
Epigenetic drift score captures directional methylation variability and links aging to transcriptional, metabolic, and genetic alterations
- Xiu Fan1,2,10,
- Qili Qian3,4,10,
- Wenran Li3,4,
- Tianzi Liu3,4,
- Changqing Zeng1,2,5,
- Peilin Jia1,2,
- Huandong Lin6,
- Xin Gao6,
- Li Jin4,7,
- Mingfeng Xia6,
- Sijia Wang3,8 and
- Fan Liu1,9
- 1China National Center for Bioinformation, Beijing 100101, China;
- 2Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China;
- 3CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200031, China;
- 4Collaborative Innovation Center of Genetics and Development, Fudan University, Shanghai 200433, China;
- 5Institute of Biomedical Research, Henan Academy of Sciences, Zhengzhou 450046, China;
- 6Department of Endocrinology and Metabolism, Zhongshan Hospital, Fudan Institute for Metabolic Disease, Human Phenome Institute, Fudan University, Shanghai 200032, China;
- 7Human Phenome Institute, Zhangjiang Fudan International Innovation Center, and Ministry of Education Key Laboratory of Contemporary Anthropology, Fulocal University, Shanghai 200433, China;
- 8Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming 650223, China;
- 9Department of Forensic Sciences, College of Criminal Justice, Naif Arab University for Security Sciences, Riyadh 11452, Kingdom Saudi Arabia
-
↵10 These authors contributed equally to this work.
Abstract
Epigenetic drift refers to the gradual and stochastic accumulation of epigenetic changes, such as DNA methylation variability, with advancing age. Although increasingly recognized for its potential role in aging biology, its extent, biological significance, and population specificity remain insufficiently characterized. Here, we present the first comprehensive epigenome-wide drift study (EWDS) in a large Chinese cohort (n = 3538), with replication in two independent Chinese (total n = 1467) and two European cohorts (total n = 956), to investigate the scale and relevance of epigenetic drift across populations. Through simulation, we identify White's test as the most powerful method among four alternatives for detecting age-associated methylation variability. Our EWDS reveals that 10.8% (50,385 CpGs) of sites on the 850 K EPIC array exhibit epigenome-wide significant drift, with 99% showing increased interindividual variability (positive drift) and 1% showing decreased variability (negative drift). Integration with single-cell RNA-seq data demonstrates that positive drift-CpGs are associated with increased transcriptional variability and upregulation in specific cell types, whereas negative drift-CpGs exhibit the opposite effect. We develop epigenetic drift scores (EDSs) to quantify individual drift burden; these scores are strongly age-associated and correlate with lipidomic profiles and clinical aging indicators. Longitudinal data confirm within-individual accumulation of drift over time. Finally, a GWAS of EDS identifies genetic determinants of drift magnitude, including heritable loci (e.g., ASTN2, SOCS5). Collectively, these findings establish epigenetic drift as a pervasive, directional, and biologically meaningful feature of human aging.
Epigenetic drift refers to the progressive, stochastic accumulation of molecular alterations across the epigenome during aging. These include changes in DNA methylation, histone modifications, chromatin remodeling, and noncoding RNAs. Collectively, such alterations disrupt gene regulatory networks, leading to transcriptional dysregulation, loss of cellular homeostasis, and increased vulnerability to age-related diseases (Li and Tollefsbol 2016; López-Otín et al. 2023). Among these, DNA methylation drift has emerged as the most extensively characterized component of epigenetic aging. It is typified by increased variance in methylation levels at specific CpG sites—termed drift-CpGs—over chronological age, distinguishing it from the linear age-associated methylation changes at epigenetic clock sites (Fraga et al. 2005; Horvath and Raj 2018; Seale et al. 2022). Importantly, epigenetic drift likely unfolds more gradually across wider temporal windows, capturing interindividual and cellular heterogeneity in a way that reflects biological aging beyond the linear progression captured by clock-based models. This stochastic accumulation is hypothesized to reflect rising intercellular and interindividual heterogeneity with age, potentially capturing biological aging more dynamically than static methylation clocks (Meyer and Schumacher 2024). Understanding and quantifying epigenetic drift may offer novel biomarkers for aging trajectories, disease susceptibility, and therapeutic interventions aimed at mitigating age-associated decline.
The etiology of epigenetic drift is multifactorial. Genetic predisposition accounts for substantial interindividual variation in baseline methylation patterns (Shah et al. 2014). In addition, longitudinal twin studies have underscored the influence of environmental factors; for instance, Tan et al. (2016) identified over 2000 CpGs exhibiting significant methylation drift over a decade, strongly implicating environmental exposures. Infectious agents, such as cytomegalovirus, have also been shown to induce widespread methylation variance in a cell composition-independent manner (Bergstedt et al. 2022). At the chromatin level, drift-CpGs are enriched in repressive Polycomb-bound regions (Slieker et al. 2016) and exhibit pronounced variability on the inactive X Chromosome (Liu et al. 2023), suggesting epigenomic compartmentalization of drift. The biological consequences of epigenetic drift are increasingly being explored. Hannum et al. (2013) showed that drift-CpGs in blood are associated with transcriptional changes, implicating drift as a potential regulator of aging pace. At the molecular level, this may manifest as altered transcriptional output and increased expression noise, disrupting cellular function.
Detecting epigenetic drift, however, requires robust statistical modeling capable of detecting changes in variance rather than mean methylation. Several approaches have been proposed. The most commonly used methods apply heteroscedasticity tests, such as the Breusch–Pagan test or double generalized linear models (DGLMs), to identify CpG sites whose methylation variance increases linearly with age (Slieker et al. 2016; Bergstedt et al. 2022; Liu et al. 2023). Complementary entropy-based metrics estimate methylome-wide disorder, capturing the global stochasticity of the epigenome (Hannum et al. 2013). These statistical frameworks serve as critical tools for identifying drift-CpGs and quantifying the systemic erosion of epigenetic fidelity.
Despite recent advances, several key challenges remain. First, there is no consensus on the optimal statistical framework for identifying drift-CpGs, with different studies applying distinct models and yielding limited concordance. Second, the generalizability of previously identified drift-CpGs is limited, as most were derived in European populations and lack validation across diverse ancestries, such as East Asians. Third, previous research has predominantly focused on CpGs with increasing methylation variance over age, whereas CpGs with decreasing methylation variance over age remain largely understudied, despite their potential biological importance. Fourth, it is still unclear whether drift-CpGs are cell type–specific or reflect a shared aging signature across hematopoietic lineages. Fifth, although entropy-based metrics have been used to estimate global methylation disorder, no standardized method currently exists to construct an epigenetic drift score (EDS) at the individual level using a finite set of CpGs. Lastly, the biological relevance of drift-CpGs, particularly their contribution to age-related complex traits and their relationship with underlying genetic architecture, remains poorly understood.
To address these challenges, we first performed a comprehensive evaluation of four commonly used statistical methods for identifying heteroscedasticity, using both computer simulations and empirical population data. We then applied this method to 735,267 CpG sites (850 K EPIC Array) measured in 3538 Chinese individuals, systematically identifying a high-confidence set of drift-CpGs. Our replication analysis was conducted in 2423 individuals from two additional Chinese cohorts and two European cohorts. Both positive and negative drift-CpGs were characterized to reveal complementary biological patterns. We further investigated whether drift-CpGs exhibit cell type specificity or represent shared signatures of hematopoietic aging by integrating single-cell RNA-seq analyses. To quantify individual-level epigenetic drift, we developed an EDS based on a finite subset of drift-CpGs, offering a standardized alternative to entropy-based metrics. Finally, we evaluated the functional relevance of EDS through association analyses with age, lipidomic profiles, and genome-wide genetic variation, uncovering potential mechanistic links between stochastic epigenetic changes and age-related complex traits.
Results
Population characteristics
An overview of the study design, detection technologies, and analytical workflow is presented in Figure 1. The discovery cohort, the National Survey of Physical Traits (NSPT), comprised 3538 Chinese individuals with a mean age of 50.2 years (SD = 12.7, range 18–83) and 37.0% male participants (Supplemental Table S1). The first replication cohort included 1060 individuals from the Chinese Academy of Sciences cohort (CAS), predominantly highly educated individuals in intellectual professions, with a mean age of 40.8 years (SD = 9.4, range 22–64) and 59.7% male participants. The second replication cohort, a longitudinal study from the Shanghai Changfeng Study (Changfeng), spanned a median follow-up period of 4 years with 407 subjects. Baseline ages ranged from 47.6 to 80.0 years, with a mean age of 61.4 years (SD = 7.4), and follow-up ages ranged from 51.9 to 84.0 years, with a mean age of 65.5 years (SD = 7.3). DNA methylation at 735,267 CpGs, overlapping between the discovery and replication cohorts, showed highly concordant distributions (Supplemental Fig. S1). For subsequent epigenome-wide analysis, we retained 469,061 CpGs after excluding those affected by mQTLs (Peng et al. 2024), those with minimal variance in age-regressed residuals (R2 < 1 × 10−5), or those significant in a multimodal distribution test (P < 1 × 10−4).

Schematic representation of the study design, detection technologies, and analytic approaches. The method marked in red text and with a checkmark (√) was selected for downstream analysis.
Positive and negative drifts enriched in different functional regions
A simulation analysis was conducted to compare the statistical power and Type-I error rates of four existing heteroscedasticity test methods: Method A: double generalized linear model (Liu et al. 2023); Method B: Breusch-Pagan likelihood ratio-based χ2 statistic (Bergstedt et al. 2022); Method C: Breusch-Pagan T-statistic (Slieker et al. 2016); and Method D: White Test (White 1980; see Supplemental Notes for details). This analysis showed that Method B was overly aggressive, whereas Method A was overly conservative. Method D showed superior performance in scenarios involving nonlinear relationships between CpG variance and age (Supplemental Fig. S2). Using the White Test, our epigenome-wide drift study (EWDS) identified 10.8% of CpG sites (50,385) as drift-CpGs (P < 1 × 10−7) (Fig. 2A,B). This number significantly exceeds findings from previous studies, including one involving 385 Swiss twin pairs using the 450K array, which identified 571 drift-CpGs (different method due to twin samples), with 229 overlapping with our list (Wang et al. 2018). Another study of 3295 European individuals from the BIOS Consortium using the 450K array and the Method C identified 6366 drift-CpGs (Fig. 2C), with 3000 overlapping with ours (Wang et al. 2018). Our number also surpassed that from the Milieu Intérieur study of 968 Europeans (Bergstedt et al. 2022), which identified 20,140 drift-CpGs from a total of 644,517 CpGs using Method B. These discrepancies are likely attributable to a combination of factors such as larger sample size, broader age range, greater CpG coverage, and enhanced statistical power. The vast majority of the identified drift-CpGs (99.0%, n = 49,877) exhibited an increase in variance with aging, termed as positive drift-CpGs (Fig. 2D), whereas a small fraction showed a significant decrease in variance, termed as negative drift-CpGs (1.0%, n = 508) (Fig. 2E). Positive drift-CpGs were highly significantly enriched in expression-repressed CpG islands (CGIs) (OR = 2.4, P < 1 × 10−300) (Fig. 2F,G), whereas negative drift-CpGs were significantly enriched in expression-active non-CGIs, such as open sea regions (OR = 2.4, P < 8.8 × 10−19), with a particularly pronounced enrichment in enhancers (OR = 10.1, P = 2.1 × 10−64). A transcription factor binding motif enrichment analysis showed that negative drift-CpGs significantly enriched at AHR-ARNT, CREB3L4, HES1, and GMEB2 (Supplemental Fig. S3).
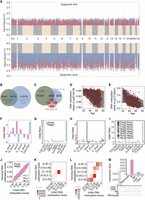
Epigenome-wide identification and annotation of drift- and clock-CpGs. (A) Manhattan plot of significant CpGs (P < 1 × 10−7), colored by drift (blue), clock (blue), or both (red) effects. (B) Venn diagram showing overlap between drift- and clock-CpGs. (C) Overlap of NSPT drift-CpGs with those from Slieker et al. (2016) and Wang et al. (2018). (D,E) Representative CpGs with age-related increase (D) or decrease (E) in methylation variance. (F,G) Genomic enrichment of positive and negative drift-CpGs (F: CpG island-related; G: gene-related features). (H,I) Genomic enrichment of drift- and clock-CpGs (H: CpG island-related; I: gene-related features). (J) Scatter plot of initial (young) versus terminal (old) methylation levels at drift-CpGs. (K,L) Heat maps of positive (K) and negative (L) drift-CpG distribution by methylation levels. (M) Upset plot showing cell type specificity of drift-CpGs across immune lineages. (PosD) positive drift-CpGs, (NegD) negative drift-CpGs, (PosC) positive clock-CpGs, (NegC) negative clock-CpGs, (NonD) nondrift CpGs, (NonC) nonclock CpGs.
Compared to the abundant age-associated CpGs identified by linear regression in the same data set, referred to here as clock-CpGs (31.2%, n = 146,497, P < 1 × 10−7), the number of drift-CpGs was substantially smaller and exhibited lower statistical significance, as expected. Nonetheless, we observed a significant overlap between drift-CpGs and clock-CpGs (23.3%, n = 34,118), indicating that methylation drift and directional age associations are not mutually exclusive. In fact, drift-CpGs were significantly more likely than non-drift-CpGs to also exhibit age-associated mean changes in methylation (OR = 5.7, P < 1 × 10−300) (Supplemental Table S2). Notably, positive drift-CpGs were more frequently associated with CpGs that also showed increasing average methylation with age (positive clock-CpGs), whereas negative drift-CpGs were more likely to coincide with CpGs that decreased in methylation with age (negative clock-CpGs; OR = 1.3, P = 7.8 × 10−3) (Supplemental Table S2). Positive drift-CpGs and positive clock-CpGs were most significantly enriched in CGIs (Fig. 2H), whereas negative drift-CpGs and negative clock-CpGs were most significantly enriched in enhancers (Fig. 2I). This concordance suggests a partial coupling between directional epigenetic aging and the age-related changes in epigenetic variability.
We examined whether changes in DNA methylation variance at CpG sites are associated with initial (young group age < mean − 2SD) and terminal (old group age > mean + 2SD) methylation levels. Specifically, we asked whether reduced variance indicates convergence toward methylation extremes (0 or 1), and whether increased variance reflects divergence from such states. This analysis identified differential drift patterns based on the direction of methylation drift (Fig. 2J–L). Positive drift-CpGs predominantly remained at intermediate methylation ranges (0.1–0.9) throughout aging, rarely converging toward either hypermethylation or hypomethylation extremes. In contrast, negative drift-CpGs typically moved from intermediate methylation states toward methylation extremes but rarely vice versa. These observations suggest fundamentally different biological mechanisms underlying positive versus negative methylation drift during aging. We hypothesize that negative drift, characterized by shifts towards methylation extremes, likely reflects more targeted biological aging processes such as cellular senescence or clonal expansions, whereas positive drift might represent broader stochastic or heterogeneous aging processes.
Epigenetic drift's cell type specificity and transcriptional impact
To investigate whether epigenetic drift-CpGs are driven by changes in blood cell–type composition, we applied the EpiDISH algorithm (Zheng et al. 2018). Among the 50,385 identified drift-CpGs, the majority (88%) did not exhibit cell type–specific methylation patterns, suggesting that most epigenetic drift occurs independently of blood cell composition (Fig. 2M). Of the 6226 CpGs (12%) that did show significant cell type specificity, the vast majority (>99%) were associated with lymphoid lineage cells. Specifically, these CpGs were predominantly enriched in B cells (82%), followed by CD8+ T cells (12%), CD4+ T cells (4%), and natural killer (NK) cells (2%), whereas <1% were linked to myeloid cells. Importantly, CD4+ T cell-specific drift-CpGs (n = 266) showed a distinct pattern, all of which exhibited negative drift with age. These findings suggest a cell type–specific suppression of epigenetic drift noise in CD4+ T cells, in contrast to the general age-associated increase observed in other cell types. Representative examples of lymphoid, myeloid, and CD4+ T cell-specific drifts are provided in Supplemental Figure S4A–C.
To investigate how age-associated DNA methylation drift influences gene expression, we integrated drift-CpG profiles with single-cell RNA sequencing (scRNA-seq) data across peripheral blood mononuclear cell (PBMC) immune cell types. Our analysis revealed statistically significant, albeit modest, shifts in gene expression profiles associated with drift-CpGs relative to the genome-wide background (Supplemental Fig. S5A,B). Specifically, genes associated with positive drift-CpGs exhibited a tendency towards higher mean expression and discernibly greater expression variability compared to the genome-wide background, suggesting a relatively more active yet potentially unstable transcriptional state. Conversely, genes adjacent to negative drift-CpGs displayed slightly but significantly lower transcriptional noise, indicating a more stable transcriptional pattern.
We further examined age-related transcriptional changes in immune cells. Although overall mean transcriptional activity showed a modest but significant increase in older individuals (Supplemental Fig. S5A,B), stratifying by drift direction revealed consistent, albeit subtle, age-associated increases in transcriptional levels for genes proximal to both positive and negative drift-CpGs. Notably, only genes near positive drift-CpGs showed increased transcriptional variance with age, whereas those near negative drift-CpGs did not.
Subsequent cell type–specific analyses confirmed that age-related transcriptional changes associated with drift-CpGs were largely nonoverlapping across immune cell types, underscoring a high degree of cell type specificity (Supplemental Fig. S5C–F). In CD4+ T cells, BASiCS analysis revealed reduced transcriptional noise in aged individuals for genes associated with positive drift-CpGs, suggesting a context-dependent noise-suppressive function. The most pronounced transcriptional perturbations in genes associated with positive epigenetic drift were observed in B cells (Supplemental Fig. S5G). Conversely, negative epigenetic drift primarily affected CD4+ T cells, driving distinct transcriptional changes in this subset (Supplemental Fig. S5H).
Robust drifts in an independent CAS cohort
In the CAS cohort of 1060 samples, 48,171 CpGs overlapped with the 50,385 significant CpGs identified in NSPT. Of these overlapping CpGs, 50.2% were replicated at a nominal significance level in CAS (P < 0.05). The significance of this replication rate (OR = 8.9) was confirmed by a Fisher's exact test (P < 2.2 × 10−16) (Fig. 3A; Supplemental Table S3). An analysis of these replicated drifts revealed that 99.9% of positive drifts were consistently positive, and 99.1% of the negative drifts remained negative. When applying more stringent significance criteria in the discovery cohort, we observed a progressive increase in replication rates, peaking at 100% for a threshold of 1 × 10−20. Notably, both positive and negative drifts exhibited complete consistency at these stringent levels.
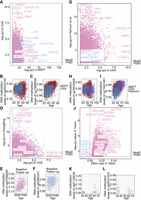
Replication of drift-CpGs in independent cohorts. (A) Replication of significant drift-CpGs in CAS, with thresholds corresponding to Bonferroni significance in NSPT and nominal significance in CAS. CpGs are colored by drift direction and selected genes are labeled. (B,C) Scatter plots of top positive (B: cg24035745) and negative (C: cg13868026) drift-CpGs validated in CAS. (D–F) Validation of drift-CpGs in the Changfeng cohort, including representative positive (E: cg27099280) and negative (F: cg03883331) examples. (G–I) Replication in Hannum et al. (2013), with representative positive (H: cg16532606) and negative (I: cg11647481) drift-CpGs. (J–L) Validation in monozygotic Danish twins (Tan et al. 2014), including top positive (K: cg21109038) and negative (L: cg08337633) drift-CpGs.
In the CAS cohort, the most significantly replicated positive drift was observed on Chr 4 for cg24035745 in FBXO2 (NSPT P = 5.1 × 10−14, CAS P = 6.8 × 10−11). Young individuals exhibited significantly smaller variance in methylation compared to mid-aged and elderly individuals (Fig. 3B). Increased expression of FBXO2 occurs with neurons’ developmental maturation (Ciceri et al. 2024). FBXO2 deficiency exacerbated deficits in motor function and enhanced neurodegeneration (Liu et al. 2020). Similarly, the most significantly replicated negative drift was identified on Chr 14 for cg13868026 in EVL (NSPT P = 6.8 × 10−59, CAS P = 8.8 × 10−7). In this case, young and mid-aged individuals showed significantly larger variance in methylation compared to elderly individuals (Fig. 3C). DNA methylation of EVL was identified as a prognostic signature in colon cancer, and promoter methylation of EVL differs between individuals and between regions of the normal colon (Yi et al. 2011). The CAS cohort also replicated the presence of other top significant negative drift-CpGs that were found in or near MAPRE2, ARPIN-AP3S2, TRAPPC9, APBB1IP, and FOXK1. Among these, methylation of FOXK1 has a known effect on the regulation of immune and metabolic functions (Fujinuma et al. 2023).
Epigenetic drifts in independent longitudinal Changfeng population
We further analyzed the CAS-replicated drift-CpGs (23,758 out of the available 24,161) within the Changfeng longitudinal cohort to determine if the methylation levels showed any drift over an approximate follow-up period of 4 years. For each individual CpG site, we performed a paired t-test. This test compared the squared deviation of the methylation level from the population mean at the baseline to the same deviation at the follow-up. In mathematical terms, it involved comparing (cpg − mean(cpg))2 at baseline with those at follow-up. Our results indicated that 48.1% (n = 11,422, P < 0.05) of the examined drift-CpGs exhibited a nominally significant difference (OR = 5.0, Fisher's test, P < 2.2 × 10−16) between these two time points. Among these nominally significant CpGs, 99.7% exhibit the same effect directions as observed in NSPT and CAS (Fig. 3D). These findings demonstrated the robustness of the drift effects at our identified sites and underscore the genuine impact of negative drifts. In Changfeng, the most significantly replicated positive drift-CpG was cg27099280 in CELF6 (P = 6.2 × 10−19) (Fig. 3E) and negative drift-CpG was cg03883331 in ZBTB18 (P = 4.0 × 10−4) (Fig. 3F) and cg22005677 in LPP (P = 0.02). The repeated highlighting of the negative drift at LPP in both the CAS and Changfeng cohorts underscores its robustness and prominence in our findings. It is important to note that both the CAS and Changfeng replication cohorts have narrower age ranges compared to the discovery cohort, which may contribute to conservative replication rates. Functionally, the CpG site in ZBTB18 is instrumental in regulating the expression of ZBTB18, a colorectal tumor suppressor gene (Bazzocco et al. 2021). Loss of its activity enhances chromatin accessibility and transcriptional adaptations that promote the phenotypic changes required for metastasis (Wang et al. 2023). This suggests that negative drifts may be involved in regulating global chromatin accessibility dysregulation in the development of age-related phenotypes.
Epigenetic drift is cross-ethnic
We further examined 14,909 of our drift-CpGs overlapping with the 450K beadchip in the data set of Hannum et al. (2013), which consisted of a mixed population of 426 Caucasian and 230 Hispanic individuals (age range: 19–101 years). A substantial proportion (76.3%, 11,378 out of 14,909) exhibited nominally significant drifting effects in the same direction (OR = 9.3, Fisher's test, P < 2.2 × 10−16) (Fig. 3G–I).
Next, we focused on a cohort of 150 pairs of monozygotic (MZ) Danish twins aged 30 to 74 years (78 male pairs and 72 female pairs) (Tan et al. 2014). Our aim was to determine whether our identified drift-CpGs could account for previously observed interindividual epigenetic variations (Planterose Jiménez et al. 2021). To this end, we categorized the MZ twins into two age groups (<50 years and ≥50 years). Using a t-test, we examined the absolute methylation level discrepancies (|MZ1 − MZ2|) between the two age groups. Among the CpGs corresponding with our drift-CpGs, a substantial proportion (49.6%, 6731 out of 13,571) showed nominally significant drift effects (Fig. 3J–L). These findings support our hypothesis that aging plays a pivotal role in the epigenomic differentiation of MZ twins.
Positive drifts related to neural system functions and negative drifts linked to immune functions
GO and KEGG functional enrichment analyses of our replicated drift-CpGs have illuminated possible mechanisms tied to both positive and negative epigenetic drift (Fig. 4). Specifically, positive drift-CpGs (n = 24,051) were markedly enriched for processes related to nervous system development (P = 7.9 × 10−41 after FDR) and neuroactive ligand-receptor interaction (P = 6.2 × 10−23 after FDR), corroborating the work of Slieker et al. (2016). Conversely, negative drift-CpGs (n = 110) showed distinct enrichment in alpha-beta T cell differentiation (P = 1.4 × 10−3 after FDR). Phenotype enrichment analysis, derived from EWAS Atlas data, revealed that positive drift-CpGs were significantly associated with phenotypes such as B acute lymphoblastic leukemia (P = 1.8 × 10−91 after FDR), hepatocellular carcinoma (P < 3.4 × 10−39 after FDR), and Helicobacter pylori infection (P = 5.4 × 10−38 after FDR). This finding aligns with earlier studies showing that increased DNA methylation variability frequently occurs at loci related to malignancy and can be predictive of cancer emergence (Landau et al. 2014; Feinberg and Levchenko 2023). In contrast, the phenotype enrichment for negative drift underscored aging as the most significantly associated trait (P = 1.3 × 10−36 after FDR), followed by Down syndrome as the second most significant (P = 2.1 × 10−24 after FDR). These patterns suggest a differential impact of positive versus negative drift on phenotype expression, with negative drift showing a pronounced association with aging processes.
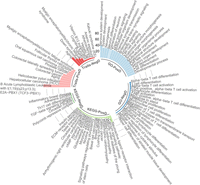
Biological and trait enrichments of directional epigenetic drift. This circular plot visualizes Gene Ontology (GO) terms, KEGG pathways, and traits enriched for positive (PosD) and negative (NegD) drift-CpGs. Enrichment score, calculated as −log10(FDR P), increases with distance from center.
EDS represents a unique aging dimension
We developed a positive epigenetic drift score (EDS_POS) using 204 independent CpGs, each more than 500 kbp apart (Supplemental Table S4). These CpGs were selected from the 49,877 positive drift-CpGs identified in NSPT, based on their smallest Fisher combined P-values across NSPT, CAS, and Hannum et al. (2013), using a nonnegative least squares regression model.
The EDS_POS ranges from 0 to 1, reflecting low to high levels of epigenetic positive drift. In both NSPT and Hannum et al. (2013), the distribution of EDS_POS was slightly right-skewed, with a mean of 0.41 (box plot: 0.21, 0.32, 0.39, 0.47, 0.69). EDS_POS exhibited a linear correlation with age, which strengthened as more CpGs were included. Specifically, Pearson's correlation increased from 0 to 0.56 as the number of CpGs grew from 1 to 100, and plateaued at 0.60 with 200 CpGs (Fig. 5A,B). The EDS_POS constructed in CAS using the same weights also demonstrated a highly consistent distribution (mean 0.39, box plot: 0.25, 0.34, 0.37, 0.43, 0.56) (Fig. 5C) and a robust age correlation (r = 0.50).
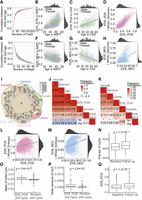
Validation and aging associations of the epigenetic drift score. (A) EDS_POS shows increasing correlation with age as more positive drift-CpGs are included. (B,C) Scatter plots of EDS_POS versus age in NSPT (B) and CAS (C), colored by gender. (D) EDS_POS significantly correlates with genome-wide positive EDS. (E–G) EDS_NEG similarly shows increasing age correlation (E) and gender-stratified associations in NSPT (F) and CAS (G). (H) EDS_NEG correlates with genome-wide negative EDS. (I) Venn diagram showing CpG overlap between EDS and four aging clocks. (J,K) Heat maps of correlations among EDS, aging clocks, and age, before (J) and after (K) age adjustment. (L,M) EDS_POS (L) and EDS_NEG (M) correlate with genome-wide methylation entropy. (N) Methylation entropy increases longitudinally over 4 years. (O–Q) EDS_POS CpGs show greater increases in methylation mean (O), variance (P), and EDS score (Q) over time compared to random CpGs.
We also found that EDS_POS increases more slowly with age in females compared to males (P = 3.7 × 10−7) (Fig. 5B). We then compared the performance of EDS_POS with a more robust but less sensitive score that included more CpGs with equal weights (11,367 positive drift-CpGs nominally replicated in both CAS and Hannum et al. 2013). This score had a lower correlation with age (r = 0.46) than EDS_POS, likely due to added noise from less significant CpGs given equal weight. However, EDS_POS still showed a high correlation (r = 0.82) with this score (Fig. 5D). This result, along with the observation that age correlation plateaued after including 200 CpGs in EDS_POS, suggests that our EDS_POS effectively captures genome-wide epigenetic drift, with 204 drift-CpGs being sufficient for this purpose.
We similarly developed a negative epigenetic drift score (EDS_NEG) based on 81 significant negative drift-CpGs identified in NSPT. The EDS_NEG ranges from 0 to 1, indicating low to high levels of negative drift (Supplemental Table S5). In NSPT, the distribution of EDS_NEG was slightly right-skewed with a mean of 0.36 (box plot: 0.23, 0.31, 0.36, 0.42, 0.57). EDS_NEG showed an increasingly strong negative correlation with age as more CpGs were included, reaching −0.38 when all 81 CpGs were included (r = −0.39 in males and r = −0.38 in females) (Fig. 5E,F). The EDS_NEG constructed in CAS using the same weights demonstrated a highly consistent distribution (mean 0.42, box plot statistics: 0.14, 0.31, 0.40, 0.50, 0.79) (Fig. 5G) and a robust correlation with age (r = −0.49). A more robust but less sensitive score, based on 508 equally weighted negative drift-CpGs, also showed a negative correlation with age at −0.34. The correlation between EDS_NEG and this 508-CpG score was 0.54 (Fig. 5H).
We further investigated into the correlations of our EDS with several previously established DNA methylation-based aging scores, including Horvath (2013), Hannum et al. (2013), Levine et al. (2018), and Dunedin (Fig. 5I; Belsky et al. 2020). The Horvath and Hannum scores represent different versions of epigenetic clocks, whereas the Levine and Dunedin scores focus more on aging and aging pace. Our hypothesis posits that epigenetic drift may reflect a distinct or complementary aspect of aging compared to other epigenetic aging scores. Indeed, in NSPT, we observed expected modest levels of correlations prior to adjusting for age (EDS_POS: 0.29∼0.63, EDS_NEG: −0.11∼−0.39) (Fig. 5J), which were further reduced after adjusting for age (EDS_POS: 0.17∼0.31, EDS_NEG: −0.05∼−0.09) (Fig. 5K). These results support our hypothesis that epigenetic drift captures aging across different dimensions and scales compared to other epigenetic scores. Note that the methylation changes associated with epigenetic drift occur over much longer timescales, typically spanning decades, in contrast to the changes observed in epigenetic clock-CpGs, which often occur within a span of several years (Supplemental Fig. S6). Consequently, the EDS and the clock score may be capturing different aspects of aging, which could explain why the EDS exhibits a lower correlation with age compared to the typically high correlation (over 0.9) observed with clock scores.
In the NSPT cohort, individual-level entropy measures showed significant age associations: positive drift entropy significantly correlated with chronological age (Pearson correlation coefficient, r = 0.39, P = 6.9 × 10−154), and negative drift entropy exhibited a significant inverse relationship (Pearson correlation coefficient, r = −0.51, P = 8.5 × 10−237). We observed moderate but significant concordance between individual entropy and population-level drift scores (Pearson correlation coefficient, EDS_POS: r = 0.25, P = 1.0 × 10−61; negative: r = 0.29, P = 6.7 × 10−70) (Fig. 5L,M). Furthermore, longitudinal analysis in the Changfeng cohort revealed a significant increase in overall epigenetic entropy over 4 years (paired t-test, P = 1.7 × 10−13), with the mean epigenetic entropy increasing from 4101 at baseline to 4172 at the 4-year follow-up, confirming the progressive nature of epigenetic drift at the individual level (Fig. 5N).
To evaluate EDS dynamics in the same individual's longitudinal aging process, we compared EDS_POS changes in methylation levels and variance from baseline to follow-up using longitudinal Changfeng data. These changes were then compared to analogous changes derived from a bootstrapped genome background, matched for the number of CpGs. The mean methylation (P = 3.6 × 10−10) and variance (P = 2.8 × 10−9) of these 204 positive drift-CpGs showed a statistically significant increase from baseline to the follow-up period (Fig. 5O,P). Applying the weights derived from the NSPT to Changfeng confirmed a significant increase in EDS from the baseline to follow-up (baseline mean EDS = 0.35; follow-up mean EDS = 0.38, P = 1.2 × 10−12) (Fig. 5Q).
EDS associated with lipid metabolites
Given the significant influence of epigenetic mechanisms on lipid metabolism (Gomez-Alonso et al. 2021) and the observation that calorie restriction can modulate both epigenetic drift and metabolic pathways (Hahn et al. 2017; Maegawa et al. 2017), we further explored the impact of our EDS on serum lipid metabolites. We analyzed data from 3037 individuals in the NSPT cohort, for whom both metabolomic and methylation profiles were available. The metabolomic data set included 351 NMR-detected lipoprotein subfractions, comprising 176 direct measurements and 175 derived values.
After adjusting for age, gender, BMI, and sampling location, EDS_POS was significantly associated with 65 lipid metabolites following FDR correction (Fig. 6A–F; Supplemental Table S6). These associations encompassed 13 high-density lipoprotein (HDL) metrics, 17 low-density lipoprotein (LDL) metrics, one intermediate-density lipoprotein (IDL) metric, and three very-low-density lipoprotein (VLDL) metrics. Among the negative associations, EDS_POS showed the strongest correlation with phospholipids in HDL-4 (P = 4.1 × 10−3 after FDR). Among the positive associations, EDS_POS was most significantly associated with triglycerides in VLDL-5 (V5TGp, P = 2.3 × 10−2 after FDR), consistent with previous studies that reported reduced VLDL levels in long-lived and younger cohorts (Lv et al. 2015). In addition to lipid metabolites, we examined the association of EDS with 17 physical and blood biochemical indicators. After adjusting for age, EDS_POS was significantly associated with six out of 17 indicators, that is, BMI, HDL, triglycerides (TG), uric acid (UA), indirect bilirubin (IBIL), and creatinine (CREA) (FDR P < 0.05). EDS_NEG did not show any significant association in lipid or blood biochemical association analyses (P > 0.05 after FDR). Notably, further adjusting for clock scores in these association analyses did not alter our findings except for those related to UA (Supplemental Fig. S7).

Associations of EDS_POS with serum lipid metabolic profiles. Forest plots display effect sizes (with 95% confidence intervals) per unit increase of relevant scores on various lipid metrics. The significance threshold for associations shown is set at FDR P < 0.05. Four epigenetic indicators are presented: EDS_POS (red), Dunedin (light blue), Levine (green), and Hannum (dark blue). (A) Associations with HDL-related traits. (B) Associations with LDL-related traits. (C) Associations with IDL- and VLDL-related traits. (D) Associations with lipid ratio metrics. (E) Associations with lipid percentage metrics. (F) Associations with other small metabolites and apolipoproteins. (ApoA1, ApoA2, ApoB) Apolipoprotein A1, Apolipoprotein A2, Apolipoprotein B, (CH) Cholesterol, (CE) Cholesteryl Esters, (FA) Fatty Acids, (FC) Free Cholesterol, (HDL) High-Density Lipoprotein, (IDL) Intermediate-Density Lipoprotein, (LDL) Low-Density Lipoprotein, (PL) Phospholipids, (PN) Particle Number, (TG) Triglycerides, (UFA/TFA) Unsaturated Fatty Acids/Total Fatty Acids ratio, (VLDL) Very Low-Density Lipoprotein. Subclass numbers indicate size-based lipoprotein subfractions. A complete list of all metabolite abbreviations and their full names is provided in Supplemental Table S6.
Extending the lipid and blood biochemical association analyses to other epigenetic scores, including Hannum, Horvath, Levine, and Dunedin (Fig. 6A–F), we identified numerous significant associations after adjusting for age. The strongest was between the Dunedin score and ApoA2 in HDL-4 (P = 1.25 × 10−8). In terms of the number of associations, the Dunedin score exhibited the most extensive associations, with 97 metabolites and 13 blood biochemical indicators, followed by EDS_POS (65 and six), Levine (16 and three), Hannum (one and two), and Horvath (zero and six). Notably, further adjusting for clock scores in these association analyses did not alter our findings (Supplemental Fig. S8). Although the metabolites and biochemical indicators are strongly intercorrelated, our results suggest that multiple epigenetic scores play complex roles in metabolic features related to lipid metabolism, kidney function, and liver function.
GWASs on EDSs identify genetic factors
We conducted separate genome-wide association studies (GWASs) for EDS_POS and EDS_NEG, analyzing 8.6 million SNPs from microarray data after imputation in 3513 individuals. No evidence of population substratification was observed, as indicated by the inflation factor (λ < 1.03). The SNP-based heritability was estimated to be moderate for both traits using GCTA (EDS_POS VG/VP = 0.29, se = 0.07; EDS_NEG VG/VP = 0.11, se = 0.07).
The GWAS for EDS_POS identified a single SNP (rs7868942) located within the ASTN2 gene on Chr 9q33.1 showing a genome-wide significant association with EDS_POS (lead SNP rs7868942, β = 0.02, P = 4.3 × 10−8) (Fig. 7A–C). The ancestral A allele of rs7868942 had a frequency of 0.95 in our sample, similar to the East Asian (EAS) population frequency of 0.94, much higher than other continental groups in the 1000 Genomes Project (AMR 0.69, AFR 0.75, EUR 0.60, SAS 0.75). ASTN2 regulates neuronal migration and synaptic strength by trafficking and degrading surface proteins and has been repeatedly implicated in autism, Alzheimer's, and other neuropsychiatric disorders (Glessner et al. 2009; Ito et al. 2023). During aging, the chromatin state of ASTN2 becomes more promoter-like and active, with a reduction in H3K36me3 and an accumulation of H3K4me3 and H3K27ac (Fig. 7D). This result is consistent with the previously proposed aging model, where reduced H3K36me3 levels within gene bodies inhibit the recruitment of KDM5B and DNMT3B to these regions, resulting in the accumulation of H3K4me3 and reduced DNA methylation, which leads to increased transcriptional noise (McCauley et al. 2021).
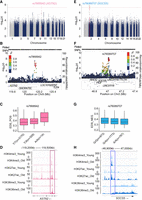
GWAS results for epigenetic drift scores. (A) Manhattan plot showing SNP associations with EDS_POS. (B,C) Locus zoom plot (B) and genotype-specific differences (C) for lead SNP rs7868942 near ASTN2. (D) Aging is associated with a more active chromatin state at ASTN2 (loss of H3K36me3; gain of H3K4me3, H3K27ac). (E) Manhattan plot showing SNP associations with EDS_NEG. (F,G) Locus zoom plot (F) and genotype-specific differences (G) for rs76089707 near SOCS5. (H) SOCS5 chromatin becomes more active with age in hMSCs, marked by increased H3K4me3 and H3K27ac enrichment (McCauley et al. 2021).
The GWAS for EDS_NEG identified 20 SNPs on Chr 2p21 near the SOCS5 gene that showed genome-wide significant associations with EDS_NEG (lead SNP rs76089707, β = −0.02, P = 1.2 × 10−8) (Fig. 7E–G). The ancestral G allele of rs76089707 had a frequency of 0.53 in our sample, closely matching the EAS population frequency of 0.52 and remaining comparable to frequencies in other continental groups from the 1000 Genomes Project (AMR 0.47, AFR 0.61, EUR 0.65, SAS 0.56). SOCS5, a cytokine signaling suppressor, negatively regulates the JAK/STAT pathway and plays a key role in balancing immune response and virus persistence (Seki et al. 2002; Kedzierski et al. 2022). While aging, T cells often experience increased activation and proliferation, leading to functional exhaustion. This exhaustion is linked to sustained JAK/STAT pathway activation, where SOCS proteins play a crucial inhibitory role (Sharma et al. 2019). During aging, the chromatin state of SOCS5 becomes more active, with an accumulation of H3K4me3 and H3K27ac, yet without a decrease in H3K36me3 (Fig. 7H). In exhausted T cells, specific CpG sites may become epigenetically stable, contributing to the long-term repression or activation of certain genes. Although these preliminary findings provide valuable insights, the modest effect sizes and the proximity of the results to the genome-wide significance threshold suggest a cautious interpretation. Further validation through replication studies and functional assays is essential to confirm and elucidate the roles of these genomic loci in epigenetic drift.
Discussion
In this study, we present the largest EWDS to date, conducted in a Chinese population and replicated in multiple Chinese and European cohorts. We systematically benchmarked statistical approaches for detecting heteroscedasticity in DNA methylation data and identified White's test as the most robust method. Applying this approach, we identified and functionally annotated over 50,000 significant drift-CpGs, which segregated into positive and negative drift categories with distinct genomic enrichments, suggesting divergent underlying biological mechanisms. Integration with single-cell RNA sequencing revealed that drift-associated CpGs exert direction-specific transcriptional effects and display marked cell type specificity. We further developed and validated composite EDS_POS and EDS_NEG to quantify individual-level drift, both of which showed strong correlations with chronological age. Notably, a positive EDS was significantly associated with lipidomic profiles, as well as metabolic and clinical health indicators. Importantly, the population-level drift-CpGs and EDS signatures were also validated longitudinally at the individual level. Finally, genome-wide association analyses identified genetic loci, including ASTN2 and SOCS5, associated with positive and negative drift variation, respectively, underscoring a heritable component. Together, these findings offer key conceptual and methodological advances in mapping epigenetic drift and underscore its significance in aging biology and age-related disease susceptibility.
Our findings reveal that both positive and negative epigenetic drift are associated with aging, yet they likely represent distinct biological processes with divergent regulatory consequences. Positive drift, characterized by increased methylation variance with age, is more prevalent genome-wide and typically occurs in CpG island-rich regions (Slieker et al. 2016). It exhibits minimal dependence on immune cell composition, suggesting a population-wide, stochastic process. Functionally, positive drift is associated with increased gene expression variability, that is, transcriptional noise, particularly in genes linked to neurological and metabolic pathways. These changes may reflect compensatory or stress-induced transcriptional activation and contribute to systemic dysregulation observed in aging, including altered lipid metabolism and blood biomarkers. Despite having a smaller effect size than traditional epigenetic clocks, we show that a subset of ∼200 key drift-CpGs can effectively summarize genome-wide stochastic methylation variability. These results support the notion that positive drift captures nonprogrammatic, nonlinear aspects of aging, potentially serving as a mechanistic substrate for the emergence of epigenetic clocks themselves through cumulative stochastic changes. In contrast, negative drift, although less common, is enriched in enhancer regions and exhibits strong CD4+ T cell-specific patterns. It is associated with reduced transcriptional noise in aged individuals and appears to reflect immune-specific regulatory stabilization, rather than general epigenomic entropy. Mechanistically, negative drift is enriched for binding sites of transcription factors such as AHR and may participate in fine-tuning immune gene regulation during aging (Salminen 2022). This is consistent with prior observations of age-associated T cell depletion, immune remodeling, and skewing of CD4+ T cells toward extreme regulatory and effector phenotypes (Dorshkind et al. 2009; Elyahu et al. 2019). Specifically, aging might reduce CD4+ T cell numbers and reshape their composition, potentially driving the emergence of distinct DNA methylation patterns linked to negative drift. Thus, whereas positive drift may reflect stochastic cellular deterioration, negative drift may represent an adaptive or programmed component of immune aging. These distinct patterns suggest that epigenetic drift is not a monolithic process but rather a bifurcated aging mechanism—stochastic (positive) versus programmed (negative)—with far-reaching implications for identifying aging biomarkers and designing targeted interventions.
Whereas some previous studies have proposed that epigenetic drift may contribute to the generation of epigenetic clocks (Meyer and Schumacher 2024), our findings suggest that epigenetic drift appears to represent a distinct dimension of epigenetic aging, primarily reflecting the accumulation of stochastic, nonprogrammatic changes over time. In contrast, epigenetic clocks are typically derived from CpGs whose methylation levels change in a coordinated and directional manner with age, likely reflecting functional, environmentally responsive, or developmentally regulated processes. This distinction is supported by the only moderate correlations we observed between the EDS and established epigenetic age estimators (Hannum et al. 2013; Horvath 2013; Levine et al. 2018; Belsky et al. 2020). Unlike clocks, which are trained to predict chronological or biological age, the EDS captures genome-wide epigenomic instability irrespective of directionality. We therefore interpret positive drift as a signature of cumulative random damage or loss of epigenetic maintenance fidelity, rather than a direct mechanistic contributor to clock formation.
Our study demonstrates that drift-CpGs and the EDS capture cumulative, age-related epigenetic variability at the individual level. Validated examples—such as positive drift at FBXO2 and LINC02716 and negative drift at FOXK1 and TCF12—highlight their relevance to transcriptional regulation and disease. Interindividual differences in EDS further support its utility in quantifying genome-wide drift and predicting aging-related traits. Notably, we found significant associations between EDS and lipid metabolism, particularly with subclasses of LDL and HDL, underscoring a potential regulatory link. These results align with prior studies on DNA methylation and lipoproteins (Gomez-Alonso et al. 2021) and suggest the need for further functional investigation into the causal role of epigenetic drift in lipid regulation. Our study suggests potential sex-specific differences in epigenetic aging, with females exhibiting a slower accumulation of positive epigenetic drift compared to males. This observation may reflect greater epigenomic stability in females against age-associated stochastic changes, potentially influenced by hormonal or chromatin-related factors. Whereas these findings may offer a partial explanation for the observed female longevity advantage, they also highlight the importance of considering sex as a biological variable in the development of aging biomarkers and potential interventions.
Our analysis focused on identifying drift-CpGs using population-level, cross-sectional DNA methylation array data. In contrast, some other studies have proposed metrics such as Shannon entropy or the proportion of intermediately methylated sites to quantify methylation heterogeneity within individual samples (Scherer et al. 2020). These entropy-based measures typically require deep bisulfite sequencing (e.g., WGBS) to resolve single-molecule differences. Although we also calculated individual-level entropy from array data, our primary objective was to model changes in methylation variance with age across the population, enabling the identification of drift-CpGs that reflect systemic epigenetic variability rather than within-sample noise. This focus is partly driven by data availability: whereas WGBS offers near-complete CpG coverage and can assess cell-intrinsic variability, the EPIC array surveys a fixed subset of CpGs and reflects averaged methylation signals across heterogeneous cell populations. Importantly, we validated the drift-CpGs identified at the population level using independent longitudinal data sets, supporting their temporal robustness. Furthermore, the EDS derived from drift-CpGs showed a strong correlation with entropy-based measures, suggesting that these two approaches, although methodologically distinct, converge on capturing common aspects of epigenetic aging.
Nevertheless, we acknowledge several limitations. First, our study lacks multitissue or large-scale longitudinal data spanning broader age ranges, which are necessary to evaluate drift dynamics across developmental and aging trajectories. Second, our analysis is based on array-derived methylation data rather than whole-genome bisulfite sequencing, potentially limiting the detection of drift in non-CpG sites and distal regulatory elements. Third, we did not integrate matched transcriptomic or additional epigenomic layers, which constrains our ability to systematically assess the downstream regulatory consequences of drift. Future studies employing multiomic, single-cell, and longitudinal designs across diverse tissues and larger cohorts will be critical to further refine and validate our model of epigenetic drift in human aging.
Methods
Study cohorts, DNA methylation, serum metabolomics, SNP microarray, and phenotyping
NSPT (Peng et al. 2024), CAS (Peng et al. 2024), and Changfeng (Li et al. 2024) cohorts have been described in detail previously and related data access information is provided in the Supplemental Notes. No new participants were recruited. In brief, all cohorts consisted of Chinese individuals, and genome-wide DNA methylation was measured using the Illumina Infinium MethylationEPIC BeadChip. Written informed consent was obtained from all participants, and each study was approved by the respective institutional review board.
Genome-wide DNA methylation in all three cohorts was profiled using Illumina MethylationEPIC BeadChips. DNA extraction and bisulfite conversion followed published protocols (Peng et al. 2024; Xia et al. 2024). Raw IDAT files were processed using minfi (NSPT) or CHAMP (CAS and Changfeng) without background correction. Quality control excluded samples with unclear gender and probes with SNPs, sex chromosome location, or high missingness. Missing values were imputed (impute.knn), Type-2 bias corrected (BMIQ), and batch effects adjusted (ComBat on M-values).
The NSPT cohort included detailed phenotypic data (e.g., height, weight, blood pressure) and 13 blood biochemical traits. Serum metabolomics on a subset was performed using a 600 MHz NMR platform (Bruker), analyzed with B.I.LISA and B.I.Quant-PS software (Wu et al. 2021). Genotyping was conducted using the Illumina Global Screening Array, followed by standard QC in PLINK and imputation with 1000 Genomes (SHAPEIT3 and IMPUTE2). After QC, 8,603,582 SNPs remained for analysis.
Statistical analyses
White method for detecting epigenetic drift-CpGs
To identify epigenetic drift-CpGs, defined as CpG sites exhibiting age-related heteroscedasticity, we employed an improved two-step regression method based on White's heteroscedasticity test (White 1980).
In the first step, we constructed a linear regression model (Eq. 1) using beta values of each CpG and age, calculating the squared residuals from the least squares regression while also correcting
for potential confounding factors such as gender, BMI, and cell composition that may affect DNA methylation levels. Cell type
proportions were estimated using the EpiDISH algorithm (Zheng et al. 2018)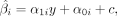 (1)
where α1i and α0i denote the estimated effect and bias of the ith CpG site, respectively. y = (y1, y2, …, yn), c = (c1, c2, …, cn), and
(1)
where α1i and α0i denote the estimated effect and bias of the ith CpG site, respectively. y = (y1, y2, …, yn), c = (c1, c2, …, cn), and 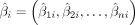 represent the age, covariates (gender, BMI, and cell composition), and estimated beta values of the ith CpG site for all samples, respectively, with n denoting the sample size. The deviation
represent the age, covariates (gender, BMI, and cell composition), and estimated beta values of the ith CpG site for all samples, respectively, with n denoting the sample size. The deviation 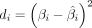 between the true beta value βi and the estimated value
between the true beta value βi and the estimated value 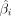 reflects the degree of epigenetic drift at CpG site i under a given age.
reflects the degree of epigenetic drift at CpG site i under a given age.
In the second step, we regressed di on age using the following: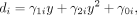 (2)
where γ1i, γ2i, and γ0i denote the estimated age effect, age square effect, and bias of the ith CpG site. To assess whether Equation 2 significantly differs from the null model with no variables, we performed a hypothesis test on the model in Equation 2 compared with a model without age effect using a χ2 statistic, and obtained the corresponding P-value.
(2)
where γ1i, γ2i, and γ0i denote the estimated age effect, age square effect, and bias of the ith CpG site. To assess whether Equation 2 significantly differs from the null model with no variables, we performed a hypothesis test on the model in Equation 2 compared with a model without age effect using a χ2 statistic, and obtained the corresponding P-value.
Simulation benchmarking for heteroscedasticity testing of drift-CpGs
To evaluate the performance of existing heteroscedasticity testing methods in detecting epigenetic drift-CpGs, we conducted a comprehensive simulation study. We generated four distinct types of synthetic DNA methylation data sets: (1) a baseline data set with no heteroscedasticity or outliers (to assess Type-I error); (2) a data set with outliers but no heteroscedasticity (to evaluate robustness to outliers); (3) a data set exhibiting linear age-related heteroscedasticity; and (4) a data set demonstrating nonlinear age-related heteroscedasticity.
We then compared the Type-I error and statistical power of four methods: Liu et al. (2023) (Method A, double generalized linear model using dglm R package); Bergstedt et al. (2022) (Method B, heteroscedastic likelihood ratio test using gamlss R package); Slieker et al. (2016) (Method C, Breusch-Pagan test); and our improved White method (Method D). Performance was evaluated based on false positive rates for Data set 1, impact of outliers in Data set 2, and statistical power for linear (Data set 3) and nonlinear (Data set 4) drift-CpGs. Detailed simulation parameters and method implementations are provided in Supplemental Notes.
Epigenome-wide drift study
Prior to epigenome-wide drift analysis, we removed 342,815 CpGs from a total of 811,876 CpGs in the discovery NSPT data set, including 284,128 DNA methylation CpGs, that is, those significantly affected by mQTLs in Chinese populations (Peng et al. 2024), 57,691 CpGs with no variation (variance < 1 × 10−5), and 996 CpGs failing the multimodal distribution test (R-diptest, P < 1 × 10−4), resulting in a total of 469,061 CpGs. Then, we conducted epigenome-wide drift identification using the White method on these 469,061 CpGs and further investigated and compared the performance of the other three methods. P values smaller than 1 × 10−7 (Bonferroni P < 0.05) were considered as epigenome-wide significant. Significant drift-CpGs were categorized into positive drift (age-increasing interindividual variability) and negative drift (age-decreasing variability). Beyond identifying drift-CpGs, our analysis comprehensively characterized their properties and regulation through exploring correlations between DNA methylation variation and initial/terminal levels (details in Supplemental Notes).
Epigenome-wide association study (EWAS) for chronological age
To identify the age-associated CpGs, termed here as clock-CpGs, we used a linear model to perform epigenome-wide association analysis based on 469,061 CpGs in the NSPT cohort with the same starting amount as the EWDS. P values smaller than 1 × 10−7 (Bonferroni P < 0.05) were considered as epigenome-wide significant. Covariates included gender, BMI, cell composition, experiment batch, the first five genetic principal components, and the first five epigenetic principal components. Genomic principal components (genomic PCs) were calculated using PLINK 1.9 based on all genome-wide SNPs. For methylation principal components (methylation PCs), we applied the prcomp function in R to the β values of 810,000 CpG sites across the genome.
Cell type–specific and single-cell RNA-seq integration
To assess cell type–specific contributions to methylation drift, we adapted the CellDMC framework (Zheng et al. 2018), modeling the interaction between age and estimated cell type proportion (estimated using EpiDISH) (Zheng et al. 2018), for significant drift-CpGs. For each CpG, age-dependent methylation drift specificity across cell types was assessed, with Bonferroni-adjusted P-values < 0.05 considered significant. To elucidate the contribution of age-related methylation drift to interindividual immune variation, we integrated population-scale epigenetic drift profiles with single-cell transcriptomic data from peripheral blood mononuclear cells in the OneK1K cohort (Yazar et al. 2022). We compared transcriptional dynamics between individuals at the extremes of the age spectrum. The BASiCS algorithm (Vallejos et al. 2015) was used to estimate age-associated changes in both transcriptional levels and noise, stratified by methylation drift direction.
Biological annotations
CpGs were mapped to the hg19 build for genomic annotations (e.g., Enhancer, TSS1500, TSS200, UTR5, 1stExon, ExonBnd, Body, UTR3, Promoter) and CpG island annotations (N_Shelf, N_Shore, Island, S_Shore, S_Shelf). Relative enrichment analysis of chromosome states and gene regions was conducted separately for positive/negative drift and clock CpGs. Significance was assessed using a hypergeometric test (P < 0.05). The formula for odds ratio calculation and detailed annotation categories are in Supplemental Notes. Transcription factor binding site (TFBS) enrichment analysis on drift-CpGs was performed using the TFmotifView web tool (https://bardet.u-strasbg.fr/tfmotifview/), extracting 10-bp sequence windows centered on each CpG site. Statistical significance was assessed using Bonferroni correction (adjusted P < 0.05). We tested drift-CpGs for enrichment of gene ontologies and KEGG pathways using the gometh function using R package “missmethyl” (v1.28.0). Using the online analysis platform EWAS atlas, we performed phenotype enrichment analysis on successfully validated drift-CpGs. To explore the distinct biological functions associated with positive and negative drift, we performed enrichment analyses separately for the sets of positive drift-CpGs and negative drift-CpGs, applying a significance threshold of 0.05 after FDR adjustment.
Replication analysis
Drift-CpGs significant in the NSPT discovery analysis (P < 1 × 10−7) were followed up with a replication analysis in the CAS cohort using the White method. Further validation was performed in the mixed Caucasian and Hispanic population assessed with a 450K beadchip, obtained from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE40279 (Hannum et al. 2013). In the longitudinal Changfeng population, CpG drift values between two time points were calculated and analyzed using paired t-tests. The stability of drift was also investigated in the twin cohort, obtained from GEO under accession number GSE61496 (Tan et al. 2014). We categorized the twins into two age groups (e.g., Age < 50 and Age ≥ 50) and used a t-test to examine if the absolute methylation level discrepancies between twin pairs differed significantly between the two groups.
Construction of epigenetic drift score
To quantify an individual's epigenetic drift burden, we developed the epigenetic drift score. This score aggregates individual-level methylation variability at robustly selected CpGs, reflecting an individual's overall drift status. First, we selected highly robust drift-CpGs, defined as those significantly associated with age-related variability in the NSPT discovery cohort and consistently replicated in both CAS and Hannum cohorts. To ensure independence, CpGs within 500-kb proximity of a more significant CpG were removed. Specific filtering criteria and the final number of independent CpGs are detailed in Supplemental Notes.
For each individual i and selected drift site j, we computed the site-specific drift magnitude (dij) as the squared deviation of methylation level from the mean, scaled by the site's standard deviation: 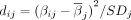 , where dij denotes the drift magnitude for individual i at site j, βij is the methylation level for individual i at site j,
, where dij denotes the drift magnitude for individual i at site j, βij is the methylation level for individual i at site j, 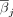 is the mean methylation level at site j, and SDj is the standard deviation of methylation at site j. To derive weights for aggregation, a nonnegative least squares regression between each site's drift score dij and the age of the individual yi was then performed, yi = α + γjdij, where α is the intercept term, and γj is the regression coefficient reflecting the correlation between drift score and age. The overall positive epigenetic drift
score
is the mean methylation level at site j, and SDj is the standard deviation of methylation at site j. To derive weights for aggregation, a nonnegative least squares regression between each site's drift score dij and the age of the individual yi was then performed, yi = α + γjdij, where α is the intercept term, and γj is the regression coefficient reflecting the correlation between drift score and age. The overall positive epigenetic drift
score 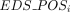 for individual i was then calculated by summing across selected sites (K), weighted by their respective nonnegative regression coefficients (γj):
for individual i was then calculated by summing across selected sites (K), weighted by their respective nonnegative regression coefficients (γj): 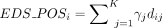 .
.
These weighting factors, derived from the NSPT and Hannum cohorts, serve as a standard reference. EDS_POS scores were subsequently range-normalized to a 0–1 scale using reference populations to project minimum and maximum possible scores. The negative epigenetic drift score (EDS_NEG) was constructed following a similar approach. This involved using significant negative drift-CpGs from the NSPT cohort and then range-standardizing the scores to a 0–1 scale (details on site selection and final number of sites in Supplemental Notes). As a validation, we also implemented an entropy-based approach (Scherer et al. 2020) to measure individual-level DNA methylation variability. Genome-wide Shannon entropy was computed for positive and negative drift-CpGs, and its concordance with EDS values was assessed (details and formula in Supplemental Notes).
Association of EDS with age, metabolome, and genetic variants
We evaluated correlations between EDS and chronological age in the NSPT and CAS cohorts and assessed EDS distributions across gender groups. In the NSPT cohort, we used linear regression to test associations between EDS and NMR-derived lipoprotein subfractions and small metabolites, adjusting for covariates (see Supplemental Notes). Significance was determined using FDR-adjusted P < 0.05. GWAS of EDS in NSPT was conducted using linear regression models in PLINK, adjusting for relevant covariates. EDS heritability was estimated using GCTA. Age-associated chromatin state changes at key loci were visualized using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) with hMSC ChIP-seq data from GEO under accession number GSE156409 (McCauley et al. 2021).
Published software and resources
Publicly available software and R packages utilized in this study include: R (V4.4.0) (R Core Team 2024), ggplot2 (Wickham 2016), diptest (https://cran.r-project.org/web/packages/diptest/index.html), missMethyl (Phipson et al. 2016), poolr (Cinar and Viechtbauer 2022), corrplot (https://github.com/taiyun/corrplot), forestplot (https://github.com/gforge/forestplot), SHAPEIT3 (O'Connell et al. 2016), IMPUTE2 (Howie et al. 2009), PLINK2.0(Purcell et al. 2007), GCTA (Yang et al. 2011), and IGV (URLs in Supplemental Notes).
Software availability
The source codes used in this study to identify and quantify epigenetic drift and to generate corresponding graphs are available at GitHub (https://github.com/Fun-Gene/EpigeneticDriftScore) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The authors thank the study participants, the general practitioners, pharmacists, Human Phenome Data Center of Fudan University, and the staff from the NSPT, CAS, and Chengfeng Study for their dedication, commitment, and contribution. We thank Mr. Yuan Cheng for valuable discussions on statistical aspects of the heteroscedasticity test. F.L. was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB38010400, XDC01000000), the Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01), the National Natural Science Foundation of China (NSFC; 81930056), the Science and Technology Service Network Initiative of Chinese Academy of Sciences (KFJ-STS-QYZD-2021-08-001, KFJ-STS-ZDTP-079), and the Naif Arab University for Security Sciences (NAUSS-23-R17, NAUSS-23-R18, NAUSS-23-R19). S.W. was supported by the National Natural Science Foundation of China (Grant No. 92249302 and 32325013), the CAS Project for Young Scientists in Basic Research (Grant No. YSBR-077), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB38020400), the Shanghai Science and Technology Commission Excellent Academic Leaders Program (22XD1424700), and the Human Phenome Data Center of Fudan University. X.G. was supported by the Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01). M.X. was supported by the National Natural Science Foundation of China (Grant No. 32371333) and the Program of Shanghai Academic Research Leader (Grant No. 23XD1423300). W.L. was supported by the National Natural Science Foundation of China (Grant No. 32200472) and the China Postdoctoral Science Foundation (Grant No. 2021M693274 and BX2021336).
Author contributions: X.F. and F.L. conceived the study. X.F. and Q.Q. developed mathematical formulations, performed data analyses, developed the computer pipeline, and visualized and interpreted results. T.L., W.L., and H.L. contributed to data collection. C.Z., P.J., H.L., X.G., M.X., L.J., S.W., and F.L. provided the data resources and obtained funding. X.F., Q.Q., and F.L. drafted the initial manuscript. All authors approved the submission of the final version of the article for publication.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280155.124.
-
Freely available online through the Genome Research Open Access option.
- Received October 25, 2024.
- Accepted July 23, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








