
APOBEC3A drives deaminase mutagenesis in human gastric epithelium
- Yohan An1,10,
- Ji-Hyun Lee2,10,
- Joonoh Lim1,3,10,
- Jeonghwan Youk1,11,
- Seongyeol Park1,12,
- Ji-Hyung Park1,4,
- Kijong Yi1,3,
- Taewoo Kim1,13,
- Chang Hyun Nam1,
- Won Hee Lee1,14,
- Soo A Oh3,
- Yoo Jin Bae5,
- Thomas M. Klompstra2,
- Haeun Lee1,
- Jinju Han1,
- Junehwak Lee6,
- Jung Woo Park6,
- Jie-Hyun Kim5,
- Hyunki Kim7,
- Hugo Snippert8,
- Bon-Kyoung Koo2,9 and
- Young Seok Ju1,3
- 1Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34051, Republic of Korea;
- 2Center for Genome Engineering, Institute for Basic Science, Yuseong-gu, Daejeon 34126, Republic of Korea;
- 3Inocras Inc., San Diego, California 92121, USA;
- 4Department of Neurology, Severance Hospital, Yonsei University College of Medicine, Seoul 03722, Republic of Korea;
- 5Department of Internal Medicine, Division of Gastroenterology, Gangnam Severance Hospital, Yonsei University College of Medicine, Gangnam-gu, Seoul 06229, Korea;
- 6Korea Institute of Science and Technology Information, Daejeon 34141, Republic of Korea;
- 7Department of Pathology, Yonsei University College of Medicine, Seoul 03722, Republic of Korea;
- 8Oncode Institute, Center for Molecular Medicine, University Medical Center Utrecht, 3521 AL Utrecht, the Netherlands;
- 9Department of Life Sciences, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea
-
↵10 These authors contributed equally to this work.
Abstract
Cancer genomes frequently carry apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC)-associated DNA mutations, suggesting APOBEC enzymes as innate mutagens during cancer initiation and evolution. However, the pure mutagenic impacts of the specific enzymes among this family remain unclear in human normal cell lineages. Here, we investigate the comparative mutagenic activities of APOBEC3A and APOBEC3B, through whole-genome sequencing of human normal gastric organoid lines carrying doxycycline-inducible APOBEC expression cassettes. Our findings demonstrate that transcriptional upregulation of APOBEC3A leads to the acquisition of a massive number of genomic mutations in just a few cell cycles. In contrast, despite clear deaminase activity and DNA damage, APOBEC3B upregulation does not generate a significant increase in mutations in the gastric epithelium. APOBEC3B-associated mutagenesis remains minimal even in the context of TP53 inactivation. Further analysis of the mutational landscape following APOBEC3A upregulation reveals a detailed spectrum of APOBEC3A-associated mutations, including indels, primarily 1 bp deletions, clustered mutations, and evidence of selective pressures acting on cells carrying the mutations. Our observations provide a clear foundation for understanding the mutational impact of APOBEC enzymes in human cells.
Large-scale cancer genome studies have revealed various mutational processes in human somatic cells (Alexandrov et al. 2020). APOBEC enzymes, originally known for cytosine deamination in the DNA and RNA against pathogens (Vieira and Soares 2013), were concluded as major endogenous mutagens in cancer (Nik-Zainal et al. 2012a,b; Lawrence et al. 2013; Roberts et al. 2013). About 75% of human cancer types, including bladder transitional cell carcinoma (98%; 381 of 389 samples), breast adenocarcinoma (83%; 759 of 915 samples), and stomach adenocarcinoma (21%; 101 of 486 samples) showed APOBEC-associated mutational patterns (Sondka et al. 2024). In parallel, the APOBEC-associated mutations are also observed in non-neoplastic normal cells, particularly within the epithelium of the bladder, bronchus, and small intestine (Lawson et al. 2020; Yoshida et al. 2020; Wang et al. 2023). Their mutational spectra, predominantly C > T and C > G base substitutions enriched in TpCpN context (with the mutated cytosine underlined), corresponds to COSMIC signatures SBS2 and SBS13 (https://cancer.sanger.ac.uk/signatures/). Of the 11 APOBEC family genes in the human genome, APOBEC3A (A3A) and APOBEC3B (A3B) have been suggested as major potential contributors to the APOBEC-associated mutations in most human cell types (Roberts et al. 2013; Chan et al. 2015), along with APOBEC1 in the small intestine (Wang et al. 2023).
The mutagenic potential of A3A and A3B has been investigated across a range of human cancer cell lines (Burns et al. 2013; Petljak et al. 2022; Carpenter et al. 2023). In doing so, APOBEC-associated mutagenic activity has been proposed to occur episodically in cancer cell line models (Petljak et al. 2019). In addition, nonhuman model systems, such as Mus musculus, Saccharomyces cerevisiae, and the cell line derived from Gallus gallus domesticus, have also been utilized for exploring the mutagenic impact of APOBEC enzymes (Chan et al. 2015; Law et al. 2020; DeWeerd et al. 2022; Durfee et al. 2023; Naumann et al. 2023). However, these systems are suboptimal, as these models carry confounding factors, such as additional oncogenic alterations or nonhuman genomic backgrounds. To isolate the pure mutagenic activity of A3A and A3B in human normal cells, we explored genomic alterations in human non-neoplastic gastric organoid lines following APOBEC upregulation, using single-cell cloning and whole-genome sequencing (WGS) (Jager et al. 2019; Pleguezuelos-Manzano et al. 2020; Youk et al. 2024) as well as duplex DNA sequencing (Hoang et al. 2016).
Results
Gastric organoids with doxycycline-inducible APOBEC genes
We established human gastric organoid lines with doxycycline-inducible expression of either A3A or A3B, referred to as hGOiA3A and hGOiA3B, respectively. In these lines, doxycycline treatment simultaneously induces (1) mCherry fluorescence, and (2) expression of the corresponding APOBEC enzyme. To this end, we integrated two cassettes into the genome of gastric organoids (1) expressing rtTA and hygromycin B resistance protein (CMV-rtTA-HygR), and (2) expressing APOBEC enzymes and fluorescence protein (TRE-APOBEC (A3A or A3B)-IRES-mCherry) using the piggyBac transposon system (Fig. 1A; Woodard and Wilson 2015; Wilson et al. 2007). Successfully engineered organoids were selected using hygromycin treatment and subsequently constructed into clonal lines. WGS confirmed their single-cell origin (Supplemental Fig. S1), the copy number and genomic positions (Supplemental Tables S1, S2), and the correctness of the reading frame of the APOBEC gene in the integrated cassettes (Supplemental Fig. S2).
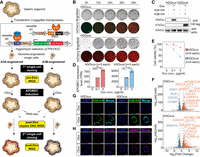
Overview of conditional APOBEC (APOBEC3A or APOBEC3B) overexpression models with human gastric organoids. (A) Experimental design of the study. Schematic illustration of genetic engineering, cloning, and sequencing (whole-genome sequencing, duplex sequencing, and bulk RNA-seq) is shown. Dox, doxycycline. (B) Changes of morphology and mCherry fluorescence during 48 h with 0.1 µg/mL doxycycline treatment; (top) hGOiA3A lines, and (bottom) hGOiA3B lines. Scale bars represent 1 mm. (C) Inducible expression of APOBEC3A (A3A) or APOBEC3B (A3B) enzymes in hGOiA3A and hGOiA3B lines, respectively, following 3 µg/mL doxycycline treatment for 48 h. (D) Expression levels of APOBEC3A (A3A) or APOBEC3B (A3B) in each line following 0.1 µg/mL and 3 µg/mL doxycycline treatment for 48 h; (left) hGOiA3A lines (n = 3 per condition), and (right) hGOiA3B lines (n = 3 per condition). Data are presented as mean ± 95% confidence interval. Statistical significance was determined using a t-test: (*) P < 0.05, (**) P < 0.005, (****) P < 0.00005. (E) Changes of viability of each line following 0.1 µg/mL and 3 µg/mL doxycycline treatment for 48 h (n = 4 per each condition). Data are presented as mean ± 95% confidence interval. (F) Differentially expressed genes upon APOBEC (A3A or A3B) overexpression following 0.1 µg/mL doxycycline treatment for 48 h; (top) hGOiA3A line, and (bottom) hGOiA3B line. adj.pval, adjusted P-value. (G) Subcellular localization of overexpressed A3A and A3B following 3 µg/mL doxycycline treatment for 48 h in each line; (left) hGOiA3A line, and (right) hGOiA3B line. Scale bars represent 50 µm. (H) γ-H2AX foci following 3 µg/mL doxycycline treatment for 48 h in each line; (left) hGOiA3A line, and (right) hGOiA3B line. Scale bars represent 50 µm.
Upon doxycycline treatment, mCherry fluorescence as well as APOBEC upregulation were clearly detected within 48 h (Fig. 1B,C). Here, we treated with 0.1 µg/mL and 3 µg/mL of doxycycline for low- and high-level induction, respectively. Gene expression levels of induced A3A or A3B ranged from 400 to 6000 transcripts per millions (TPMs), representing a several hundred-fold increase over endogenous levels (Fig. 1D; Supplemental Fig. S3). Of note, endogenous APOBEC transcripts minimally contributed <0.2% of the total APOBEC transcripts (Supplemental Table S3).
Single-cell transcriptome data from various cancer types indicated that the range of APOBEC expression levels observed in our models were comparable to those observed during episodic bursts in many cancer types, including lung adenocarcinoma, head-and-neck squamous cell carcinoma, triple-negative breast cancer, esophageal adenocarcinomas, and squamous cell carcinomas (Supplemental Fig. S4; Puram et al. 2017; Karaayvaz et al. 2018; Wu et al. 2018; Maynard et al. 2020). Unfortunately, expression levels in gastric adenocarcinoma could not be explored due to the lack of available single-cell Smart-seq data.
Following A3A or A3B induction, the viability of the organoid lines was substantially compromised, suggesting a detrimental impact of APOBEC upregulation on cell survival (Fig. 1E). After APOBEC induction, differentially expressed genes included those involved in the cell cycle (e.g., CDKN1A and CDC20) and immune response (e.g., CXCL8 and CCL20) in both the hGOiA3A and hGOiA3B models (Fig. 1F; Amador et al. 2007; Wu et al. 2007; Cazzalini et al. 2010; Matsushima et al. 2022). The induced APOBEC enzymes predominantly localized to the nuclei and led to an increase in γ-H2AX foci, suggesting APOBEC-induced DNA damage, such as replication stalling or double-strand breaks (DSBs), as previously reported (Fig. 1G,H; Supplemental Fig. S5; Burns et al. 2013; Green et al. 2016).
APOBEC3A, not APOBEC3B, induces genomic mutations
To assess the mutagenic impacts of A3A and A3B, we investigated acquired mutations in the hGOiA3A and hGOiA3B lines after 48 h of doxycycline treatment. On average, the hGOiA3A lines treated with 0.1 µg/mL and 3 µg/mL doxycycline exhibited 267 and 2448 SBS2/13 base substitutions genome-wide, respectively (0.1 µg/mL: 95% CI, 107–505; 3 µg/mL: 95% CI, 1052–3708) (Fig. 2A,B,F; Supplemental Tables S4, S5). In contrast, the hGOiA3B clones showed a negligible number of APOBEC-associated mutations following induction (0.1 µg/mL: 95% CI, 0–1) (Fig. 2A,D,F; Supplemental Tables S4, S5).
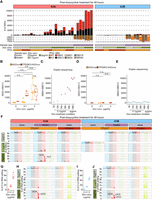
Mutational impact of APOBEC-associated single base substitutions in DNA and RNA. (A) Mutational burden of SNVs following APOBEC overexpression (A3A or A3B), measured by whole-genome sequencing of the clones and duplex DNA sequencing. The number of SNVs measured by the duplex DNA sequencing was normalized per diploid genome; (left) hGOiA3A lines, and (right) hGOiA3B lines. (B) Mutational burden of APOBEC-associated SNVs in the hGOiA3A and TP53KO-hGOiA3A clone sequencing. The number of A3A-associated SNVs (SBS2+SBS13) in hGOiA3A and TP53KO-hGOiA3A clones under each condition. Statistical significance was determined using a t-test: (*) P < 0.05, (n.s.) not significant. (C) Number of A3A-associated SNVs (SBS2+SBS13; normalized per diploid genome) in BotSeqS results for hGOiA3A lines under each doxycycline treatment condition. Black lines represent 95% confidence intervals based on a Poisson distribution. (D) Mutational burden of APOBEC-associated SNVs in the hGOiA3B and TP53KO-hGOiA3B clone sequencing; the number of A3B-associated SNVs (SBS2+SBS13) in hGOiA3B and TP53KO-hGOiA3B clones under each condition. Statistical significance was determined using a t-test. (E) Number of A3B-associated SNVs (SBS2+SBS13; normalized per diploid genome) in BotSeqS results for hGOiA3B lines under each doxycycline treatment condition. Black lines represent 95% confidence intervals based on a Poisson distribution. (F) Mutational burden and spectra of APOBEC-associated SNVs in each experimental condition. The number of SNVs in BotSeqS results were normalized per diploid genome; (left) hGOiA3A lines, and (right) hGOiA3B lines. (G) Number of C > U RNA editing in bulk RNA-seq in hGOiA3A lines (n = 3 per condition), normalized per 3.1 Gb of mapped bases. (H) Spectra of RNA editing in trinucleotide contexts in hGOiA3A lines. (I) Number of C > U RNA editing in bulk RNA-seq in hGOiA3A lines (n = 3 per condition), normalized per 3.1 Gb of mapped bases. (J) Spectra of RNA editing in trinucleotide contexts in hGOiA3B lines.
To minimize potential detection bias in our single-cell cloning system, where proliferative cells are preferentially sequenced, we employed duplex DNA sequencing (Hoang et al. 2016), which precisely captures nonclonal mutations across the entire cell population, including those confined to single cells that could not proliferate further. The mutational burden per diploid genomes from the duplex DNA sequencing revealed a time-dependent increase in A3A-associated mutations following doxycycline treatment, ultimately resulting in higher levels than those observed from the clones (0.1 µg/mL: 95% CI, 4590–6038; 3 µg/mL: 95% CI, 9316–10,077) (Fig. 2A,C; Supplemental Table S6). In contrast, but in line with our observation from the clones, APOBEC-associated base substitutions in hGOiA3B lines remained minimal in duplex sequencing (0.1 µg/mL: 95% CI, 136–352; 3 µg/mL: 95% CI, 79–133) (Fig. 2A; Supplemental Table S6). Of note, unlike hGOiA3A, we did not detect A3B-associated mutations even at earlier time points (Fig. 2E), suggesting that the absence of mutations in hGOiA3B is unlikely to be due to the negative selection against hypermutated cells. Our findings overall indicate that A3B alone does not act as a major mutator, at least in human normal gastric epithelium.
Despite the absence of fixed mutations in hGOiA3B models, the A3B enzyme in cell lysates was fully functional, exhibiting strong cytosine deamination activity for extracellular DNA oligonucleotides in vitro (Fig. 3A). Denatured genomic DNA fragments exposed to recombinant A3B enzymes underwent near-complete deamination of unmethylated (non-CpG) cytosines ([∼99%]) upon contact with the enzyme (Fig. 3B,C). This activity was further confirmed in the duplex DNA sequencing, where lysates from hGOiA3B induced a substantial number of C > T artifacts during library preparation, particularly in the 5′ head region (∼50 bp) of the sequencing reads with a clear positive correlation with the doxycycline concentration, as reported previously (Supplemental Fig. S6A,B; Abascal et al. 2021). Finally, the increase in γ-H2AX foci in the hGOiA3B following doxycycline induction (Fig. 1H; Supplemental Fig. S5) also indicates DNA damage consistent with A3B activity. Collectively, our findings strongly indicate that gastric organoids either efficiently repair A3B-induced lesions or possess intrinsic mechanisms that suppress A3B-associated cytosine deamination in the nucleus.
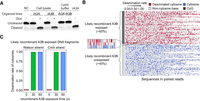
Deamination activity of APOBEC3B in vitro. (A) Cytosine deaminase activities of cell lysates from hGOiA3A and hGOiA3B lines following 3 µg/mL doxycycline treatment for 48 h. NC, negative control; rA3A, recombinant A3A. (B) Sequences in randomly selected 100 read pairs in recombinant APOBEC3B-exposed cell-free denatured genomic single stranded DNA for 60 sec. (C) Deamination rate of unmethylated cytosines in recombinant A3B-exposed cell-free DNA fragments. 0, negative control; 30, DNA exposed for 30 sec; 60, DNA exposed for 60 sec.
To further investigate whether inactivation of tumor suppressor genes accelerates APOBEC-associated mutagenesis, we generated the hGOiA3A and hGOiA3B lines carrying biallelic truncating mutations in TP53 (hereafter referred to as TP53KO-hGOiA3A and TP53KO-hGOiA3B, respectively) (Fig. 1A). In the TP53KO-hGOiA3A lines, we observed a similar burden of APOBEC-associated mutations as that observed in the hGOiA3A lines (0.1 µg/mL: 95% CI, 785–1917; 3 µg/mL: 95% CI, 152–3091) (Fig. 2A,B; Supplemental Table S5). The TP53KO-hGOiA3B lines exhibited a lack of APOBEC-associated mutations consistent with the hGOiA3B lines (0.1 µg/mL: 95% CI, 11–19; 3 µg/mL: 95% CI, 2–12) (Fig. 2A,D; Supplemental Table S5). Taken together, these findings imply that the inactivation of TP53 does not promote APOBEC-associated mutagenesis in gastric organoids.
Despite the increased γ-H2AX foci following A3A or A3B overexpression (Fig. 1H; Supplemental Fig. S5), which often indicate DNA DSBs, we did not observe a remarkable increase of structural variations (SVs) (Supplemental Table S5) in all the clones, including TP53KO-hGOiA3A and TP53KO-hGOiA3B lines. These findings indicate that APOBEC-induced DSBs or replication stalls are efficiently repaired within gastric organoids.
Both APOBEC3A and APOBEC3B induce C > U RNA editing
Despite their contrasting mutagenic impact on DNA, both enzymes exhibited C > U RNA editing events. In RNA sequencing normalized to a total base count of 3.1Gb, both the hGOiA3A and hGOiA3B lines showed ∼eightfold and 30-fold increases in C > U RNA editing sites, following 0.1 µg/mL and 3 µg/mL doxycycline treatment for 48 h, respectively (Fig. 2G,I; Supplemental Fig. S7A; Supplemental Table S7).
The spectra of C > U RNA editing deviated from SBS2 associated with DNA mutations and also varied between A3A and A3B (Fig. 2H,J). Briefly, (1) both A3A and A3B exhibited reduced specificity for the UpCpU context (TpCpT in DNA). However, (2) A3A displayed enhanced specificity for the UpCpG context (TpCpG in DNA), and (3) A3B showed increased specificity for contexts other than UpCpN (non-TpCpN in DNA).
Further de novo decomposition of RNA editing spectra revealed three RNA editing signatures, referred to as RNA-SBSA, RNA-SBSB, and RNA-SBSC, corresponding to A3A-, A3B- and adenosine deaminase RNA specific (ADAR)-associated A-to-I RNA editing, respectively (Supplemental Fig. S8A,B). Although RNA-SBSA was overall consistent with an A3A-associated RNA-editing spectrum previously reported (Martínez-Ruiz et al. 2023; Fixman et al. 2024), RNA-SBSB was slightly different from one for A3B established from Mus musculus (Alonso de la Vega et al. 2023). Spectra in pentanucleotide contexts further showed that A3A-associated RNA editing was preferentially enriched in ApUpCpApN (ApTpCpApN in DNA) and ApUpCpGpN (ApTpCpGpN in DNA) contexts, whereas A3B-associated editing did not show such an enrichment (Supplemental Fig. S9).
The C > U RNA editing sites were frequently recurrent, particularly among the sites identified in the low doxycycline concentration (Supplemental Fig. S7B). Of the 16,574 C > U RNA editing sites detected across all samples, 7593 (45.8%) were recurrent or observed in more than one sample (Supplemental Fig. S7C–E; Supplemental Table S8), suggesting that these were C > U RNA editing hotspots. Of these, 3769 and 2769 were exclusively identified in hGOiA3A and hGOiA3B lines, respectively (Supplemental Fig. S7D,F).
A3A-associated RNA editing hotspots were enriched in specific sites in the secondary stem-loop structure of RNAs, including the 3rd, 4th, and 4th positions of the 3-bp, 4-bp, and 5-bp RNA-loop structures, respectively, as reported previously (Supplemental Fig. S7G; Jalili et al. 2020). Further, we found an enrichment in specific positions of the larger loops (Supplemental Fig. S7G). In contrast, A3B-associated RNA editing hotspots did not show site preferences in the RNA-loop structures (Supplemental Fig. S7G). The lower context stringency in A3B may be caused by the structural differences of α1/loop-1 and β-2 residues of the CD2 domain (Kim et al. 2023).
Characteristics of APOBEC-associated mutations
Using the pure A3A-mediated mutational profiles from hGOiA3A lines, we examined the detailed characteristics of A3A-associated mutational signatures. These analyses could not be applied to A3B due to the near absence of A3B-associated mutations in this study (Supplemental Fig. S10A–C). A3A-associated mutations in the TpCpA context were 2.7× more abundant following pyrimidine bases (YpTpCpA) compared to purine bases (RpTpCpA) (Fig. 4A). Of note, the RpTpCpA sequence context has been reported as a preferred motif for A3B-associated mutations in the Saccharomyces cerevisiae and in vitro model (Chan et al. 2015; Sanchez et al. 2024). The YpTpCpA preference in our study closely mirrors the enrichment level observed in human cancer tissues carrying APOBEC-induced hypermutations (YpTpCpA/RpTpCpA = 2.5 in cancers with SBS2/13 burdens >5000 genome-wide), indicating that A3A could be a key enzyme of the APOBEC-associated hypermutations in most human cancers.
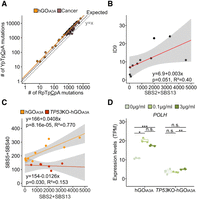
Characteristics of A3A-associated mutational signatures. (A) Context preference of A3A between YpTpCpA and RpTpCpA context in hGOiA3A lines and APOBEC-associated mutations in hypermutant cancer samples. Only PCAWG cancer samples with a combined APOBEC-associated clonal mutational burden (SBS2+SBS13) greater than 5000 were selected (n = 63) among eight cancer types with a high prevalence of APOBEC mutational activity: lung adenocarcinoma (n = 15), breast adenocarcinoma (n = 12), bladder urothelial carcinoma (n = 11), head-and-neck squamous cell carcinoma (n = 13), lung adenocarcinoma (n = 6), uterine corpus endometrial carcinoma (n = 3), esophageal adenocarcinoma (n = 2), and stomach adenocarcinoma (n = 1). Dashed black line, expected; orange line, hGOiA3A; dark brown line, cancer. (B) Correlation between A3A-associated base substitutions and ID9 contributing indels among hGOiA3A lines. (C) Associations between A3A-associated (SBS2 and SBS13) and age-associated (SBS5 and SBS40) SNVs among hGOiA3A lines and TP53KO-hGOiA3A clones. (D) Changes in POLH gene expression (translesion synthesis DNA polymerase) following A3A induction in hGOiA3A and TP53KO-hGOiA3A lines.
Further, in line with previous reports from cancer (DeWeerd et al. 2022), indels attributable to the COSMIC ID9 signature (Sondka et al. 2024) showed a suggestive positive correlation with the number of A3A-associated base substitutions in hGOiA3A clones, one per 333 SBS2/13 base substitutions (P-value = 0.051) (Fig. 4B). Of note, base substitutions attributable to clock-like mutational signatures (SBS5 and SBS40) demonstrated a positive correlation with the burden of A3A-associated base substitutions (SBS2 and SBS13) (Fig. 4C), suggesting that A3A-induced genomic damage indirectly promotes error-prone DNA repair processes across the genome. However, this association was absent in hGOiA3A clones carrying TP53 truncating mutations (Fig. 4C). We speculate that this difference reflects differential DNA repair pathways according to the activities of TP53 (Kim et al. 2016). Notably, among the polymerases encoded in the human genome, increased transcription of DNA polymerase eta (POLH), which is involved in translesion synthesis (TLS) (Choi and Pfeifer 2005; Delbos et al. 2005), was exclusively observed in clones with functional TP53 (Fig. 4D; Supplemental Fig. S11). Previously, REV1, a component of the TLS pathway, was shown to contribute to the generation of SBS5 and SBS40 in cancer cell lines (Petljak et al. 2022). Collectively, this suggests that the TLS machinery may contribute to the mutational processes underlying SBS5 and SBS40.
Clusters of APOBEC-associated mutations
In cancer genomes, APOBEC-induced localized hypermutation events are frequently observed (Nik-Zainal et al. 2012a; The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). Across the mutations detected in our hGOiA3A and TP53KO-hGOiA3A lines, ∼5% of the 29,650 acquired base substitutions were clustered within 1 kbp, which is ∼100-fold higher than expected by chance (Fig. 5A). Such clustered mutation events can be classified as either omikli and kataegis, based on the density (Bergstrom et al. 2022b). Overall, we detected 615 omikli and 109 kataegis events. The observed kataegis events ranged from four to 22 base substitutions (median = 5). The absence of correlation between the clustered mutations in this study and complex genomic events indicate that those were driven by the pure activity of A3A. For instance, SV-associated kataegis, which consists of ∼36% of kataegis events in cancer genomes (Nik-Zainal et al. 2012a; The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020; Bergstrom et al. 2022b), were rare in our data (2.8%; 3 out of 109 events). Similarly, the kataegis in our clones were independent of other known kataegis-inducing genomic events, such as anaplastic DNA bridges (Maciejowski et al. 2020) and extrachromosomal DNA (ecDNA) (Bergstrom et al. 2022b).
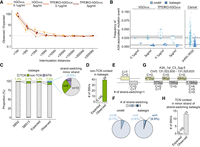
Landscape of APOBEC3A-associated clustered mutation. (A) Observed-to-expected ratios of the intermutational distances between SNVs in each clone. Data are presented as mean ± standard error. (B) Frequencies of clustered mutation events (omikli and kataegis) among A3A-associated base substitutions in clones and cancer genomes. (C) Proportions of classic (in TpCpN context) and nonclassical mutations (in non-TpCpN and NpTpN) in SBS2, SBS13, and kataegis regions in the clones. (D) Comparison of expected and observed number of SNVs in the non-TpCpN context within kataegis regions. (E) Schematic representation of strand-switching kataegis. (F) Strand-switching frequencies of omikli and kataegis across classes of clustered mutation events. (G) A complex strand-switching kataegis involving five switches found in clone A3A_1st_C3_3µg-5. (H) Enrichments of minor strand mutations in the TpCpN context in strand-switching kataegis events.
The relative frequencies of omikli and kataegis remained consistent at 2.4% (95% CI: 1.7%–3.1%) and 0.4% (95% CI: 0.22%–0.58%) of all A3A-associated mutation events, respectively, with each isolated and clustered mutation counted as a single event (Fig. 5B; Supplemental Table S9). These ratios were more or less constant among clones, regardless of the doxycycline concentration or TP53 mutational status. Notably, the frequencies of omikli and kataegis were comparable to the SV-unrelated omikli and kataegis frequencies in cancers (Fig. 5B; Supplemental Table S9).
Within kataegis regions, cytosine alterations did not always occur in the TpCpN context. We observed that ∼7% of cytosine substitutions occurred in non-TpCpN context (39 out of 556 mutations) (Fig. 5C), about twofold higher than expected on SBS2 and SBS13 signatures (7.00% vs. 3.33%; χ2 test, P < 0.005). Our data indicated that these non-TpCpN mutations are not independent to but are part of kataegis for two reasons: (1) non-TpCpN mutations were 267-fold more abundant than observed in the background regions (Fig. 5D); and (2) non-TpCpN mutations within kataegis regions were completely phased with other classical TpCpN mutations on the same allele (27 out of 27), where only 50% phasing would be expected by chance. This suggests that DNA repair mechanisms for cytosine deamination in non-TpCpN contexts are less accurate in the mutagenesis of clustered mutations.
Of the 615 omikli and 106 kataegis events (excluding three kataegis events associated with rearrangements), ∼5% (35 omikli and 5 kataegis events) exhibited strand-switching of the mutated cytosines between parental and daughter strands during replication (Fig. 5E,F). In one kataegis event, composed of nine base substitutions (in clone A3A_1st_C3_3µg-5), we observed five strand-switching events (Fig. 5G). Phasing analysis revealed that clustered mutations on both DNA strands occurred on the same allele (26 out of all 26 informative events). Additionally, the mutational spectrum of the minorly contributing strand was predominantly composed of mainly C > T or C > G mutations in the TpCpN context (62%; 13 out of 21 mutations) (Fig. 5C), implying that all the mutations were APOBEC-associated. Besides, the rate of TpCpN mutations in the minor strand within strand-switching kataegis regions was 124-fold higher than randomly expected (Fig. 5H). Although the underlying mechanism remains unclear, our findings indicate that strand-switching of the A3A enzyme should be possible when generating clustered mutations during replication (Fig. 5F).
Epigenetic contexts associated with A3A-associated mutations
Mutational processes are often influenced by the epigenetic contexts of the genome (Otlu et al. 2023). Of the 14 genomic features examined, three features (replication timing, local transcription level, and H3K27me3) showed potential associations with local A3A-associated mutational burdens (Fig. 6A). Of the three features, replication timing and local gene transcription level demonstrated consistent trends correlating with A3A-associated mutation rates (Supplemental Table S10).
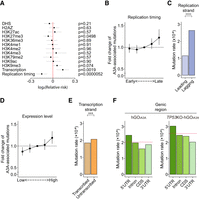
Genomic and epigenomic distribution of APOBEC3A-associated mutations. (A) Correlations between epigenetic markers and A3A-associated substitutions. (B) Fold change of mutation rates of A3A-associated SNVs across genomic regions grouped by replication timing. Data are presented as mean ± 95% confidence interval. (C) Mutation rates on the leading and lagging DNA strands during replication. Statistical significance was determined using a χ2 test: (****) P < 0.00005. (D) Fold change of mutation rates of A3A-associated SNVs across genomic regions grouped by transcription. Data are presented as mean ± 95% confidence interval. (E) Mutation rates on the transcribed and untranscribed DNA strands during transcription. Statistical significance was determined using a χ2 test: (***) P < 0.0005. (F) Mutation rates across subgenic regions (5′-UTR, introns, protein coding sequences [CDSs], and 3′-UTR) in hGOiA3A clones (left) and TP53KO-hGOiA3A clones (right). Red dashed line, average genome-wide mutation rate.
For the replication timing, the latest-replicating regions showed a 1.26-fold higher rate of A3A-associated mutation compared to the earliest-replicating regions (Fig. 6B) as previously reported (Kazanov et al. 2015). This may be attributed to the DNA repair mechanisms, including base excision repair, which are particularly active in early-replicating regions consisting of open chromatin (Amouroux et al. 2010; Rhind and Gilbert 2013). Further, the A3A-associated mutation rate in the lagging strand of DNA replication was 1.26-fold higher compared to the leading strand (Fig. 6C), which presumably originated from more frequent exposure of single-stranded DNA (ssDNA) induced by Okazaki fragments in the lagging strand (Wu et al. 2020), similar to previous elucidation (Hoopes et al. 2016). In addition, mutation rates in genic regions were correlated with expression levels (Fig. 6D), showing a 1.37-fold higher mutation rate in actively transcribed genomic regions compared to silent genic regions, consistent with the previous studies (Nordentoft et al. 2014; Kazanov et al. 2015). Of note, the lagging strand and highly transcribed genes tend to be more frequently single-stranded than the leading strand and silent genes (Okazaki et al. 1968; Gnatt et al. 2001), which may make them more susceptible to A3A-induced DNA damage, respectively. The results demonstrated that DNA regions with frequent ssDNA exposure have a higher chance of being damaged by A3A. In line with this observation, the nontranscribed genic strand was mutated 1.13-fold more frequently than the transcribed strand (Fig. 6E), consistent with a previous report (Saini et al. 2017).
Compared with noncoding sequences, protein-coding sequences showed much lower mutation rates, at 0.79-fold the genome average, suggesting a selective pressure against mutations that could alter amino acid-changing mutations (Fig. 6F). Of note, in TP53-inactivated clones (TP53KO-hGOiA3A clones), mutation rates in protein-coding regions slightly increased to 0.834-fold of the genome average, potentially due to reduced negative selection pressures in the absence of functional TP53.
Discussion
Our study clearly demonstrated the qualitative and quantitative mutational impact of A3A and A3B in human non-neoplastic cells using a gastric organoid culture system. Previous studies have highlighted the mutagenic potential of A3B in various model systems (Chan et al. 2015; Carpenter et al. 2023; Durfee et al. 2023; Dananberg et al. 2024). In contrast, a recent study suggested only a modest contribution of A3B mutagenesis in human cancer cell lines (Petljak et al. 2022). To our knowledge, our study is the first to directly assess the mutagenic activity of A3B in human normal cells.
The gastric organoid model offered distinct advantages based on three key criteria: (1) biological relevance, supported by the frequent occurrence of APOBEC-associated mutations in gastric cancers; (2) experimental feasibility, owing to its robust proliferative capacity under culture conditions; and (3) the availability of standardized protocols for genetic manipulation (Fujii et al. 2015; Gaebler et al. 2020). For example, despite the high prevalence of APOBEC-associated mutations in breast and lung cancers, the corresponding normal epithelial cells are suboptimal for this study due to their limited proliferative capacity in organoid culture as well as their poor compatibility with genetic engineering.
When A3A was induced by 3 µg/mL of doxycycline in hGOiA3A lines, transcription was activated for ∼2–3 days, reaching peak expression levels of ∼800 TPM. Under these conditions, we detected ∼2500 A3A-induced base substitutions in proliferative clones, suggesting that ∼1000 base substitutions could be acquired in a day. These findings support the notion that a transcriptional burst of A3A can also lead to a massive number of mutations in normal gastric epithelium as well, consistent with previous observations in cancer (Petljak et al. 2019). However, the frequency of episodic A3A upregulation per se is unknown in human normal gastric epithelium. According to conventional wisdom, APOBEC overexpression is thought to be associated with viral infection. However, publicly available transcriptomic data from SARS-CoV-2-infected human gastric organoids revealed no substantial upregulation of A3A or A3B (Supplemental Fig. S12; Giobbe et al. 2021).
Our models also indicated increased mutational burdens of SBS5 and SBS40 proportional to the overall burden of A3A-associated mutations, suggesting that base substitutions attributable to other than SBS2 and SBS13 can also be indirectly promoted in APOBEC upregulation. These signatures were significantly enriched in late-replicating regions and correlated with multiple epigenomic features, including replication timing and transcriptional activity, in line with previous reports (Supplemental Fig. S13A,B; Sondka et al. 2024).
Although this study successfully evaluated the mutagenic activity of A3A and A3B in human normal cells, several technical limitations warrant consideration. First, the study was primarily conducted using gastric epithelial cells, and we cannot exclude the possibility that A3B may exhibit substantial mutagenic activity in other cell types. Second, the endogenous copies of A3A and A3B were not inactivated in the hGOiA3A and hGOiA3B models, raising the possibility that a minor fraction of the observed APOBEC-associated mutations and RNA editing sites may have originated from native enzymes. Third, the inclusion of additional control models, such as catalytically inactive mutants (e.g., A3A-E72A and A3B-E255A) (Carpenter et al. 2023) or APOBEC-enzyme inhibitors, would further clarify the mechanisms underlying APOBEC overexpression. Finally, our genomic analyses were conducted using an earlier version of the human reference genome (GRCh37), due to the dependency of our somatic mutation calling pipeline using previously constructed large-scaled unmatched normal sample matrix based on the GRCh37 sequences (Park et al. 2021). Nonetheless, our findings remain consistent in the benchmark analysis with the latest reference (GRCh38) (Supplemental Fig. S14).
Methods
Material availability
Organoids established in this study will be available under a material transfer agreement. To do so, please contact the lead author (ysju{at}kaist.ac.kr).
Human normal gastric samples
Normal gastric tissues were obtained via endoscopic biopsy from a female undergoing routine screening. The protocol for this study was approved by the Institutional Review Board of Yonsei University Gangnam Severance Hospital (3-2018-0207) and KAIST (KH2022-211).
Human stomach organoid culture
Organoid culture methods and media compositions were adopted from previous research with slight modifications (Bartfeld et al. 2015). Wnt3A- and R-Spondin 1-conditioned media were produced with the HEK293 cell line producing Afamin-Wnt3a (Mihara et al. 2016) and the Cultrex HA-R-Spondin 1-Fc 293T cell line (Trevigen, #3710-001-01).
Tissues were incubated in TrypLE (Gibco, #12604013) at 37°C for 30 min, then dissociated into clusters of 10–15 cells by pipetting. After washing with PBS twice and centrifugation at 300g for 5 min at 4°C, pellets were resuspended in cold Matrigel (Corning, #BDL356231) and seeded in 12- or 24-well plates (Merck). Following a 10-min incubation at 37°C in 5% CO2, 0.5–1 mL of prewarmed culture medium was added. Medium was changed every 2–3 days. Organoids were passaged every 2 weeks using Cell Recovery solution (Corning, #354253) and Accutase (Stemcell Technology, #07922) (see Supplemental Methods; Supplemental Table S11).
Preparation of vectors for transfection
Two vectors were purchased: (1) CMV-rtTA-HygR vector (Addgene, #102423) and (2) CRISPR-Cas9 vectors containing gRNA sequence for TP53 (Addgene, #121917). To generate the pPB-CMVmin-APOBEC (A3A or A3B)-IRES-mCherry vectors, either the APOBEC3A (NM_145699.4) or APOBEC3B (NM_004900.4) sequence was cloned into the vector backbone containing a Tet-on system (pPB-TRE-CMVmin-IRES-mCherry) (see Supplemental Methods; Lee et al. 2022).
Transfection of organoids
Transfection methods were adopted from previously reported protocols (Fujii et al. 2015; Gaebler et al. 2020). A mixture of three kinds of vectors was utilized: (1) pPB-TRE-APOBEC (A3A or A3B)-IRES-mCherry; (2) pPB-CMV-rtTA-HygR; and (3) piggyBac transposase. Organoids resuspended in Opti-MEM (Gibco, #31985062) or BTXpress buffer (BTX) were mixed with a vector cocktail. Electroporation was performed using a previously described program from the literature (see Supplemental Methods; Supplemental Table S12; Fujii et al. 2015). Selection was carried out for 1 week with 1 µg/mL hygromycin (InvivoGen, #ant-hg-1). Single-cell cloning was performed using FACSAriaII (BD Biosciences), followed by manual picking of organoids derived from single cells by pipetting (Youk et al. 2021).
Doxycycline treatment
A doxycycline (Sigma-Aldrich, #D9891-1G) stock solution was prepared by dissolving doxycycline in DMSO. Prior to treatment, dissociated 10k viable cells were seeded in 24-well plates. After 7 days, doxycycline solution was added to the medium, and the organoids were incubated with doxycycline.
Capturing fluorescent images
mCherry fluorescence following doxycycline treatment was visualized using a fluorescence microscope (LEICA, DMi8). Fluorescent images were captured using Las X programs, and brightness/contrast adjustments were applied using the same program.
Preparation of cell lysates
Organoids treated with 3 µg/mL doxycycline were harvested using Cell Recovery solution (Corning), followed by one wash with PBS. The isolated cell pellets by centrifugation were resuspended in lysis buffer containing 25 mM HEPES (pH 7.9), 10% glycerol, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM MgCl2, 1 mM ZnCl2, RNase A (0.2 mg/mL; Thermo Fisher Scientific, #EN0531), and 1× protease inhibitors (Thermo Fisher Scientific, #877885). Cell lysates were then sonicated, incubated on ice for 30 min, and centrifuged at 13,000 rpm at 4°C for 10 min. The supernatant was collected, and protein concentration was measured with Qubit Protein and Protein Broad Range (BR) Assay kits (Thermo Fisher Scientific, #Q33211).
Western blotting
Lysates were prepared by mixing samples 1:1 with Laemmli Sample Buffer (Bio-Rad, #161-0737), followed by denaturation at 95°C for 5 min. Proteins were separated by SDS-PAGE on a Mini-PROTEAN TGX pre-cast 12% gel (Bio-Rad, #4561044) in SDS running buffer (Higene, #PB151-10h) and transferred to an Immobilon PVDF membrane (Millipore, #IPFL00010) via overnight wet transfer in Tris/Glycine buffer (Bio-Rad, #1610771).
Primary antibodies included anti-HA.11 Epitope Tag Antibody (1:5000; BioLegend, #951514) and anti-α-Antin-1 (1:1000; Sigma-Aldrich, #A2066). HRP-conjugated secondary antibodies were used at 1:2000 dilution: goat anti-mouse IgG-HRP for anti-HA (Santa Cruz, #sc-2005) and goat anti-rabbit IgG-HRP for anti-actin (Santa Cruz, #sc-2004) (see Supplemental Methods).
Whole transcriptome sequencing library construction
RNA was isolated during DNA extraction for BotSeqS libraries with AllPrep DNA/RNA Mini kit (QIAGEN, #80204), following the manufacturer's instructions. Libraries were constructed with the NEBNext Ultra II Directional DNA Library Prep kit for Illumina (NEB, #E7760) and the QIAseq FastSelect -rRNA HMR kit (QIAGEN, #334388), according to the manufacturer's instructions. Libraries were sequenced on the NovaSeq 6000 with paired-end sequencing.
Calculating RNA expression levels
Bulk RNA-seq reads were aligned to GRCh37 using STAR2 v2.6.1d (Dobin et al. 2013). TPM and read counts were calculated with RSEM v1.3.1 (Li and Dewey 2011). Differential expression gene analysis was conducted using the DESeq2 package in R (Love et al. 2014).
Calculation of the ratio between endogenous APOBEC mRNA and overexpressed APOBEC mRNA
Sequence differences between the endogenous and overexpressed mRNA were utilized for counting (see Supplemental Methods). The proportion of endogenous mRNA was estimated by calculating the ratio of supporting reads.
Analysis of APOBEC3A and APOBEC3B expression levels in single cancer cells with public scRNA data
Publicly available single-cell RNA-seq data sets, generated with a Smart-seq library, from lung adenocarcinoma, triple negative breast cancer, esophageal adenocarcinoma, and esophageal squamous cell carcinoma were analyzed using the same pipeline applied to our bulk RNA-seq data. Epithelial cell populations were first identified following the tutorial workflow of the Seurat R package (Satija et al. 2015), without applying a cell filtering step. Within the epithelial population, cancer cells were distinguished based on the presence of large-scale copy number variations (CNVs) inferred using the inferCNVpy Python package (https://infercnvpy.readthedocs.io/en/latest/index.html). CNV profiles were calculated using fibroblast and endothelial cell populations as reference nonmalignant cells. For the head-and-neck squamous cell carcinoma data set, where raw data were not available, we utilized a publicly provided summary table containing TPM values and annotated cell types.
Viability assay of organoids
Organoid viability was assessed using the Celltiter-Glo 3D Assay kit (Promega, #G9681), following the manufacturer's instructions (see Supplemental Methods). Viability was calculated as the percentage of luminescence in doxycycline-treated samples related to the average luminescence of control groups.
Immunostaining and imaging of organoids
Whole-mount staining of human gastric organoids was performed as previously described (van Ineveld et al. 2020). Briefly, organoids were fixed with 4% paraformaldehyde (PFA) after depolymerizing the 3D matrix using ice-cold Cell Recovery solution (Corning). After washing with 0.1% PBS-Tween-20 and blocking with organoid washing buffer (0.1% Triton X-100, 0.02% SDS, 0.2% bovine serum albumin [BSA] in 1× PBS), immunolabeling was performed with mouse anti-HA tag antibody (1:50; Santa Cruz, #sc-7392) and rabbit anti-phospho-histone H2A.X antibody (1:400; CST, #2577S) to detect HA-tagged APOBEC proteins and DNA damage, respectively. Secondary antibodies included goat anti-mouse Alexa Fluor 488 (1:500; Thermo Fisher Scientific, #A-11001) and donkey anti-rabbit Alexa Fluor 647 (1:500; Thermo Fisher Scientific, #A32795). Nuclei were counterstained with DAPI (1:1000; Sigma-Aldrich, #D9542). After washing, FUnGI clearing solution was added to the organoids, which were then mounted between two coverslips with a 0.25-mm-deep iSpacer (SunJin Lab, #IS213). Imaging was performed at least 1 h after slide preparation. Imaging was performed with a Leica Stellaris 8 Confocal Microscope. Alexa Fluor 488, Alexa Fluor 647, and DAPI signals were obtained using either 40× or 63× objectives, with a digital zoom factor of one- to seven-fold. The X/Y resolution was set to 1024 × 1024 pixels. Images were processed and analyzed using Adobe Photoshop.
Standard whole-genome sequencing alignment
According to the manufacturer's protocol, DNA from clonal organoids was extracted using a DNeasy Blood & Tissue kit (QIAGEN, #69506), and libraries were constructed with TruSeq DNA PCR-Free Library Prep kits (Illumina, #20015963). Whole-genome sequencing was performed on the NovaSeq 6000 with a mean 30× depth of coverage. Adapter sequences in the FASTAQ files were removed by cutadapt software (Martin 2011). For WGS and BotSeqS, reads were aligned to the human reference genome GRCh37 using BWA-MEM v0.7.17 (Li and Durbin 2010). Further processing, including sorting, marking duplication, and indel realignment, was conducted with SAMtools v1.9 (Li et al. 2009), Picard v2.1.0 (McKenna et al. 2010), and GATK tools v3.8.0 (McKenna et al. 2010). For in vitro deamination results, Bismark v0.23.0 (https://felixkrueger.github.io/Bismark/) was utilized for the mapping process.
BotSeqS library construction
Libraries constructed with TruSeq DNA PCR-Free Library Prep Kit (Illumina) were utilized for the BotSeqS libraries construction. The construction of BotSeqS libraries was based on the previous literature with slight modifications (Hoang et al. 2016). Briefly, after the quantification of DNA libraries with the KAPA Library Quantification Kit Illumina Platforms (Roche, #KK48247), libraries equivalent to 4 pg of DNA were amplified with primers having a Y-adaptor sequence with a phosphorothioate bond(*) at the 3′ end from IDT:
Forward: 5′-AATGATACGGCGACCACCGAG*A-3′
Reverse: 5′-CAAGCAGAAGACGGCATACGA*G-3′
PCR was performed for 20 cycles, following the protocol of the KAPA Library Amplification kit (Roche). Libraries were sequenced as paired-end sequencing (2 × 151 bps) on the NovaSeq 6000.
Calling copy number variations
Copy number variations were accessed by Sequenza (Favero et al. 2015). CNVs in segments smaller than 1 Mbp were considered false-positives. After removing these short-segment CNVs, final CNV profiles were obtained through a second run of the Sequenza.
Confirming non-neoplasticity in primary human gastric organoid
Germline variant calling was performed using the germline mutation calling mode in GATK v4.0.12.0 (McKenna et al. 2010). The primary call set was first filtered using in-house scripts based on the pysam module in Python (Li et al. 2009). Given the relevance of cancer driver genes, the functional impact of variants was evaluated. The absence of CNVs in the normal gastric organoids was also confirmed (Supplemental Fig. S15).
Detection of somatic mutations
To detect single nucleotide variants and indels in clonal organoids, GATK Mutect2 v4.1.9 (McKenna et al. 2010) and Strelka2 v2.9.2 (Kim et al. 2018) were utilized. Bulk whole-genome sequencing of first single-cell cloned lines, hGOiA3A, TP53KO-hGOiA3A, hGOiA3B, and TP53KO-hGOiA3B organoids, were utilized as matched normal for calling somatic mutations in doxycycline-treated organoids. False-positive variants in each call set were filtered out with in-house Python scripts annotating information within BAM files with the pysam module (see Supplemental Methods). The filtered call sets from both Mutect2 and Strelka2 were merged, and the union call set was utilized for downstream analysis. To exclude mutations generated during the culture of first single-cell cloned lines, recurrent somatic mutations observed across multiple samples were removed.
For BotSeqS, VarScan2 v2.3.9 (Koboldt et al. 2012) was used to increase the sensitivity. Similarly, in-house Python scripts were utilized to remove false-positive calls (see Supplemental Methods). Unlike with standard whole-genome sequencing, overexpressed A3B induced the significantly increased C > T artifacts in the 5′ head region during the BotSeqS library construction process (Supplemental Fig. S6A,B). Thus, the false-positive SNVs located within 100 bp of the 5′ end or 3′ end, considering reference strand, were removed. Additionally, rare variants from HEK293 cells, which were used to generate the conditioned medium, were observed. Thus, variants observed at least three times in the HEK293 BAM file were removed from the final call set.
Calculation of mutation rate in BotSeqS
Unlike standard WGS, the effective covered region was calculated for each BAM file. First, each read was evaluated using in-house scripts that considered DNA strand orientation and applied the following criteria to isolate effective DNA fragments: (1) median mapping quality >20; and (2) total depth of each type of reads ≥3. Only regions where both F1R2 and F2R1 reads were aligned were included in the covered region calculation. To account for the exclusion of mutations located within 100 bp of the extreme of read-ends during variant filtering (considering the reference genome strand), the total length of the covered region was adjusted by multiplying it by [(151 × 2–110)/(151 × 2)]. Mutation rates were calculated by dividing the number of observed mutations by this adjusted covered length. To normalize mutational burdens to the standard genome length, mutation rates were then multiplied by dividing the number of observed mutations by this adjusted covered length. To normalize mutational burdens to the standard genome length, mutation rates were then multiplied by the total genome length excluding repeat regions (3,041,373,115 bp) and further doubled.
Analysis of mutational signatures
Mutational signature analysis of single base substitutions (SBSs) and small indels were carried out using the nonnegative least squares method. The mutational signature was represented by 96 patterns of SBSs and 83 patterns of small indels (Alexandrov et al. 2020). Prelearned catalogs of mutational signatures in COSMIC (Sondka et al. 2024) were used to fit individual samples with a known set of signatures for each tissue type. SBS2 and SBS13 (known APOBEC-associated signatures) were included for all cases.
Calling RNA editings
VarScan2 was utilized to identify RNA editing. WGS of first single-cell cloned lines served as the paired normal reference. RNA editings were filtered with in-house Python script with the pysam module (see Supplemental Methods). To compare the RNA editing counts across samples, the number of RNA editings was normalized by a total base count. The total base count was calculated using SAMtools, considering only reads with mapping and base quality >20. The lowest total base count (3.1 Gb) was used as the normalization baseline. After calculating the normalization factor, the adjusted total depth of variant position and variant read counts were filtered with the same criteria. Recurrent RNA editing sites were defined as those observed in at least one sample treated with 0.1 µg/mL or 3 µg/mL doxycycline with the normalized callset.
Analysis of RNA editing signatures
RNA editing signatures were obtained by a modified version of the mutational signature extraction method described in previous studies (Alexandrov et al. 2013; Youk et al. 2024). Briefly, nonnegative matrix factorization (NMF) was utilized to disentangle an individual RNA editing spectrum based on a notion of mixed spectra (see Supplemental Methods; Cichocki et al. 2006; Roux et al. 2015). A total of 18 samples (three samples each of 0 µg/mL, 0.1 µg/mL, and 3 µg/mL APOBEC3 exposure with hGOiA3A and hGOiA3B) were analyzed by splitting into two subsets: A3A and A3B sets.
Analysis of secondary structure of RNA editing sites
The secondary structures of RNA editing sites were predicted as described previously (Jalili et al. 2020). Briefly, a 41-bp sequence centered on each RNA editing site in the canonical mRNA was used to assess secondary structure potential. Stem strength was calculated as 3 × G/C pair + 1×A/T pair in stem. Among candidate structures, the most probable one was selected based on the following hierarchical criteria: (1) highest stem strength; (2) greatest number of G/C pairs in the stem; and (3) smallest loop size.
Calling structural variations
Structural variations were identified using DELLY v0.7.6 (Rausch et al. 2012). Raw calls were filtered using in-house scripts from our previous reports (Lee et al. 2019). The final call set was manually reviewed by the Integrative Genomics Viewer (Robinson et al. 2011).
DNA deaminase activity assay
The cytosine deaminase activity assay against DNA was conducted based on the previous literature with slight modifications (Buisson et al. 2019; Sanchez and Buisson 2025). Briefly, a total volume of 50 µL was prepared, containing either 8 µL of normalized cell lysates or recombinant APOBEC3A (1:8 dilution) from an NEBNext Enzymatic Methyl-seq kit (New England Biolabs, #E7120S), and 42 µL of reaction buffer. The reaction buffer consisted of 20 pmol DNA oligonucleotide, 50 mM Tris (pH 7.5), 1.5 U of uracil DNA glycosylase (New England Biolabs, #M2080S), RNase A (0.1 µg/mL; Thermo Fisher Scientific, #EN0531), and 10 mM EDTA. The DNA oligonucleotide was synthesized by Integrated DNA Technologies with the sequence: 5′-(6-FAM)GCAAGCTGTTCAGCTTGCTGA-3′.
The reaction mixture was incubated at 37°C for 40 min, followed by the addition of 0.5 µL of 10 N NaOH and further incubation at 95°C for 40 min. Then, 50 µL of formamide was added, and the mixture was incubated at 95°C for 10 min, followed by cooling at 4°C for 5 min. For analysis, 5 µL of each sample were mixed with an equal volume of Gel Loading Buffer II (Thermo Fisher Scientific, #AM8546G) and incubated at 95°C for 5 min. Separation of the DNA oligonucleotides was performed on a Novex TBE-Urea Gel, 15% (Invitrogen, #EC6885BOX) by electrophoresis at 150 V for 60 min. Imaging was performed using the ibright CL750 Imaging System (Invitrogen).
In vitro deamination of DNA with recombinant APOBEC3B
The NEBNext Enzymatic Methyl-seq (EM-seq) kit (NEB, #E7120S) was utilized, following the manufacturer's protocol, for DNA library construction with slight modifications. Ten picograms of input DNA were utilized for the library construction. For the deamination steps, recombinant APOBEC3B, synthesized by EUPROTEIN INC., was used. The deamination step with APOBEC enzymes were conducted for 30 and 60 sec. Library amplification steps followed the BotSeqS library construction.
Detection of cytosine deamination by recombinant APOBEC3B in vitro
The BotSeqS variant calling pipeline was utilized with slight modifications. Among the criteria, the distances from read ends were excluded. Initially, all filtered mutations were collected. Among mutations in grouped DNA, only those located in DNA fragments where only one strand was mapped were counted. To calculate the genome-wide mutation rates, the BotSeqS pipeline was utilized, with the exception that cytosines or guanines were counted in a strand-specific manner. Because original methylated CpG sites were preserved during the library construction, both total base counts and mutation counts in CpG contexts were excluded from the analysis. To calculate mutation rates in DNA fragments containing at least one C > T variant, the genomic ranges of such fragments were identified using BEDTools (Quinlan and Hall 2010), considering the strand orientation. After this step, cytosines or guanines outside of CpG contexts were counted.
Analysis of 4-bp context mutations
SNVs from 10 hGOiA3A samples (excluding control samples) and six TP53KO-hGOiA3B samples were utilized in the analysis. Among 567 PCAWG samples across eight cancer types with a high prevalence of APOBEC mutational activity—breast adenocarcinoma (BRCA; n = 195), esophageal adenocarcinoma (ESAD; n = 97), stomach adenocarcinoma (STAD; n = 68), head-and-neck squamous cell carcinoma (HNSC; n = 56), lung squamous cell carcinoma (LUSC; n = 47), uterine corpus endometrial carcinoma (UCEC; n = 44), lung adenocarcinoma (LUAD; n = 37), and bladder urothelial carcinoma (BLCA; n = 23)—only samples with a combined clonal APOBEC-associated mutational burden (SBS2+SBS13) greater than 5000 were selected for this analysis (n = 63). This subject included: LUSC (n = 15), BRCA (n = 12), BLCA (n = 11), HNSC (n = 13), LUAD (n = 6), UCEC (n = 3), ESAD (n = 2), and STAD (n = 1). Only clonal mutations were utilized for the analysis.
Calling clustered mutations
SigProfilerClusters (Bergstrom et al. 2022a), a Python module, was utilized to identify clustered mutations. FlexMix, R package (Leisch 2004), was utilized to classify the identified clustered mutations into omikli and kataegis. Because the tool determines the intermutation distance threshold through simulations that randomly distribute SNVs, the total number of SNVs influences the detection rate of clustered mutation events. To correct for the number of clustered mutation events, simulations were conducted (see Supplemental Methods; Supplemental Fig. S16). The “drm” function in the drc, R package (Ritz et al. 2019), was utilized for the analysis. To compare clustered mutation events, 146 samples from the PCAWG database were selected based on the following criteria: (1) nine cancer types showing high prevalence of APOBEC mutational signatures; and (2) samples with fewer than 500 SBS2+SBS13 were excluded in both our samples and PCAWG cancer samples to avoid bias.
Analysis of mutation rates based on epigenetic marker
Relative risk of mutation rates between signal and nonsignal regions was analyzed for each epigenetic marker, based on previous studies (Supek and Lehner 2017; Nam et al. 2023). Genome-wide signals for each marker, including replication timing, were downloaded from Roadmap Epigenomics Consortium for eight cell types (E017, E114, E117, E118, E119, E122, E125, and E127). Fold-enrichment signals were averaged, and regions with values <1 were defined as bin0 (nonsignal); all others were classified as signal-detected. SNVs were counted in each region, and relative risks were calculated, accounting for 3-bp genomic context.
For APOBEC-associated mutations, cytosines in the TCN context were considered as background, and C > T and C > G substitutions at these sites were counted. For SBS5 and SBS40, all thymine bases except within TCN context were used as the reference, and T > A, T > G, T > C, C > T, and C > G substitutions were counted. C > A mutations were excluded to avoid overlapping signals with SBS1 and SBS18.
For replication timing and H3K27me3, signal-detected regions were further divided into four equal-length bins to assess fold-enrichment. For transcriptional activity, TPM values in each base were derived from RNA-seq of doxycycline-treated organoids (3 µg/mL, 48 h). Using the “hg19_refGene.txt” file from ANNOVAR (Wang et al. 2010), only genic regions were analyzed. Genes with TPM = 0 were defined as bin0; TPM > 0 regions were binned by quartiles (bin1 = 0.05, bin2 = 1.73, and bin3 and bin4 = 9.68). To account for interactions among transcription, H3K27me3, and replication timing, enrichment analysis was performed using the glm.nb() function from the MASS R package (Venables and Ripley 2003) as described in previous research (Supek and Lehner 2017).
Analysis of mutation rates based on genomic location
SNVs located in previously described mappable regions were utilized throughout the analysis. Gene annotations from the “hg19_refGene.txt” file were utilized to match the additional information of position of SNVs. Classification of subgenic regions (5′-/3′-UTR, protein coding sequence [CDS], and intron) and transcription strand orientation was also based on this gene information. All merged genic regions were used as reference for discrimination of genic and intergenic regions.
For subgenic mutation rate comparisons, only non-overlapping genic regions and CDS regions flanked by introns were used, following a previously reported approach (Frigola et al. 2017).
Comparison of single nucleotide variants (SNVs) between reference genome versions
SNVs were additionally identified using human GRCh38 genome sequence. Coordinates of SNVs based on GRCh37 were converted to GRCh38 using BCFtools/liftover for comparative analysis (Supplemental Fig. S14; Genovese et al. 2024).
Publicly available data sets
Publicly available whole-genome sequencing data were utilized to demonstrate copy number variation in the normal human gastric organoid. WGS of blood from HC05 sample was utilized as the unpaired normal sample (obtained from the European Genome-phenome Archive [EGA; https://ega-archive.org] under accession number EGAS00001006213) (Nam et al. 2023). To compare the 4-bp-context preference and frequency of clustered mutation, SNV calls from the ICGC/TCGA Pan-Cancer Analysis of Whole-Genome (PCAWG) Consortium were utilized for the analysis. The callset data are available for download at https://docs.icgc-argo.org/docs/data-access/icgc-25k-data#relocated-icgc-25k-data.
We analyzed publicly available Smart-seq-based single-cell RNA-seq data sets from five cancer types: (1) lung adenocarcinoma (obtained from the NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession number PRJNA591860) (Maynard et al. 2020); (2) head-and-neck squamous cell carcinoma (BioProject: accession number PRJNA401654; Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/]: GSE103322) (Puram et al. 2017); (3) triple negative breast cancer (BioProject: PRJNA485423; GEO: GSE118390) (Karaayvaz et al. 2018); and (4) esophageal adenocarcinoma and esophageal squamous cell carcinoma (BioProject: PRJNA401501) (Wu et al. 2018).
In addition, we analyzed publicly available whole transcriptomic sequencing data of SARS-CoV-2-infected human gastric organoid (BioProject: PRJNA643724; GEO: GSE153698) (Giobbe et al. 2021) for the correlation between viral infections and expression of APOBEC family genes in human gastric organoid.
Quantification and statistical analysis
All statistical analyses were performed with R version 4.1.3 (https://www.npackd.org/p/r/4.1.3). A two-tailed one-sample t-test was used to evaluate P-values for comparing APOBEC-associated SNVs and expression levels between the groups. Linear regressions were conducted using the basic “lm” function in R to analyze the association among APOBEC-associated SNVs, ID9, SBS5, and SBS40. A χ2 test was utilized to evaluate P-values for comparing replication strand bias and transcription strand bias. A 95% confidence interval was used to determine the statistical range of continuous data.
Data access
All raw and processed sequencing data generated in this study have been submitted to the Korean Nucleotide Archive (KoNA; https://kbds.re.kr/KRA) under accession number KAP240815. Essential in-house scripts used in this study are available on Zenodo (https://doi.org/10.5281/zenodo.12771074) and as Supplemental Code.
Competing interest statement
Y.S.J. is a genomic cofounder of Inocras Inc. All other authors declare no competing interests.
Acknowledgments
The authors thank Youngwon Cho (Epithelial Biology Center, Vanderbilt University Medical Center) for help with the cell viability test, Myungsuk Choi (KAIST) for technical help for organoids, and Mia Petljak (New York University) for her valuable advice and feedback on the manuscript. This work was supported by the Young Investigator Grants from the Human Frontier Science Program (RGY0071/2018 to Y.S.J., B.-K.K., and H.S.); the National Research Foundation of Korea (Leading Researcher Program NRF-2020R1A3B2078973 to Y.S.J.); the Korea Bio Data Station (N24NM016-24 to J.L. and J.W.P.); and the Institute for Basic Science (IBS-R021-D1 to B.-K.K.).
Author contributions: Y.S.J., B.-K.K., and H.S. designed the experiments. J.-H.K. provided human gastric epithelium samples from biopsy. Y.J.B. established organoids from primary human gastric tissues. J.L. and J.W.P. helped with basic bioinformatic works (alignment and calling somatic variants). J-H.L. and J.Y. conducted vector cloning. Y.A. and T.K. conducted the transformation of organoids, with J-H.L. and J.Y. providing advice. Y.A. performed organoid culture, clonal expansion, and DNA/RNA extraction. B.-K.K. and H.K. provided training in organoids culture technologies. Y.A. performed cell viability analysis. J-H.L. performed immunohistochemistry. S.A.O. conducted in vitro deamination experiments and most of the library construction including WGS and RNA-seq. Y.A., J.-H.P., and K.Y. conducted library construction for duplex sequencing analysis. Y.A. conducted most bioinformatic analyses, including alignment, mutation calling, duplex sequencing analysis, and analysis of expression levels with bulk RNA-seq data, with S.P., J.Y., and Y.S.J. providing advice. Y.A. and Jo.L. conducted quantitative and statistical data analysis. Y.A. and Jo.L. conducted mutational signature analysis including RNA editing. W.H.L. helped with the construction of the pipeline for analysis of duplex sequencing. Y.A. and C.H.N. conducted epigenomic analysis of variants and comparing variants based on two reference genomes, GRCh37 and GRCh38. H.L., J.H., and T.M.K. conducted the western blotting experiment.
Footnotes
-
↵Present addresses: 11Division of Hematology and Medical Oncology, Department of Internal Medicine, Seoul National University College of Medicine and Seoul National University Hospital, Seoul 03080, Republic of Korea; 12Stanford Cancer Institute, Stanford University School of Medicine, Stanford, California 94305, USA; 13Center for Neurogenetics, Brain and Mind Research Institute, Weill Cornell Medicine, New York, New York 10044, USA; 14Department of Radiation Oncology, Yonsei Cancer Center, Heavy Ion Therapy Research Institute, Yonsei University College of Medicine, Seoul 03722, Republic of Korea
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280338.124.
-
Freely available online through the Genome Research Open Access option.
- Received December 12, 2024.
- Accepted August 22, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








