencing (HTS) technologies, studies have revealed that somatic SVs can drive malignant phenotypes by altering the expression or function of oncogenes (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020; Cosenza et al. 2022). Additionally, certain somatic SVs within tumors have been used to make therapeutic decisions, such as the structural rearrangements involving the ALK gene in nonsmall cell lung cancer and the TRA leading to BCR-ABL1 fusion in chronic myeloid leukemia (Soda et al. 2007; Hochhaus et al. 2017a,b). However, limited by the short read lengths of HTS, the current understanding of somatic SVs in HCC is still incomplete.
Long-read sequencing (LRS) technology, represented by Oxford Nanopore Technologies (ONT) and Pacific Bioscience (PacBio), can produce continuous reads longer than 10,000 bp. These reads can completely span across genomic repetitive segments and complex genomic variant regions. Thus, LRS is progressively being applied to studies on genomic SVs in tumors. For instance, utilizing nanopore sequencing, a complex SV named “cancerous local copy-number lesions” (CLCLs) has been identified within lung cancer, and the precise junctions of these complex SVs are difficult to identify using short-read sequencing (Sakamoto et al. 2020). Although previous studies have revealed distinct mechanisms in the generation of germline and somatic SVs in liver cancer by analyzing complete breakpoint sequences derived from long reads, the genome-wide landscape of somatic SV characteristics within HCC has yet to be fully elucidated (Fujimoto et al. 2021). A recent study revealed notable occurrences of somatic SVs within cirrhotic tissues (Brunner et al. 2019). Given that cirrhosis from any etiology is the strongest risk factor for HCC, this further underscores the importance of deciphering somatic SVs in elucidating the pathogenesis of HCC.
Results
Detection of somatic SVs using LRS
We performed a multisite sampling of tumor tissues and adjacent nontumor tissues from five patients diagnosed with HBV-positive HCC. The nontumor tissues were categorized as proximal nontumor (located within proximal 1 cm of the tumor) and distal nontumor (>1 cm distal to the tumor), with both exhibiting fibrosis. Blood samples collected before surgical resection were used as matched normal controls. In total, 27 tumor tissue samples, 10 matched nontumor liver samples (comprising six proximal and four distal), and five matched blood samples were obtained from the five patients (Fig. 1A). Detailed clinical information of included patients is presented in Supplemental Table S1.
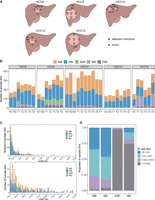
Identification of somatic SVs through multiregion LRS. (A) Schematic diagram showing the sampling locations for five patients. Blue dots represent adjacent nontumor tissues. Red dots represent tumor tissues. (B) Bar plots depicting the number of somatic SVs across individual samples, including INSs, DELs, DUPs, INVs, and TRAs. (C) Stacked plots showing the distribution of somatic SV lengths are categorized into two ranges: (50 bp, 1 kb) and (1 kb, 10 kb), stratified based on SV types. (D) The proportion of different SV size ranges within each somatic SV type.
We generated long-read whole-genome sequencing data from all samples using the PromethION (ONT) platform. To obtain high-quality sequencing data, we excluded reads with lengths <1 kb and mean base quality lower than 7. The mean N50 of long reads was 22.41 kb (19.22 kb ∼ 31.02 kb) after quality control (Supplemental Fig. S1A; Supplemental Table S2). The average sequencing depth reached 36.72 × (30.50× ∼ 44.22×) for tumor and nontumor tissues, 22.19× (19.84× ∼ 25.34×) for blood samples (Supplemental Fig. S1B). Considering the relatively high single-base error rate of LRS, short-read whole-genome sequencing using the Illumina NGS platform was additionally conducted on all samples to facilitate the detection of somatic SNVs.
Utilizing LRS, we developed a rigorous in-house bioinformatics pipeline to identify somatic SVs and HBV DNA integration events, facilitating the characterization of SV types associated with HBV integration (Supplemental Fig. S1C). Clean long reads were aligned against a custom reference genome consisting of the human genome and 21 HBV genomes using minimap2. Subsequently, the alignment results from paired samples were merged. The widely used variant caller Sniffles2 was used to cluster the long reads supporting the same SV. In instances where supporting reads for SVs span both the HBV and human genomes in alignments, SV types were re-inferred based on the alignment patterns of human sequences flanking the breakpoints. After conducting quality control and manual inspection, we identified an average of 253 somatic SVs across all samples (range from 122 to 405, a median of 252, Fig. 1B). This count notably exceeds previous reports based on short-read sequencing, where only ∼20% of HCC cases displayed somatic SV counts >200 (Fujimoto et al. 2016; Li et al. 2020b). These results underscore the superior sensitivity of LRS in the detection of SVs (Aganezov et al. 2020; Xu et al. 2023).
The number of somatic SVs rapidly decreases as the SV length increases, consistent with previous studies (Beyter et al. 2021). We identified two distinct peaks, located at ∼170 bp and 320 bp (Fig. 1C). The predominant types of SVs at these peaks were INS and DEL, accounting for 98.47% and 99.17%, respectively. To further characterize these sequences, we performed sequence annotation using RepeatMasker. The SVs ∼170 bp were predominantly composed of satellite elements (Supplemental Fig. S2A), which are involved in the maintenance of chromosomal integrity and have been found to be overexpressed in various cancer types (Ting et al. 2011; Ho et al. 2017). In contrast, the sequences near 320 bp are primarily composed of SINEs (Supplemental Fig. S2B), which are associated with SV mechanisms associated with the mobilization of active transposable elements (Kolomietz et al. 2002; Robberecht et al. 2013). Notably, somatic INSs and DELs were significantly shorter in length compared to somatic DUPs and INVs, with the majority of INVs and DUPs exceeding 10,000 bp (Wilcoxon rank-sum test, P-value < 2.2 × 10−16, Fig. 1D).
The mean variant allele frequency (VAF) of somatic SVs across all samples was 0.14. Notably, the VAF of somatic SVs in the adjacent nontumor samples from each patient was significantly lower than that in the tumor samples (Supplemental Fig. S3A), a phenomenon also observed in somatic SNVs in this project (Supplemental Fig. S3B) and prior studies (Huang et al. 2017; Strandgaard et al. 2020). This indicates that the fibrotic liver tissue adjacent to the tumor does not reflect a normal genomic state and has undergone an accumulation of low-frequency somatic mutations.
Somatic SVs are prominent in adjacent nontumor tissues
A recent study revealed that cirrhotic livers exhibit a higher mutation burden compared to normal livers, with the most significant divergence observed in SVs (Brunner et al. 2019). To explore the process of somatic mutation accumulation in HCC, we compared the burden of mutation between adjacent nontumor tissues and tumor tissues. The genome was segmented into 500 bp bins, and the number of bins harboring each mutation type was determined. We observed a significantly lower count of somatic SNVs in the adjacent nontumor liver samples compared to the tumor samples (Wilcoxon rank-sum test, P-value = 5.7 × 10−9, Fig. 2A). Besides, there were also fewer somatic CNV events in the adjacent nontumor liver samples, with 50% of these samples exhibiting no CNVs. However, somatic SVs were prominent in adjacent nontumor tissues, with no significant difference observed when compared to tumor tissues (Fig. 2A). These results indicate that somatic SVs follow a distinct development process compared to somatic CNVs and somatic SNVs during the progression of HCC.
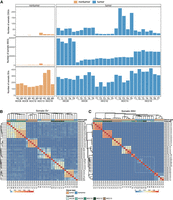
Somatic SVs are prominent in adjacent nontumors. (A) Bar plots showing the number of somatic mutations (classified into SVs, SNVs, and CNVs) in adjacent nontumor and tumor tissues from all patients. (B) Heatmap depicting the hierarchical clustering of 37 tumor and adjacent nontumor samples based on Jaccard indexes calculated from somatic SVs. (C) Heatmap depicting the hierarchical clustering of 37 tumor and adjacent nontumor samples based on Jaccard indexes calculated from somatic SNVs. The white dashed box outlines a cluster consisting of adjacent nontumor samples.
To compare the mutation profiles across different sampling sites, hierarchical clustering of all samples was performed on mutation matrices of 500 bp for both somatic SNVs and SVs across all samples. The clustering analysis of somatic SNVs revealed that tumor samples grouped according to their patient origin, whereas nontumor samples formed a distinct outgroup, separate from the tumor cohort (Fig. 2B). This separation likely arises from the significantly lower number of somatic SNVs in adjacent nontumor tissues compared to tumor tissues (an average of 851 vs. 24,234), leading to their clustering based on their mutual dissimilarity to the tumors. In contrast, clustering analysis based on somatic SVs revealed a distinct pattern, with somatic SVs clustering according to patients, and both tumor and nontumor samples clustered together within individual (Fig. 2B). Due to the absence of somatic CNVs in half of the adjacent nontumor samples, we directly compared the mutational profiles of somatic CNVs across different sampling sites within the same patient (Supplemental Fig. S4A–E). Notably, there was a high degree of consistency in CNV changes among various tumor sampling sites. For instance, all five tumor sampling sites from HCC8 exhibited amplification of the entire Chromosome 2 and 5, despite the absence of CNV alterations in the adjacent nontumor samples (Supplemental Fig. S4A). These findings underscore the similarity of nontumor tissues to tumors in terms of somatic SVs, while highlighting a marked distinction in terms of somatic CNVs and SNVs.
To further examine the similarities in mutations among samples in detail, we performed pairwise comparisons across different sampling sites (Supplemental Fig. S5A–E). The proportion of shared mutations between adjacent nontumor and tumor tissues exhibited significant differences among the categories of somatic SVs, SNVs, and CNVs. Specifically, in both nontumor and tumor samples, the proportion of shared somatic SVs between adjacent nontumor and tumor tissues was significantly higher than that of somatic SNVs (P-value = 5.3 × 10−8 and 2.2 × 10−16, t-test, Supplemental Fig. S6A). Notably, ∼12.98% of somatic SVs detected in the tumors were also present in the adjacent nontumor tissues, whereas somatic SNVs accounted for only 0.45%. There were no shared somatic CNVs between adjacent nontumor and tumor tissues. This observation aligns with the current understanding of tumor evolution, indicating that the acquisition of CNVs occurs late in clonal expansion (Li et al. 2020a), representing a consequence of the final malignant transformation of genomic stability.
To further understand tumor evolution, we constructed phylogenetic trees for each patient based on the mutation matrix of 500 bp windows, and branch evolution patterns correlated with tumor heterogeneity were observed in all patients (Supplemental Fig. S7A). Notable branches shared between tumor and adjacent nontumor tissues were observed on the phylogenetic tree based on somatic SVs, whereas no such pattern was evident in the case of somatic SNVs (Supplemental Fig. S7B). These results suggest that somatic SVs may represent the primary mutation type in the context of chronic liver disease during the early stages of HCC progression.
HCC is characterized by somatic INS and DEL at an early stage
The adjacent nontumor tissue in HCC is not entirely normal (Carlessi et al. 2023; Chen et al. 2024b). A transcriptomic study examining HCC alongside seven other tumor types has demonstrated that adjacent nontumor tissues exhibit a unique intermediate state between health and tumor (Aran et al. 2017). To investigate the role of shared SVs between adjacent nontumor and tumor tissues in HCC pathogenesis, we categorized somatic SVs into three distinct categories within individual: shared (present in both adjacent nontumor and tumor tissues), T-specific (exclusive to tumor tissues), and N-specific (exclusive to adjacent nontumor tissues). For each patient, we found that the shared somatic SV categories exclusively comprised DELs and INSs, whereas TRAs were unique to the T-specific category (Fig. 3A). Although DUPs and INVs were present in both T-specific and N-specific categories, the relative proportions varied significantly among patients. For instance, HCC9 exhibited a high proportion of DUPs, whereas HCC13 had a higher proportion of INVs. Notably, although HCC9 lacked DUPs in the adjacent nontumor tissue, there was a high consistency among DUPs across different tumor sites, with ∼68.46% being mutually shared among samples (Supplemental Fig. S8A). In contrast, although INVs were present in the adjacent nontumor tissue of HCC13, no shared INVs were identified between the tumor and adjacent tissues (Supplemental Fig. S8B). The accumulation of INVs in the tumor of HCC13 was localized to Chromosome 9, suggesting that this chromosome may have experienced a catastrophic event, such as chromothripsis (Supplemental Fig. S8C). Moreover, the percentage of DUPs, INVs, and TRAs within the T-specific category exhibited a significantly higher prevalence than those in the N-specific category (chi-square test, P-value < 0.01). These specific types of SVs are frequently associated with chromosomal rearrangements in tumor evolution. These results indicate a significant difference in the types of SVs between adjacent nontumor tissues and tumors, suggesting that the tumor genome has undergone more complex genomic alterations.
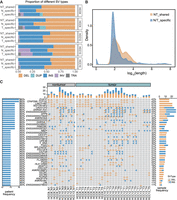
The characteristics of shared somatic SVs between adjacent nontumor and tumor tissues. (A) The frequency of each somatic SV type in different categories was shown: shared (in both adjacent nontumor and tumor tissues), T-specific (only in tumor tissues), and N-specific (only in adjacent nontumor tissues). (B) The length distribution of shared somatic SVs between adjacent nontumor and tumor tissues, as well as the SVs unique to either adjacent nontumor or tumor tissues. (C) An overview of genes overlapped by recurrently shared somatic SVs in adjacent nontumor and tumor tissues across patients. The left bar chart shows the mutation percentage across patients. The right bar chart displays the mutation percentage across samples.
We further investigated the differences in somatic INSs and DELs between the shared and N/T-specific categories. Notably, the lengths of somatic INSs and DELs shared between adjacent nontumor and tumor samples were significantly greater than those exclusive to either category (Fig. 3B, Wilcoxon rank-sum test, P < 2.2 × 10−16). Additionally, the shared somatic INSs and DELs exhibited a higher prevalence of complex repetitive sequences and satellite elements, whereas nonrepetitive and simple repeat sequences were less represented (Supplemental Fig. S9A,B).
Next, we analyzed the landscape of shared somatic SVs between tumor and nontumor samples within individual. Notably, we identified a subset of recurrent somatic SVs, present in two or more patients, affecting a total of 42 genes (Fig. 3C). These include tumor-related genes such as PGA5 and DPP6 (Shen et al. 2020; Choy et al. 2021; Munkhjargal et al. 2023), as well as newly identified candidate genes like EVA1C, which is present in four out of five patients. EVA1C is a membrane protein-encoding gene that plays a role in inflammatory and immunobiological processes (Hu and Qu 2021). The SV observed on the EVA1C gene manifests as a DEL event spanning ∼160 bp in length. Moreover, this DEL is consistently located in close proximity across different samples, with breakpoint variations confined within 10 bp. Short-read sequencing in the samples failed to detect this DEL, primarily due to the tandem-repeated Alu sequences present within the corresponding region of the reference genome (Supplemental Fig. S10A,B).
To assess the functional impact of these recurrent somatic SVs, we conducted long-read RNA sequencing (RNA-seq) on samples HCC10_T7 and HCC10_N1 (Supplemental Table S3). Transcript loss was observed for EVA1C, GSTM1, and GSM2 in samples harboring DELs (Supplemental Fig. 11A,B), despite the DEL in EVA1C localized exclusively to an intronic region. Additionally, qPCR validation revealed that the expression level of the EVA1C gene in samples with DEL was significantly lower than in samples without DEL (Supplemental Fig. S11C). Furthermore, based on gene expression and clinical data obtained from the KM plotter database (Menyhart et al. 2018), survival analysis showed that the low expression of EVA1C was associated with poor survival of patients (Supplemental Fig. S11D, P = 0.0055). These findings indicate that somatic SVs shared between tumors and adjacent nontumor tissues have the potential to induce abnormal expression of genes associated with cancer.
HBV integrations induce the formation of SVs
Viral DNA integration into the host genome can lead to the malignant transformation of tumors, resulting in an increased burden of mutations near the integration breakpoints (Zapatka et al. 2020). HBV infection is frequently linked to proliferative HCC, which is characterized by chromosomal instability (Villanueva 2019). To further explore the connection between somatic SVs and HBV integrations, we developed a novel pipeline to comprehensively identify HBV integration events using LRS. We primarily identified two categories of long reads for detecting HBV integration events. First, chimeric reads that mapped simultaneously to the human genome and the HBV genome. Secondly, due to disparities in minimap2 alignment, certain HBV sequences were either mapped as INSs or remained unmapped due to clipping (Supplemental Fig. S12A). Basing on these two kinds of reads, we identified an average of 10 HBV integration breakpoints in the tumor samples (range from 0 to 37, median: 8) and five in the adjacent nontumor samples (range from 0 to 15, median: 4), which was significantly more than previous short-read data sets (Sung et al. 2012; Álvarez et al. 2021). Furthermore, 61.38% of HBV DNA integrated into the human genome exceeded 500 bp in length. (Supplemental Fig. S12B). In short-read sequencing, aligning these insert sequences larger than the DNA library fragment size is challenging, and complete insert sequences cannot be detected (Craven et al. 2022; Rajaby et al. 2023).
Within the 335 identified HBV integration breakpoints, 85.67% (287/335) were derived from HBV integration events fully captured by long reads, characterized by HBV integration sequences flanked by human sequences on both sides within the same read. These HBV integration events provided an opportunity for further investigation into the specific patterns of HBV integration into the HCC genome. Based on the alignment positions, orientations, and order of human sequences flanking the HBV sequences, we inferred the integration patterns of HBV (Supplemental Fig. S12C–G). We observed that simple INS of HBV accounted for only 20.20% of events, whereas the majority of HBV integrations resulting in chromosomal rearrangements within the HCC genome. Among these genomic alterations, TRAs were the predominant type, constituting 44.80%, followed by INVs (20.48%), DUPs (8.62%), DELs (4.12%), and complex SVs (1.78%). Notably, the complex SVs identified were all DUP-INV events, originating from five distinct tumor sampling sites in HCC9. These rearrangements were characterized by HBV integration resulting in a fold-back INV, with human sequences on both sides of the integration site being inverted and duplicated (Fig. 4A). Moreover, a 76.23 Mb copy number amplification (Chr 7: 124,879,000–48,644,500) was observed 779 bp downstream from the HBV integration site, which is consistent with the breakage-fusion-bridge (BFB) INV model.
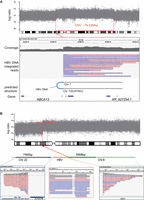
Inferring SV types based on HBV DNA integration patterns. (A) In sample HCC9_T3, an HBV DNA INS induces a fold-back INV in the host DNA. The top panel illustrates the CNV increase occurring ∼76.23 Mb downstream from the integration site. Sequencing data demonstrate that long reads encompass the sequences flanking the HBV integration, revealing identical breakpoints and reverse DUP of the adjacent sequences. (B) An example of HBV DNA integration resulting in a TRA (HCC13_T2). The Integrative Genomics Viewer (IGV; Robinson et al. 2011) plot below illustrates the alignment of long reads to Chr 22, HBV, and Chr 8. The plot above depicts the concordance of its breakpoint on Chr 8 (red dashed line) with a breakpoint of ∼5.36 Mbp CNV (gray shade).
Next, we further explored the relationship between HBV integration breakpoints and CNV boundaries. We found significant differences in the proximity of various HBV-induced SV types to CNV boundaries. INSs were located further from CNV boundaries, with all INS breakpoints situated more than 100 kb away from the CNV borders. In contrast, except for five cases of complex (DUP-INV) events, TRAs and DUPs were found closer to the CNV boundaries, with 6.4% of TRA breakpoints located within 1 kb of the CNV boundary (Supplemental Fig. S13A). For instance, within the HBV integration event observed in HCC13_T3, ∼2914 base pairs (bp) of HBV sequence integrated into the HCC genome, giving rise to a TRA event that linked Chr 8: 31,957,777 and Chr 22: 50,545,277. Notably, a CNV is located 214 bp downstream from the TRA breakpoint on Chromosome 8. This CNV corresponds to a 5.36 Mb gain on Chromosome 8p12 (Chr 8: 31,957,991–37,314,348), and this CNV region harbors several tumor-associated genes, such as NRG1, UNC5D, and KCNU1 (Fig. 4B; Repana et al. 2019). These findings indicate that HBV-induced SVs may play a role in CNV formation and directly affect critical oncogenes involved in tumorigenesis.
In addition, in comparison to a random genomic background, we found a significant enrichment of somatic SVs in proximity to HBV integration sites (two-sided binomial test, P-value < 0.01, Supplemental Fig. S13B). The above findings suggested that the HBV integration was closely related to the formation of SVs within the HCC genome.
Multisite shared HBV-induced SVs emerge as distinctive signatures of HCC
Based on the HBV-induced SVs identified above, we explored their breakpoint location characteristics within the HCC genome. Among all samples, it was observed that 56.2% of breakpoints were shared across multiple samples (Supplemental Fig. S15A). It is noteworthy that these breakpoints were shared among tumor samples, with no shared breakpoints detected in the adjacent nontumor tissues (Supplemental Fig. S15B). Specifically, we observed a pronounced aggregation of HBV-induced SV breakpoints within tumors sampled from diverse regions of the same patient, whereas HBV-induced SV breakpoints in adjacent nontumor tissues exhibited a stochastic distribution (Fig. 5A). Furthermore, annotation of the HBV genome revealed that the HBV-induced SVs shared across the five samples consistently harbored similar HBV integration sequences, with an average sequence similarity of 96.78% (Fig. 5B). These integration sequences were derived from proximal loci within the HBV genome, with an average breakpoint position difference of 35 bp (Supplemental Fig. S14A). For instance, all five tumor sampling sites in HCC9 revealed a consistent TRA linked Chr 2: 74,273,672 and Chr 6: 70,300,819, with the integrated HBV sequences derived from the HBV Hbx gene (HBV:1544-1481). These findings suggest that HBV DNA integration into the liver occurs randomly, whereas integration at specific loci may lead to hepatocellular carcinogenesis.
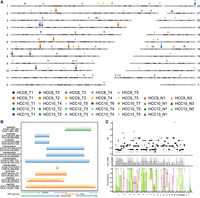
Integration breakpoints of HBV DNA in human and HBV genomes. (A) Integration breakpoints of HBV DNA within the human genome. The same color scheme represents the same patient. Circular points represent tumor samples, and triangular points represent adjacent nontumor samples. (B) A schematic diagram of shared HBV-induced SVs at all tumor sampling sites in the patients. The horizontal line represents the relative position of the integrated HBV sequence within the HBV genome. (C) In HCC13, the clonal status of HBV integration sites was in the context of somatic SVs (top panel), with green indicating early clonal events. The red asterisk symbol represents the shared HBV integration sites. The middle panel represents the CNV status, and the lower panel displays the inferred mutation timeline.
Given the PCR-free nature of nanopore sequencing, each read is likely to represent a distinct single-cell origin. The probability of the same HBV DNA integration event occurring independently in multiple cells is minimal. Therefore, the shared HBV-induced SVs observed across different tumor sampling sites may indicate a common clonal origin for these tumor regions. To elucidate the clonal characteristics, we examined the mutational timing of the shared HBV-induced SVs within individuals. VAF analysis revealed a significantly higher VAF of HBV-induced somatic SVs that shared within tumors compared to those appearing independently (Supplemental Fig. S15C, Wilcoxon rank-sum test, P-value = 1.8 × 10−24). To further assess the cancer cell fraction (CCF) of these shared HBV-induced SVs, we modified the existing algorithm SVclone (Cmero et al. 2020) to suit LRS data. By integrating established timing algorithms MutationTimeR (Gerstung et al. 2020), these shared HBV-induced SVs were identified as early clonal events, preceding copy number gains (Fig. 5C).
HBV-induced SVs influence genes associated with tumorigenesis
Next, we studied the potential effects of HBV-induced SVs on genes, including those genes directly intersecting with breakpoints or located closest to the breakpoints. Pathway enrichment analysis of the relevant genes revealed their significant association with key HCC pathways, such as Proteoglycans in cancer, PI3K/AKT signaling pathway, and viral carcinogenesis (Supplemental Fig. S16A, P-value < 0.01). In addition, we found that shared HBV-induced SVs within individuals impact a total of 24 genes, including genes associated with tumorigenesis, such as SLC4A5, ABC13, NRG1, and SORCS1 (Fig. 6A). In addition, compared to breakpoints occurring independently, breakpoints of shared HBV-induced SVs were significantly concentrated in gene bodies and their flanking 5 kb regions (Supplemental Fig. S16B, chi-square test, P-value = 2.8 × 10−5). This finding suggests that shared HBV-induced SVs were more likely to have an impact on gene regulation and expression. Notably, among these shared HBV-induced SVs, TRAs account for 71.43%, highlighting the evolutionary advantage of this type of SV.
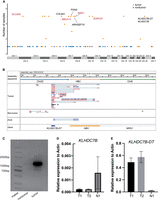
Genes disrupted by HBV-induced SVs. (A) Manhattan plot showing the genes affected by HBV-induced SVs. Tumor samples are represented by orange points, and adjacent nontumor samples are represented by blue points. Tumor genes annotated by COSMIC or NCG6 are marked in red. (B) IGV plot depicting the fusion transcript formed between the KLHDC7B-DT gene and the HBV DNA sequence. The top track represents the assembled TRA sequence, and the bottom track illustrates gene annotations for the TRA sequence. The middle tracks include long-read RNA-seq from both tumor and adjacent nontumor samples, and Sanger sequencing reads for the fusion transcript PCR product. (C) The PCR products of fusion transcript were detected by agarose gel electrophoresis. (D) The relative expression levels of KLHDC7B determined using RT–PCR in samples with and without the HBV-induced TRA. (E) The relative expression levels of KLHDC7B-DT determined using RT–PCR in samples with and without the HBV-induced TRA.
We next investigated the functional consequences of HBV-induced TRAs on genes located near the integration breakpoints. Among these genes, NRG1, KLHDC7B, and KLHDC7B-DT, which have been implicated in various cancers (Maillet et al. 2018; Li et al. 2021; Yahiro et al. 2021), were directly affected by the TRAs. In HCC13, a shared HBV-induced TRA was detected across five sampling sites, with breakpoints located within the intronic region between the first and second exons of NRG1, as well as upstream of KLHDC7B (620 bp) and KLHDC7B-DT (2359 bp). Although several previous studies have suggested their relevance to cancer (Martin-Pardillos and Cajal 2019; Rosas et al. 2021), mutations within these genes have not been identified in the context of HCC. Thus, we further investigated the impact of the HBV-induced TRA on the expression of genes situated on either side. Long-read RNA-seq was performed on both the HCC13_T1 tumor sample and its corresponding adjacent nontumor tissue. To obtain high-quality sequences, we conducted local assembly of the TRA using reads supporting this integration events. By aligning RNA reads spanning the integration breakpoints to the assembled TRA sequence, we identified a fusion gene between the KLHDC7B-DT gene and the inserted HBV sequence in the tumor sample. However, no evidence of this fusion gene was detected in the adjacent nontumor sample (Fig. 6B). Subsequent PCR validation confirmed the presence of this fusion gene in the tumor sample and its absence in the adjacent nontumor sample (Fig. 6C). Additionally, quantitative PCR (qPCR) revealed that the expression levels of KLHDC7B in samples with HBV integration were significantly reduced, whereas KLHDC7B-DT expression was markedly increased (Fig. 6D,E). These findings suggest that HBV integration may disrupt the promoter of KLHDC7B, leading to decreased expression of this gene. Furthermore, qPCR demonstrated that HBV integration led to reduced expression of NRG1 in the tumor (Supplemental Fig. S16C), potentially attributed to the integration disrupting the structural integrity of the gene. Overall, these findings indicate that HBV-induced TRA can directly impact genes adjacent to integration sites, including the formation of fusion genes and disruption of gene expression.
Discussion
Somatic SVs play a pivotal role in HCC, yet their accurate identification using short-read HTS poses challenges. To overcome limitations related to intratumor heterogeneity (ITH) and refine the estimation of the mutational landscape, we employed multisite sampling in tandem with long-read nanopore sequencing for the comprehensive genome-wide analysis of somatic SVs. We depicted the characteristics of somatic SVs in both adjacent nontumor and tumor tissues, revealing somatic SVs as early genomic alterations that may contribute to the complexity of HCC development. Furthermore, we elucidated that HBV integration serves as a frequent driver of somatic SVs, particularly in the context of interchromosomal TRAs. Our findings indicated that shared HBV-induced SVs represent early clonal events in the progression of HCC, with the potential to directly impact the transcription of genes relevant to tumorigenesis.
Building on these findings, it is important to note that although our study provides compelling insights into the role of somatic SVs in HCC, some of the results require additional validation to establish their broader significance. Specifically, we observed that the proportion of somatic DUPs, INVs, and TRAs was significantly higher in patients with a history of smoking compared to nonsmokers. However, this observation warrants further validation through the inclusion of additional patient samples to improve statistical robustness. Utilizing multiple tumor sampling sites, we identified that HCC13 exhibits the highest number of somatic INVs in tumor sites T1, T2, and T5, which markedly differ from those at T3 and T4. The morphological characteristics of HCC13 indicate that T1, T2, and T5 are spatially distinct from T3 and T4, suggesting that the differing INV patterns may reflect their clonal origins. This highlights the potential of somatic SVs as biomarkers for pathological subtyping, offering insights into the clonal evolution of tumors and their underlying mechanisms. Further investigation into these patterns could enhance our understanding of tumor heterogeneity and its implications for personalized therapeutic strategies.
Recent studies have shown that adjacent nontumor tissues are not healthy and may harbor molecular features associated with tumors (Carlessi et al. 2023; Zhu et al. 2023; Chen et al. 2024b). A recent study has found that the complexity of HCC arises during the progression to chronic liver disease and subsequent malignant transformation (Brunner et al. 2019). However, to our knowledge, the association of genomic mutation features between adjacent nontumor and tumor tissues remains to be further elucidated. In this study, we observed a comparable prevalence of somatic SVs in adjacent nontumor samples and tumor samples, contrasting with the distinct patterns observed for somatic SNVs and CNVs. This could be attributed to the instability of the host genome caused by HBV DNA integration (Zhao et al. 2016). The number of somatic SNVs and somatic CNVs in adjacent nontumor tissues was significantly lower than that observed in tumor tissues, suggesting that these alterations may share similar developmental processes in HCC (Huang et al. 2017). Notably, the differences in somatic CNVs observed between HCC and adjacent nontumor tissues are consistent with findings from a study on urothelial cell carcinoma (Li et al. 2020a), suggesting that this may represent a general feature of tumor evolution.
Furthermore, a considerable proportion of somatic SVs were found to be shared between the adjacent nontumor and tumor tissues. INSs and DELs have emerged as shared SV types between tumor and adjacent nontumor tissues, potentially exerting a direct influence on genes integral to normal hepatic function, including TCP10L and EVA1C. A possible explanation is that these somatic SVs may arise from premalignant intermediate state cells or tumor cells within the adjacent nontumor tissues (Aran et al. 2017; Carlessi et al. 2023). Additionally, a significant proportion of the INSs and DELs consisted of transposons and other repetitive sequences, indicating that these elements may play an important role in tumorigenesis. The challenge of accurately mapping such repetitive sequences using short-read sequencing likely accounts for the increased detection of somatic SVs in our study. Given that these repetitive sequences are often refractory to accurate mapping using short-read sequencing, this could explain the higher number of somatic SVs detected in our study. However, the functional implications of these shared somatic SVs between adjacent nontumor and tumor tissues require further investigation, particularly from the perspectives of transcriptomics and epigenetics.
The integration of the HBV was considered a major factor driving the development of HCC. Studies had shown that HBV integration could lead to an increased burden of mutations in the genomic region within tens to hundreds of kilobases around the integration sites (Zapatka et al. 2020). However, the specific characteristics of HBV integration patterns were not yet clearly understood. Taking advantage of LRS, we could detect the complete sequence of HBV integration. Our findings indicated that HBV integration demonstrated a propensity toward generating somatic SVs linked to chromosomal rearrangements, particularly interchromosomal TRAs, whereas simple INSs made up only ∼20.20% of the occurrences. Notably, shared HBV-induced SVs emerged as distinctive signatures of HCC, exhibiting a clustering tendency proximal to various cancer-associated genes. However, we did not detect HBV integration in the promoter region of TERT, despite previous studies reporting its presence in ∼30% of HCC cases (Chen et al. 2024a). This discrepancy may stem from limitations in our sample size or sequencing depth. In comparison to adjacent nontumor tissues, HBV DNA integration in HCC leads to a higher incidence of gene fusion events (Zhuo et al. 2021). Using long-read RNA-seq, we confirmed the formation of fusion transcripts between HBV and human genes. Previous studies have suggested that HBV DNA methylation could influence the expression patterns of viral genes (Fernandez et al. 2009; Mirabello et al. 2012). However, we did not observe significant differences between adjacent nontumor and tumor tissues in the methylation status of integrated HBV DNA sequences. This highlights the need for more advanced techniques, such as Cas9-targeted nanopore sequencing (Goldsmith et al. 2021), to enhance the detection sensitivity of HBV DNA and facilitate a deeper exploration of its potential epigenetic regulation.
In summary, our comprehensive analysis of somatic SVs in HCC, including those induced by HBV integration, shed light on the early genomic alterations and clonal events contributing to HCC development. The intricate relationship between viral integration and somatic SVs emphasizes the importance of considering these factors in understanding the molecular landscape of HCC. This study extends our understanding of somatic SVs in HCC, providing valuable insights into their characteristics and significance in early tumorigenesis.
Methods
Long-read whole-genome sequencing
Tumor, nontumor, and blood samples from each patient were frozen in liquid nitrogen for DNA isolation. Genomic DNA from each sample was extracted with the MagAttract HMW DNA Kit (QIAGEN). DNA libraries were constructed using the SQK-LSK109 library preparation kit (ONT, UK) following the manufacturer's instructions. Then, the prepared libraries were sequenced on the PromethION sequencer (ONT, UK). Raw sequencing data were basecalled using Guppy 3.2.8 with the default parameters during sequencing.
Short-read whole-genome sequencing
Genomic DNA was isolated from fresh-frozen tumor, nontumor, and blood tissues using the DNeasy Blood & Tissue Kit (QIAGEN) according to the manufacturer's protocol. The purity and integrity of DNA were assessed via agarose gel electrophoresis. The DNA concentration was measured using Qubit 2.0 (Invitrogen). The DNA was fragmented to ∼350 bp using the Covaris ultrasonicator. The library was constructed using the established Illumina paired-end protocols. Following library quality control, sequencing was performed using the Illumina NovaSeq 6000 platform (Illumina).
Identification of somatic SVs
First, clean reads were obtained by excluding those with an average base quality < 7 and a length < 1000 bp. Then, minimap2 (v.2.17) (Li 2018) was used to align the clean reads to a custom reference genome (composed of GRCh38 (hg38) and 21 HBV genomes [Yan et al. 2015; Zheng et al. 2021]), with the following parameters: minimap2 -N 10 -p 0.3 -ax map-ont –MD. To calculate somatic SVs, blood samples were used as normal controls, and the BAM files from paired tumor or adjacent nontumor samples were merged with the BAM file from the blood sample after tagging. The merged files were then processed with Sniffles (v.2.0.6) (Smolka et al. 2024) to identify SV breakpoints and discern supporting reads, with the following parameters: Sniffles -i bam_merge -v vcf –tandem-repeats -t 36 –minsupport 1 –mapq 20 –min-alignment-length 500 –minsvlen 50 –output-rnames. All SVs with supporting reads ≥3 and a length ≥ 50 bp were identified. If all supporting reads originated from tumor or adjacent nontumor samples, and none from the normal control, the SV was categorized as a candidate somatic SV.
Next, we rigorously filtered the candidate somatic SVs. ONT sequencing data from the liver tissues of five healthy individuals (Pascarella et al. 2022), along with data from 63 healthy individuals from the HPRC cohort (Liao et al. 2023), were obtained (Supplemental Table S4). Together with the five blood samples from this study, SVs were identified using Sniffles with the following parameters: Sniffles -i bam_merge -v vcf –tandem-repeats -t 36 –minsupport 3 –mapq 20 –min-alignment-length 500 –minsvlen 50. Additionally, SVs derived from a cohort of 405 individuals from the Chinese population (Wu et al. 2021), also sequenced via nanopore technology, as well as SVs from the HGSVC database (Ebert et al. 2021), were incorporated. These SVs were used to construct a panel of normals (PON), which served to filter out false positives in somatic SV calls. Such false positives could arise due to insufficient sequencing depth in paired normal samples, biases associated with ONT technology, or tissue-specific SVs in normal liver tissue. Any candidate somatic SVs present in the PON were excluded. We also removed SVs occurring in unreliable alignment regions, including those where over 50% of reads in the SV region had mapping quality < 20, and SVs with VAF below 0.01. Furthermore, using GenomeView (Abeel et al. 2012) to visualize SVs, we manually inspected all candidate somatic SVs. Following the methodology employed in a previous study (Fujimoto et al. 2021), candidate somatic SVs displaying matching breakpoints in corresponding normal samples were discarded from further analysis.
VCF merging of SVs and identification of shared somatic SVs
The Jasmine (v1.1.5) software was used to merge the VCF files of somatic SVs across different samples, incorporating PON filtering and identification of shared SVs. This tool is specifically designed for SV detection from LRS, considering factors such as the type, length, breakpoint positions, and sequence context of the SVs. Jasmine compares and merges SV calls across samples by representing variants as points in a multidimensional space and constructing a proximity graph based on their breakpoints and lengths. We employed Jasmine with the following parameters: jasmine file_list out_file –ignore_strand. The default parameters in Jasmine merge SVs of the same type if they are within 100 bp of each other and have length differences of <50%. SVs that could be merged are considered as shared SVs.
Detection of HBV integrations and HBV-induced SVs
Although the alignment results of long reads against the customized reference genome already contain partial information concerning HBV integration, notably the presence of chimeric reads aligning to both HBV and human genomes, our examination revealed that certain HBV DNA sequences present on some long reads failed to align to the 21 HBV reference genomes. These HBV sequences either received low alignment scores in minimap2, resulting in being labeled as INS, or were too short and directly clipped without alignment. Based on these characteristics of reads, we developed a workflow for detecting HBV integration using LRS, mainly consisting of the following steps: (1) extraction of chimeric reads, (2) extraction of INS sequences and clipped sequences from all long reads, (3) realignment of INS and clipped sequences to the custom reference genome using BLAST, and (4) determination of HBV DNA integration breakpoints.
Benefiting from the advantages of LRS, long reads can simultaneously contain HBV DNA sequences and their adjacent human sequences. We utilized these spanning reads to further deduce the structural consequences of HBV DNA integration on the human genome. Based on the alignment direction, position, and order of human sequences flanking HBV DNA, five types of HBV-induced SVs were identified. INS denotes direct INS of HBV DNA into the human genome without altering the flanking human sequences. DEL corresponds to the loss of human DNA segments caused by HBV DNA integration. DUP refers to the presence of duplicated human DNA segments flanking the HBV DNA integration breakpoint. INV occurs when the human DNA adjacent to one side of the HBV DNA integration breakpoint is reversed. TRA involves human sequences flanking HBV DNA originating from different chromosomes.
Identification of somatic SNVs and CNVs
The clean reads were aligned to the human reference genome GRCh38 (hg38) using Burrows–Wheeler Aligner (v.0.7.17) (Li 2013), with the following parameters: BWA-MEM -M -Y -t 38 -R. SAMtools (v.1.9) (Li et al. 2009) was employed for sorting and indexing the resulting BAM files. PCR duplicate reads were marked using the MarkDuplicates module of the Genome Analysis Toolkit (GATK, v.4.1.2.0) (McKenna et al. 2010). Local realignment and base quality recalibration were conducted using the GATK BaseRecalibrator and ApplyBQSR modules.
Somatic SNVs were called from paired tumor and matched blood samples using GATK Mutect2, followed by variant filtering using FilterMutectCalls. Next, annotation of the filtered somatic SNVs was performed via ANNOVAR (Wang et al. 2010). To ensure high-quality somatic SNVs, the following quality control criteria were applied: (1) the variants supported by at least 7% of total reads in tumor sample and <2% of total reads in normal sample were retained. (2) The variants with sequencing depths <8 in tumor sample or <6 in normal sample were excluded. (3) Only variants with more than three mutation reads in the tumor were considered. Additionally, potential germline variants were filtered out by excluding variants present in the 1000 Genomes Project database with a minor allele frequency ≥0.001.
Somatic CNVs were detected in paired tumor and normal samples using FACETS (v0.6.2) (Shen and Seshan 2016), alongside evaluation of tumor purity and ploidy. The analysis was performed with the following parameters: cnv_facets -t tumor_bam -n normal_bam -vcf snps –snp-nprocs 30 –depth 15 4000 –cval 25 400 –nbhd-snp 500. Copy numbers characterized by a total copy number of 2 and a lesser copy number of 1 were subsequently filtered out, as this is consistent with the normal diploid copy number state. To focus on large-scale CNV alterations, any CNV smaller than 100 kb in length was removed from the analysis.
Phylogenetic tree construction and clustering analysis
The genome was divided into 500 bp bins, and binary presence/absence matrices were constructed based on SV breakpoints and SNVs. A distance-based neighbor-joining method was then applied to construct the phylogenetic trees. The resulting phylogenetic trees were subsequently visualized using the ggtree (Yu et al. 2018) package.
For clustering analysis, Jaccard indices were computed pairwise between samples. The resulting matrix of Jaccard indices was subjected to hierarchical clustering, using the pheatmap (v1.0.12) package in R (R Core Team 2023) with the “Pearson's correlation” parameter.
Timing of somatic SVs
SVclone (v1.1.2) (Cmero et al. 2020) is a computational tool designed to infer the CCF of SV breakpoints based on short-read sequencing data. To adapt SVclone for SVs detected from nanopore sequencing, several modifications were implemented: (1) SV supporting reads (including split and spanning reads), along with normal reads crossing the SV breakpoints, were utilized to calculate the variant allele fraction (VAF) of SV breakpoints. (2) As each long read in nanopore sequencing represents a single DNA molecule and does not overestimate the number of normal reads in cases of DNA gain, the step of adjusting the number of normal reads was omitted. By integrating somatic SNVs, somatic CNVs, tumor purity, and ploidy, SVclone performed CCF estimation and clustering using a coclustering mode. Next, clonality of somatic SVs was determined based on the somatic CNVs and SVclone clusters using the timing analysis algorithm MutationTimeR (Gerstung et al. 2020). This step assigned somatic SVs to four different timing categories, include early clonal, clonal not specified, late clonal, and subclonal.
Long-read RNA-seq
RNA from tumor and nontumor tissues was extracted using TRIzol Reagent (Invitrogen) according to the standard instructions. The RNA concentration was determined using Qubit RNA HS Assay Kit (Thermo Fisher Scientific), and the purity and integrity were assessed by NanoDrop One, Qubit, and agarose gel electrophoresis. The cDNA library was prepared according to the SQK-DCS109 and EXP-NBD104 protocol (ONT). Then, the cDNA library was sequenced on a PromethION machine (ONT). Basecalling from raw data was performed in batches using Guppy software 3.2.8.
PCR validation of human–HBV fusion expression
Primers were designed to target the KLHDC7B-DT gene and HBV DNA sequences identified in the long-read RNA-seq (Supplemental Table S5). A total of 35 ng SMART cDNA was mixed with 1 μL of forward primer, 1 μL of reverse primer, 12.5 μL of 2 × Taq Master Mix (Vazyme), and nuclease-free water to a total volume of 25 μL. The thermal cycling protocol was as follows: 3 min at 95°C for initial denaturation, 35 cycles of 15 sec at 95°C, 15 sec at 58°C, and 4 min at 72°C, followed by 5 min at 72°C. Next, DNA was electrophoresed on a 1% agarose gel and sent to Youkang Bio for first-generation sequencing.
qPCR validation of gene expression
cDNA was prepared from 1 μg of RNA using the PrimeScript RT reagent Kit with gDNA Eraser (TAKARA) following the manufacturer's protocol. The qPCR primers were designed using Primer3Plus. Each pair of primers was designed to target exons to avoid amplification of genomic DNA. Oligos for qPCR are provided in Supplemental Table S5. For each sample, three replicates were performed using iTaq universal SYBR Green supermix (Bio-Rad) following the manufacturer's instructions. The thermal cycling protocol on the CFX96 Real-Time System (Bio-Rad) was as follows: 3 min at 95°C for initial denaturation, 41 cycles of (10 sec at 95°C, 30 sec at 60°C, and 15 sec at 72°C), followed by a melt curve from 65°C to 95°C in 0.5°C increments. Actin beta was employed as the internal control, and the Cq value was used for quantitative analysis. The experiment was repeated three times.
Data access
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center (CNCB-NGDC Members Partners 2024), China National Center for Bioinformation/Beijing Institute of Genomics, and Chinese Academy of Sciences (GSA-Human; https://ngdc.cncb.ac.cn/gsa-human/) under accession number HRA007177. All code used is open source and available at GitHub (https://github.com/tianfuzeng/ONT_SV) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Ranlei Wei from the Laboratory of Omics Technology and Bioinformatics, West China Hospital of Sichuan University, and Kefei Yuan from the West China Hospital of Sichuan University for their technical assistance. This work was supported by grants from the National Natural Science Foundation of China (82173383 to D.X., 32200508 to L.X., and 82202260 to H.L.), the Sichuan Province Science and Technology Program (2022NSFSC1553 to L.X.), the 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYYC23024 to D.X.), and China Postdoctoral Science Foundation (2022TQ0221 to H.L.).
Author contributions: T.Z., H.L., and L.X. were responsible for the experimental design, execution, and data analysis. T.Z. wrote the paper, with contributions from all other authors. Y.H. and J.Z. were responsible for RNA sequencing and validation. S.Y., Y.L., and X.L. were responsible for data downloading, organization, and processing of raw data. D.X. was responsible for supervision of research, data interpretation, and manuscript preparation.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279617.124.
-
Freely available online through the Genome Research Open Access option.
- Received May 23, 2024.
- Accepted January 30, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








