
Common and specific gene regulatory programs in zebrafish caudal fin regeneration at single-cell resolution
- Yujie Chen1,2,3,7,
- Yiran Hou1,2,3,7,8,
- Qinglin Zeng1,2,3,
- Irene Wang1,2,
- Meiru Shang1,2,
- Kwangdeok Shin4,
- Christopher Hemauer1,2,9,
- Xiaoyun Xing1,2,3,
- Junsu Kang4,
- Guoyan Zhao1,5,6 and
- Ting Wang1,2,3
- 1Department of Genetics, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 2The Edison Family Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 3McDonnell Genome Institute, Washington University School of Medicine, St. Louis, Missouri, USA;
- 4Department of Cell and Regenerative Biology, School of Medicine and Public Health, University of Wisconsin Madison, Madison, Wisconsin 53705, USA;
- 5Department of Neurology, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 6Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, Missouri 63110, USA
-
↵7 These authors contributed equally to this work.
Abstract
Following amputation, zebrafish regenerate their injured caudal fin through lineage-restricted reprogramming. Although previous studies have charted various genetic and epigenetic dimensions of this process, the intricate gene regulatory programs shared by, or unique to, different regenerating cell types remain underinvestigated. Here, we mapped the regulatory landscape of fin regeneration by applying paired snRNA-seq and snATAC-seq on uninjured and regenerating fins. This map delineates the regulatory dynamics of predominant cell populations at multiple stages of regeneration. We observe a marked increase in the accessibility of chromatin regions associated with regenerative and developmental processes at 1 dpa, followed by a gradual closure across major cell types at later stages. This pattern is distinct from that of transcriptomic dynamics, which is characterized by several waves of gene upregulation and downregulation. We identified and in vivo validated cell-type-specific and position-specific regeneration-responsive enhancers and constructed regulatory networks by cell type and stage. Our single-cell resolution transcriptomic and chromatin accessibility map across regenerative stages provides new insights into regeneration regulatory mechanisms and serves as a valuable resource for the community.
The zebrafish (Danio rerio) serves as an excellent model organism for studying complex tissue regeneration processes owing to its extraordinary capacity to regenerate a wide range of tissues after injury (Gemberling et al. 2013). Upon caudal fin amputation, the adult zebrafish promptly initiates wound closure via epidermal cell migration, which forms a protective barrier and creates a permissive environment for fin regeneration. Subsequently, multiple types of differentiated cells at the proximal region undergo dedifferentiation, migration, and proliferation, whereas a few resident progenitor cells also become activated. Both sources of less differentiated cells migrate to the wound site and undergo lineage-restricted differentiation, eventually recreating the lost fin parts (Knopf et al. 2011; Tu and Johnson 2011; Singh et al. 2012; Pfefferli and Jaźwińska 2015; Ando et al. 2017).
Tissue regeneration is governed by a complex interplay of genetic and epigenetic mechanisms (Goldman and Poss 2020). Tissue regeneration enhancer elements (TREEs) and regeneration-responsive enhancers (RREs) are two terms used to describe cis-elements that modulate gene expression during the regenerative process. For example, enhancers located upstream of lepb and inhbaa respond to injuries in zebrafish fins and hearts (Kang et al. 2016; Thompson et al. 2020; Wang et al. 2020a). Additionally, specific transcription factors (TFs) including fosl1a and vdra are essential in orchestrating the regenerative response (Lee et al. 2020; Chen et al. 2021). However, most previous genomic studies were conducted at the bulk tissue level or with a lineage focus (Kang et al. 2016; Lee et al. 2020; Thompson et al. 2020; Wang et al. 2020a), which may obscure the nuanced contributions of cell-type-specific regulatory elements and molecular networks activated during regeneration. The recent advancement of single-cell genomic technology has empowered the dissection of transcriptome and chromatin accessibility at the single-cell level, thus allowing for an in-depth exploration of the cellular dynamics of tissue regeneration.
Several studies have utilized single-cell/nucleus RNA sequencing (sc/snRNA-seq) to elucidate cell-type contributions, identify cell-type/lineage-specific marker genes, and characterize transitions of cellular transcriptional states during zebrafish caudal fin regeneration (Hou et al. 2020; Jiang et al. 2021; Tang et al. 2022; Lewis et al. 2023; Cudak et al. 2024). Similar approaches have been applied in other regenerative contexts, such as the zebrafish heart (Hu et al. 2022) and axolotl limb regeneration (Gerber et al. 2018; Leigh et al. 2018). Although these studies have generated crucial transcriptomic resources, there are still relatively few investigations that bridge the gap between transcriptomic changes and epigenetic landscape dynamics during regeneration (Wang et al. 2020b; Jimenez et al. 2022; Lyu et al. 2023; Dong et al. 2024). As a result, a clear gap persists in understanding the interplay between transcriptomic and epigenetic modifications in regenerating cells.
In this study, we aimed to bridge this gap by using the zebrafish caudal fin regeneration model. By employing chromium single-cell multiome technology from 10x Genomics, we captured the transcriptomic and epigenomic dynamics simultaneously in the same cells at single-cell resolution of regenerating fins at 1, 2, 4, and 6 days postamputation (dpa). Specifically, we identified both common and cell-type-specific regeneration-responsive gene (RRG) programs and cis-regulatory elements, as well as key TFs associated with stages and cell types during regeneration, enhancing our understanding of tissue regeneration.
Results
Single-nucleus multiomic profiling of zebrafish caudal fin regeneration
To delineate transcriptomic and regulatory dynamics at the single-cell level within the regenerating fin, we performed paired snRNA-seq and single-nucleus assay for transposase-accessible chromatin using sequencing (snATAC-seq) with the 10x Genomics chromium single-cell multiome platform. Nuclei from both pre- and postinjury caudal fins were isolated for analysis. We sampled regenerating fins at 1, 2, 4, and 6 dpa to capture cellular states during the stages of blastema formation, outgrowth, and maintenance (Fig. 1A; Pfefferli and Jaźwińska 2015).
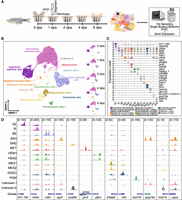
Cell-type identification in regenerating zebrafish caudal fins. (A) General experimental design. Collection of zebrafish caudal fin tissues was conducted at preinjury and 1, 2, 4, and 6 dpa for subsequent library preparation and sequencing. For all samples, we first amputated the distal half of the caudal fin. For the 0 dpa samples, we collected the first two segments of the fin rays immediately after amputation. For the 1, 2, 4, and 6 dpa samples, we collected the regenerated caudal fin tissue. This experimental workflow was partially created using bioRender (https://www.biorender.com/). (B) After quality control, cells were visualized using weighted nearest neighbor (WNN) uniform manifold approximation and projection (UMAP). (Left) Cells colored by cell types; (right) UMAPs separated by time points and colored by cell types. (C) Marker gene expression. Violin plots of log-normalized RNA expression of cell-type marker genes, with colors representing different cell types. (SE) Superficial epithelial, (IE) intermediate epithelial, (BE) basal epithelial, (EM) epidermal mucous, (MET) metaphocyte, (HEM) hematopoietic, (MES) mesenchymal, (ENDO) endothelial, (PGM) pigment, (UN1) unknown-1, (UN2) unknown-2. (D) Genome browser views showing the aggregated and normalized snATAC-seq signals at promoter regions (±1 kb from transcription start site) of selected cell-type marker genes. Tracks are colored with the same color code as in B and C.
After quality control, a total of 83,281 high-quality transcriptome profiles were obtained (Methods) (Supplemental Table S1A). Through unsupervised clustering and leveraging known cell-type-specific marker gene expression, we identified six previously reported major cell types (Hou et al. 2020; Wang et al. 2020a; Tang et al. 2022; Lewis et al. 2023). Specifically, they were superficial epithelial (SE) cells (krt1-19d, cldn23a), intermediate epithelial (IE) cells (cldna, tp63), basal epithelial (BE) cells (cldni, fras1), epidermal mucous (EM) cells (agr2, pvalb8, cldnh), hematopoietic (HEM) cells (ptprc, mpeg1.1, tnfrsf9b), and mesenchymal (MES) cells (msx1b, tnfaip6, prrx1b, cdh11) (Fig. 1B,C; Supplemental Fig. S1A; Supplemental Table S1B). Compared with our previous single-cell RNA-seq study (Hou et al. 2020), this work greatly increased the number of cells assayed (83,282 vs. 18,388). As a result, we were able to identify additional cell types, including pigment (PGM) cells (trpm1b, gpr143), endothelial (ENDO) cells (kdrl, fli1a), and metaphocytes (METs; grn2, cdh28, mpeg1.1) (Supplemental Table S1B). METs, a recently characterized non-hematopoiesis-derived immune cell type in zebrafish, were captured for the first time in a single-cell multiomic study of fin regeneration (Lin et al. 2020; Zhao et al. 2023; Zhou et al. 2023). We also discovered two unannotated cell clusters, each with unique marker gene expression, resembling tuft cells (unknown cells-1: si:ch211-213d14.1, gng13a) and neuromast cells (unknown cells-2: fndc7a, stm) (Supplemental Table S1B). Notably, cldnh was highly expressed in EM, MET, and the two unannotated cell clusters (Fig. 1C). The majority of cell-type annotations were supported by both marker gene expression and the marker gene promoter region accessibility (Fig. 1C,D). Each cell type was also characterized by specifically expressed genes, accessible peaks, and enriched motifs (Supplemental Fig. S1B–D; Supplemental Table S1C–E). All cell types were present in the preinjury fin and across all stages of regeneration, with no new cell populations identified after injury (Fig. 1B; Supplemental Fig. S1E,F; Supplemental Table S1A). Our subsequent in-depth analysis focused on major cell types (e.g., SE, BE, IE, EM, HEM, MES) that had sufficient nuclei numbers and modality information.
Dynamic regulation of common processes across cell types during fin regeneration
One of the most fundamental questions to address is the following: Do different cell types deploy the same or distinct regulatory programs for their regenerative transcriptional reprogramming? An initial pairwise comparison to identify differentially expressed genes (DEGs) between time points for each major cell type revealed dynamic gene expression across all stages and cell types (Fig. 2A; Supplemental Table S2A–F). To identify regeneration gene expression programs common to all cell types, we combined all DEGs from pairwise comparisons within each cell type and identified 781 genes that exhibited expression changes in at least one comparison in each cell type (Fig. 2B). Consensus k-means clustering divided these genes into two modules based on the gene expression change relative to 0 dpa. Predominantly, genes in shared module 1 were downregulated after injury, whereas those in shared module 2 were upregulated (Fig. 2C). These genes were enriched in several processes critical for regeneration in various regeneration model systems and species (Supplemental Table S2G), including regulation of cell population proliferation (GO:0042127), extracellular matrix organization (GO:0030198), morphogenesis of an epithelium (GO:0002009), and inflammatory response (GO:0006954) pathways (Sehring and Weidinger 2020). For example, genes such as btg2 and klf4, associated with the regulation of cell population proliferation, were clustered in shared module 1. btg2 is known for its antiproliferative roles during zebrafish early embryogenesis (Sakaguchi et al. 2001), and klf4 inhibits myoblast proliferation in mouse skeletal muscle development (Cai et al. 2023). Genes in shared module 2, such as mdka, inhbaa, nrp1a, and anxa1c in regeneration (GO:0031099), are documented to facilitate tissue regeneration across various species (Fig. 2C,D; Supplemental Fig. S2C; Jaźwińska et al. 2007; King et al. 2012; Gobbetti and Cooray 2016; Lowe et al. 2019; Ang et al. 2020; Nagashima et al. 2020; Grivas et al. 2021).
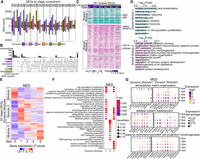
Common and cell-type-specific gene expression programs during fin regeneration. (A) Bar graphs showing the number of DEGs across stages comparisons within each cell type. Colors indicate cell types. The stage DEGs were identified between two stages. For example, “0 vs 1” means that the comparison was between 0 dpa and 1 dpa. Bars above and below the x-axis represent the number of upregulated and downregulated DEGs at 1 dpa, respectively. Similar methodology applies to other pairwise comparisons. (B) An upset plot illustrating unique and shared stage-DEGs among cell types. Stage DEGs identified within a cell type were pooled. The horizontal bar graph on the left shows the number of unique DEGs per cell type. The vertical bar graph on the top indicates the number of overlapped genes in each category. (C) A heatmap displaying log2 fold changes compared with 0 dpa of cell-type-shared DEGs, which were clustered into two modules via k-means clustering. Representative genes involved in cell proliferation and regeneration were highlighted. (D) Top GO terms enriched in each shared module in C. (E) Heatmaps showing scaled expression of all stage DEGs identified within MES, with genes clustered into three dynamic modules. Heatmaps of the other major cell types are in Supplemental Figure S2A. (F) Top 10 GO terms enriched in each dynamic modules identified in MES (E). (G) Dot plots of scaled gene expression across stages in MES showing selected genes identified from GO terms shared in MES dynamic module 1 and dynamic module 3. The GO terms are labeled on the top of each group, with dynamic module 1 genes on the left and dynamic module 3 genes on the right. Dot size indicates the percentage of cells expressing a specific gene, and dot color represents the scaled expression level.
To further elucidate gene and pathway dynamic regulation within each cell type during fin regeneration, we performed consensus k-means clustering based on the Z-scores of expression levels within each cell type, which grouped stage-wise DEGs into three dynamic modules. Each module shared similar expression dynamics across all cell types (Fig. 2E; Supplemental Fig. S2A). Genes in dynamic module 1 were initially highly expressed in preinjury cells and exhibited a rapid downregulation at early regeneration stages but gradually increased their expression at later stages. Genes in dynamic module 2 were induced quickly at 1 dpa and then gradually returned to preinjury levels at later stages. In contrast, genes in dynamic module 3 exhibited a pattern of gradual upregulation after amputation. In MES cells specifically, some genes continued to be upregulated even at 6 dpa. We performed Gene Ontology (GO) enrichment analysis to investigate whether pathways shared across all cell types (Fig. 2D; Supplemental Table S2H) were regulated with similar or distinct dynamics in different cell types (Supplemental Fig. S2B). Most of the pathways were regulated with similar dynamics across all major cell types, usually with one or two exceptions. For example, regulation of cell population proliferation (GO:0042127) was downregulated across all major cell types except SE at the early stage in dynamic module 1, represented by btg2 and klf4 (Supplemental Fig. S2C). In dynamic module 2, modification-dependent macromolecule catabolic process (GO:0043632) and ATP metabolic process (GO:0046034) were shared across most major cell types (Supplemental Fig. S2B,C). In dynamic module 3, morphogenesis and developmental-related pathways (e.g., GO:0120039, GO:0048589), important for regenerate outgrowth (Pfefferli and Jaźwińska 2015), were activated across all major cell types, with many also enriched in MES dynamic module 1 (Supplemental Fig. S2B,C). We then compared the top GO terms enriched in MES dynamic modules. Indeed, several processes were shared between MES dynamic module 1 and dynamic module 3, such as skeletal system development (GO:0001501), extracellular matrix organization (GO:0030198), and plasma membrane bounded cell projection morphogenesis (GO:0120039) (Fig. 2F,G; Supplemental Table S2I). This might indicate that a group of genes (i.e., Fig. 2G, dynamic module 1) regulating these processes needs to be downregulated at early regeneration stages to allow cells dedifferentiating and entering cell cycle, whereas another group of genes (i.e., Fig. 2G, dynamic module 3) regulating the same processes needs to be activated at the late regeneration stage to facilitate redifferentiation and outgrowth. Taken together, these findings revealed a tight and dynamic regulation of common processes across cell types during regeneration.
Chromatin regions linked to regenerative and developmental processes show rapid increase of accessibility at early stage of regeneration
We next investigated the regulatory mechanisms that orchestrate the transcriptional reprograming in the regenerative cell types. We examined chromatin accessibility dynamics throughout the fin regeneration by identifying differentially accessible regions (DARs) between the stages within each cell type (Fig. 3A; Supplemental Table S3). Global chromatin accessibility changes exhibited distinct kinetics compared with gene expression changes. Specifically, more DARs were observed between the preinjury and the regenerative stages than across the regenerative stages. For instance, in MES, approximately 3000 open DARs were identified in the comparison between preinjury and any regenerative stages, whereas almost no DARs were identified between the 2 versus 4 dpa and 4 versus 6 dpa comparisons.
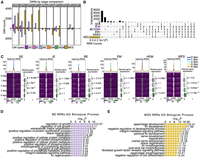
Chromatin regions linked to regenerative and developmental processes show rapid increase of accessibility at early stage of regeneration. (A) Bar graphs display the number of DARs identified between two time points within each cell type during regeneration. Each color represents a different cell type. The stage DARs were identified between any two time points. For example, “0 vs 1” means that the comparison was between 0 dpa and 1 dpa, the number of DARs more open at 1 dpa was shown in the bar graph above the x-axis, and the number of DARs more closed at 1 dpa was shown in the bar plot below the x-axis. Similar methodology applies to other pairwise comparisons. (B) An upset plot shows shared and unique stage DARs across major cell types. All stage DARs compared to 0 dpa, referred as regeneration-responsive regions (RRRs), were combined within each cell type to generate this plot. The horizontal bar graph on the left shows the number of unique stage DARs in each cell type, and the vertical bar graph represents the number of overlapped stage DARs in each category. (C) Aggregated snATAC-seq signals by time point over 5 kb flanking regions centered on opening RRRs of each cell type. Smoothed average signals for each stage are plotted at the top of each heatmap group, with the lines colored by stage. Box plots on the right of each heatmap represent the log2 fold change of expression levels of genes nearest to RRRs across time points, measured across different time points compared with 0 dpa. The P-values were calculated using a Wilcoxon rank-sum test for each two time points. (D,E) Enrichment analysis using the tool GREAT identified processes associated with RRRs in BE cells and MES cells, respectively.
To streamline the analysis, we aggregated the regeneration-opening chromatin regions (regions with increased chromatin accessibility postinjury) identified by the pair-wise comparisons (i.e., 0 vs. 1 dpa, 0 vs. 2 dpa, 0 vs. 4 dpa, 0 vs. 6 dpa) for each cell type, and we designated them as opening regeneration-responsive regions (opening RRRs). Similarly, we combined the regeneration-closing chromatin regions (regions with decreased accessibility postinjury) from all pair-wise comparison of each regenerative stage to 0 dpa and designated them as closing regeneration-responsive regions (closing RRRs). Most of these RRRs were cell type specific (Fig. 3B), with the majority (∼70%) located in intronic and intergenic regions (Supplemental Fig. S3A). Our examination of accumulated signals from RRRs of major cell types at each time point indicated that opening RRRs were predominantly open at 1 dpa and gradually closing in all major cell types at the subsequent time points. Alongside this trend, there was a gradual increase in the expression of genes adjacent to these opening RRRs (Fig. 3C). This pattern underscores a dynamic interplay between chromatin accessibility and gene expression during the regeneration process. The initial opening of RRRs at 1 dpa seems to set the stage for subsequent gene activations that are crucial for the regeneration progression. In contrast, the closing RRRs exhibited a different dynamic in which the chromatin accessibility gradually decreased in the first 2 days but slowly increased at later stages. Except for MES, the nearest genes to these RRRs did not exhibit significant expression changes (Supplemental Fig. S3B).
To further investigate whether alterations in chromatin accessibility correlated with gene expression, we first compared RRGs, identified as DEGs from stage comparisons to 0 dpa, with the RRR-nearest genes in the six major cell types. Up to 40% of the RRR-nearest genes were also RRGs (hypergeometric test, P-value 6.25 × 10−41–5.72 × 10−208) (Supplemental Fig. S3C), with the majority of these RRRs located in nonpromoter regions (Supplemental Fig. S3C,D). For all upregulated and downregulated RRGs in BE and MES, their promoter regions (±1 kb around the transcription start site [TSS]) exhibited similar signal intensities before and after injury globally (Supplemental Fig. S3E,F). This signal stability at the promoter regions suggests that the promoters of RRGs remain largely accessible, and other regulatory mechanisms may influence gene activation or repression. Genes near RRRs with increased peak signals generally had higher expression levels than those near RRRs with decreased peak signals (Supplemental Fig. S4A). Similarly, ATAC-seq peaks adjacent to upregulated RRGs tended to show higher signals than those near downregulated RRGs (Supplemental Fig. S4B). These results indicate that, in general, the increased chromatin accessibility of RRRs was correlated with increased gene expression during regeneration.
The essential tissue types for regenerating fins include wound epidermis and blastema (Pfefferli and Jaźwińska 2015). As the primary constituents of these tissues are BE and MES cells, we focused our functional analysis of RRRs on these two cell types using the GREAT tool (Fig. 3D,E; McLean et al. 2010). Many pathways relevant to fin regeneration were enriched in RRRs of both cell types, including opening RRRs located near lef1 (Poss et al. 2000), mdka (Ang et al. 2020; Thompson et al. 2020; Weinberger et al. 2024), msx1b (Nechiporuk and Keating 2002), inhbaa (Jaźwińska et al. 2007; Wang et al. 2020a), and prdm1a (Supplemental Table S3; Truong et al. 2023). Furthermore, these RRRs exhibited a higher level of sequence conservation, as measured by conservation scores (phastCons) and phylogenetic p-scores (phyloP), than did randomly shuffled regions, indicating higher purifying selection pressures on these sequences (Supplemental Fig. S3G,H). This result suggests that these evolutionarily conserved regulatory sequences might play important roles in regulating RRG expression during zebrafish fin regeneration.
Identification of common and cell-type-specific regeneration-responsive cis-regulatory elements
Our analysis identified both DEGs and DARs that are associated with fin regeneration and appendage development. To understand how cis-regulatory elements regulate their target genes during fin regeneration, we conducted a peak-to-gene link analysis (Stuart et al. 2021) by treating the full data set as bulk and also treating individual cell types as bulk. In total, 124,513 unique peak-to-gene links were identified for 82,429 peaks and 17,310 genes (Supplemental Table S4A). In all peak-to-gene link sets, the majority of linked peaks were linked to their nearest genes (Supplemental Fig. S5A). Additionally, most of these peak-to-gene links were uniquely identified within each cell type (Supplemental Fig. S5B), consistent with a previous study (Ober-Reynolds et al. 2023) that concluded that many cell-type-specific enhancer–gene links could be missed if relying only on the pseudobulk of the full data set.
To better understand how enhancers regulate their target genes, we ranked genes and peaks according to the number of their peak-to-gene links (Supplemental Fig. S5C,D). Within each cell type, the majority of genes were linked with more than one peak, whereas most peaks were linked with only one gene. We defined the genes with more than five linked peaks as highly regulated genes as those exceeding the inflection point (“elbow”) (Supplemental Fig. S5C), as described previously (Ma et al. 2020; Ober-Reynolds et al. 2023). To uncover the functions of these genes, we conducted k-means clustering analysis of the linked peaks associated with highly regulated genes (Fig. 4A). This analysis yielded six distinct clusters of peaks, with each predominantly enriched in a specific cell type. The genes linked to each peak clusters were enriched for cell-type-specific GO terms, such as skeletal system development (GO:0001501) in MES, actin cytoskeleton organization (GO:0030036) in IE and EM, and cell migration (GO:0016477) in BE and HEM (Fig. 4B; Supplemental Table S4B). Additionally, we performed GO term enrichment analysis on the top 3000 genes with only one correlated peak per major cell type. The top GO terms were less cell type specific, underscoring the complexity of regulatory networks and suggesting that highly regulated genes are more likely to be associated with key cell-type-specific functions.
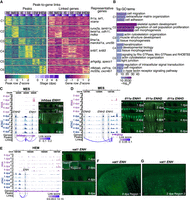
Characterization of common and cell-type-specific cis-regulatory elements in regeneration. (A) Heatmaps display chromatin accessibility (left) and gene expression (right) for peak-to-gene links, with peaks clustered using k-means clustering (k = 6, C1 through C6 represent the six clusters) across cell types and stages. For each cluster, selected highly regulated genes (with more than five linked peaks) relevant to the GO terms in B are displayed next to the gene expression heatmap. (B) Representation of enriched top GO terms associated with the highly regulated genes linked to each peak cluster in A. (C,D) On the left, WashU Epigenome Browser views show aggregated and normalized snRNA-seq signals (pink tracks) and snATAC-seq signals (blue tracks) across time points surrounding genes inhbaa and il11a in MES cells, respectively. Regions of candidate RREs are shaded in gray. Below these signal tracks, the loops illustrate peak-to-gene linkages identified in MES cells. On the right, in vivo enhancer reporter assay results for these enhancer regions are shown across various regenerating stages from F0 zebrafish. The white arrows indicate amputation site. (E) Similar to C and D but presenting a WashU Epigenome Browser view around the gene vat1 in the HEM cells. Candidate RRE is highlighted in gray shades. On the right, in vivo enhancer reporter assay results for this enhancer region are shown across various regenerating stages from F0 zebrafish. The white arrows indicate amputation site. (F,G) Two zoomed-in views from the 2 dpa regenerating fin shown in E.
To improve the likelihood of identifying RREs, we homed in our peak-to-gene links to only including connections between RRRs and RRGs. We obtained a total of 10,906 RRR–RRG pairs between 7435 RRRs and 3342 RRGs (Supplemental Table S4C). Our analysis recovered RREs known to be associated with regeneration-essential genes, and we were able to validate additional novel RREs through in vivo enhancer assays. We selected 10 regions to validate their enhancer activity during regeneration. Nine out of 10 candidates displayed enhancer activities in regenerating fin along with our two positive controls, whereas no activity was observed from the two negative controls (Fig. 4C–G; Supplemental Figs. S6–S8; Supplemental Table S4D). For example, inhbaa is a gene crucial for zebrafish fin and heart regeneration (Jaźwińska et al. 2007; Dogra et al. 2017). We identified two peaks, one (inhbaa-ENH2) of which was previously suggested to be active in the blastema of regenerating zebrafish fins (Fig. 4C; Supplemental Fig. S6A; Wang et al. 2020a). Additionally, we discovered inhbaa-ENH1 with enhancer activity during fin regeneration, which we validated using an in vivo enhancer reporter assay (Fig. 4C). Unlike inhbaa-ENH2, the activity of inhbaa-ENH1 was more enriched along the regenerating fin rays during 1–6 dpa. This observation aligns with the aggregated ATAC signal detected predominantly in MES (Supplemental Fig. S6A; Fig. 4C).
il11a has been implicated in promoting cellular reprograming and minimizing scarring after tissue injury in Xenopus laevis and D. rerio (Tsujioka et al. 2017; Allanki et al. 2021). We identified six peaks linked to il11a expression using the whole data set, three of which were identified in the upstream intergenic region of il11a in MES (Fig. 4D; Supplemental Fig. S6B). The region il11a-ENH2, with ATAC-seq signal present in all six major cell types, was previously reported to exhibit enhancer activity during larval zebrafish tail regeneration (Kang et al. 2016). The aggregated ATAC signals of il11a-ENH1 was more specific to the regenerating MES, whereas that of il11a-ENH3 was detected in multiple major cell types, predominantly in BE and MES (Supplemental Fig. S6B). Transgenic assays for il11a enhancers exhibited results consistent with the ATAC signal distribution. il11a-ENH1 and il11a-ENH2 transgenic fish displayed fluorescent reporter signals in MES. Both il11a-ENH2 and il11a-ENH3 showed enhancer activity in wound epidermis. Although signals in wound epidermal were observed for both il11a-ENH2 and il11a-ENH3 initially, our transgenic assay demonstrated their activities at the later stages were distinct as il11a-ENH2 and il11a-ENH3 activities eventually were restricted near the amputation site and at the distal tip, respectively (Fig. 4D). These results confirm that different enhancers of the same gene can exhibit cell-type specificity (Kang et al. 2016).
To demonstrate that our framework also works for other cell types, we tested in vivo enhancer activity for three HEM-specific RREs near HEM dynamic module 2 genes (intronic regions of vat1 and akap6; a proximal upstream region of gda) and two general RREs (enhancers near genes prdm1a and pcdh12) (Fig. 4E–G; Supplemental Figs. S7, S8). Notably, the akap6-ENH peak-to-gene link was only identified with the HEM subset but not with the full data set (Supplemental Fig. S7A,F). This also suggests that most enhancers are cell type specific and the necessity of identifying peak-to-gene links within cell types. Additionally, TF binding motif footprints of TFs known for their function of governing HEM cell fate and immune responses, including Stat1, interferon regulatory factors (e.g., Irf2), and Cebpa, were detected within HEM-specific enhancer regions (Supplemental Fig. S7E–H; Dai et al. 2016; Glass and Natoli 2016; Campbell et al. 2021; Daniel et al. 2023). In summary, our identification of enhancers and their in vivo validation added further support to the concept that enhancers responsive to regeneration exhibit activity that is both cell type specific and position specific.
Identification of key regulatory factors of fin regeneration through stage-specific gene regulatory network construction
To elucidate how cis-regulatory elements orchestrate gene expression changes in major cell types, we integrated peak-to-gene links, TF motif footprint signals, gene expression correlation, and motif activity–TF expression correlation following previous methods (Lyu et al. 2021, 2023). This approach enabled the construction of gene regulatory networks (GRNs) of major cell types for each stage (Methods) (Supplemental Table S5). Our analysis identified 544 TFs with motif footprints in peaks linked to targeted genes, with 70 TFs shared across all cell types. Notably, several TFs critical for tissue regeneration were ranked at the top across all major cell types by degree centrality, including multiple AP-1 and ETS family TFs (e.g., fosl1a, fosab, fosl2, jund, jun, junba, junbb, fosb, jdp2b, elf1, elf3) (Supplemental Fig. S9A). These factors are involved in various cellular processes crucial for regeneration, such as cell growth, proliferation, differentiation, and apoptosis (Shaulian and Karin 2002; Oikawa and Yamada 2003; Hess et al. 2004). Enrichment of AP-1 and ETS motifs in regions gaining accessibility postinjury has been observed in zebrafish fin and heart regeneration (Beisaw et al. 2020; Lee et al. 2020; Wang et al. 2020a; Ji et al. 2023; Jia et al. 2023; Tamaki et al. 2023). There were also cell-type-specific TFs with known function ranked at top by degree centrality, such as lef1 in BE, runx2a in MES, and spi1 in HEM (Supplemental Fig. S9A; Poss et al. 2000; Brown et al. 2009; Campbell et al. 2021). Focusing on the MES population (Fig. 5), we observed motifs with differential chromVAR motif activity (Schep et al. 2017) across various stages. For example, AP-1 family motifs, including FOS and Jun, showing higher global activity at 1 dpa, as well as RUNX2 and Gli motifs, are crucial for osteoblast differentiation (Shimoyama et al. 2007; Knopf et al. 2011; Wehner and Weidinger 2015). The majority of motifs had higher chromVAR activity scores at 2 dpa, and several associated TFs are known to be essential for tissue regeneration and development, including PRRX1, MSX, LIM, MEF2C, ZIC, and HOX family TFs (Fig. 5A,B; Akimenko et al. 1995; Carlson et al. 2001; Karlstrom et al. 2003; Poss et al. 2003; Arnold et al. 2007; Tzchori et al. 2009; Sedykh et al. 2017; Hamada et al. 2019; Lin et al. 2021; Yoshioka et al. 2021; Qin et al. 2023). For example, in zebrafish fin regeneration, genes encoding TFs, such as msx1b, msx2b, and msx3, are induced in blastema cells. TF MEF2C is recognized for its role in bone development in mice; LIM homeobox TFs such as LHX2 and LMX1B are implicated in limb patterning and outgrowth processes in mice. Target genes of these TFs—such as spp1, pth1r, wnt10a, fn1a, and bmp2b targeted by Runx2; col11a2; and runx2a targeted by Mef2c—have been identified as key players in skeletal and bone development (Fig. 5C,D; James et al. 2006; Komori 2010, 2022; Lawrence et al. 2018; Wu et al. 2022). In conclusion, our construction of GRNs not only identified known TFs that are shared across cell types or expressed in a cell-type-specific manner to regulate fin regeneration but also generated a comprehensive list of candidate TF–target interactions. This work serves as a valuable roadmap for future experimental designs aimed at investigating the functions of lineage-specific TF regulators within their respective cell types.

Gene regulatory network construction in the MES cells. (A) A heatmap displays differential activity of motifs over time in pseudobulk MES cells, with particular motifs of interest marked on the side. Motifs critical for bone development are indicated in red. (B) A dot plot illustrates the expression of TF genes and their degree centrality over time, corresponding to the motifs presented in A. TF genes critical for bone development are indicated in red. (C) A scatter plot shows the top targets of Runx2a at the regenerating stages. Targets were ranked by grnboost2 TF–target importance scores. The y-axis represents grnboost2 score; the x-axis shows log2 fold change of target gene expression compared with 0 dpa. (D) Example views from the WashU Epigenome Browser of Runx2 and Mef2c footprints located in regions that become more accessible during regeneration, along with their nearby targeted genes. Blue tracks are aggregated and normalized snATAC-seq signals across time points; pink tracks are aggregated and normalized snRNA-seq signals across time points. The TF bound track shows predicted TF binding footprints. Shaded areas highlight DARs that gained accessibility during regeneration.
Identification of lineage-specific TFs in MES population
Given the pivotal role of MES cells in blastema formation and outgrowth, we sought to better understand how TFs drive MES lineages during regeneration. We initially performed unsupervised subclustering of the MES cells and identified DEGs across the subclusters. Subsequently, we annotated the subclusters based on identified marker genes for each subpopulation that have been documented in the literature (Fig. 6A–C; Supplemental Fig. S10A; Gavaia et al. 2006; Tornini et al. 2017; McMillan et al. 2018; Hou et al. 2020; Tang et al. 2022; Lewis et al. 2023; Cudak et al. 2024). The MES population was separated into two principal populations distinguished by cdh11 expression, an osteoblast lineage marker (Hou et al. 2020; Tang et al. 2022; Cudak et al. 2024), and and2, expressed in nonbone cells (König et al. 2018; Hou et al. 2020; Cudak et al. 2024). The cdh11-high (cdh11+) MES population comprised subclusters MES_0, MES_1, MES_2, MES_3, MES_4, and MES_5. MES_0, with higher mgp and lower sost expression (Supplemental Fig. S10A), predominantly consisted of cells from 0 dpa, 4 dpa, and 6 dpa (Fig. 6B), representing the mature bone cell population. MES_1 exhibited high expression of tnfaip6, mki67, and pcna (Supplemental Fig. S10A), indicating proliferating MES cells. MES_2 and MES_3, marked by high runx2a and mmp9 expression (Fig. 6C), represented cells at the preosteoblast stage. MES_4 and MES_5, with higher expression of col10a1a, sp7, ifitm5, bglap, and spp1, were maturing osteoblasts and osteocytes. MES_3 were joint cells, confirmed by higher expression of lmx1ba, pthlha, and evx1 (Fig. 6C; Supplemental Fig. S10A; McMillan et al. 2018). The cdh11-low (cdh11−) MES, encompassing subclusters MES_6, MES_7, MES_8, MES_9, MES_10, MES_11, and MES_12, was predominantly composed of fibroblasts. Within this population, MES_7 and parts of MES_6, identified at 0 dpa with higher sost and lower mgp expression, represented mature fibroblast cells. MES_8 and MES_9, marked by hapln1a and tph1b (Fig. 6C), were the cells positioning in the more proximal central blastema region (Govindan and Iovine 2014; Tornini et al. 2017; Cudak et al. 2024). MES_10 and MES_11, enriched with robo3, were cells at the distal blastema (Fig. 6C; Lewis et al. 2023). MES_11 was also marked by mmp13a, lfng, and fgf10a (Fig. 6C; Supplemental Fig. S10A), indicating the distal-most blastema (Cudak et al. 2024). MES_12, with high loxa expression (Fig. 6C), was cells at the distal peripheral blastema region adjacent to wound epidermis (Cudak et al. 2024). MES_6 and MES_0, expressing high fgf10a, fhl1a, rgs5a, mfap5, and abi3bpb (Fig. 6C; Supplemental Fig. S10A), were the distal differentiated fibroblast cells located near the osteoblasts (Lewis et al. 2023; Cudak et al. 2024). Notably, MES_0 and MES_6 were colabeled with abi3bpb and mfap5 (Fig. 6C), indicating that these cells were at the transitional zone between the distal fibroblast blastema and osteoblasts, as described in a recent study (Cudak et al. 2024). We then performed pseudotime analysis for the cdh11+ MES and cdh11− MES populations, respectively, to infer cell lineages starting from the cluster with 1 dpa cells and exhibiting higher tnfaip6 and mki67 expression (Supplemental Fig. S10B,C; Hou et al. 2020). Three lineages were inferred from both the cdh11+ MES and cdh11− MES populations. The inferred lineages aligned with the actual regeneration status (Fig. 6B; Supplemental Fig. S10B,C). Several lineage marker genes were associated with lineage-specific chromatin accessibility signals (Fig. 6D; Supplemental Fig. S10D–G). For example, four intronic peaks of cdh11 were more accessible in osteoblast MES subclusters (MES_0–MES_5), indicating lineage-specific regulation of marker genes (Fig. 6D).
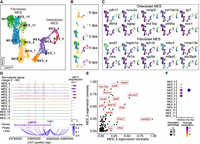
MES sublineages and lineage-specific regulatory networks. (A) A UMAP plot shows MES subclusters. Cells are colored by subclusters. (B) A set of UMAP plots divided from A by time points. (C) A set of UMAP plots with embedded RNA expression of marker genes. (D) A genome browser view generated with Signac CoveragePlot displays pseudobulk snATAC-seq signals for each subcluster around the cdh11 gene. Annotations at the bottom indicate the transcript region, ATAC-seq peaks, and peak-to-gene links. Violin plot on the right side shows the normalized RNA expression levels of cdh11 across each cluster. Tracks and violin plots are colored by subclusters. (E) A scatter plot shows the top TFs in the MES_3 and MES_5 subclusters, ranked by their eigenvector centrality scores. (F) A dot plot showing RNA expression of mef2cb and lmx1ba across subclusters.
Next, we utilized CellOracle (Kamimoto et al. 2023) to infer key TFs and their regulatory networks within MES subclusters (Supplemental Fig. S11A; Supplemental Table S6). Across all clusters, top-ranking TFs, such as fosl1a and elf3, are known to be crucial for fin regeneration and extracellular matrix organization in zebrafish (Lee et al. 2020; Sarmah et al. 2022). A pairwise comparison between MES_3 and MES_5, two clusters with bifurcating differentiating trajectories, revealed two lineage-specific TFs, mef2cb and lmx1ba (Fig. 6E). mef2cb was predominantly expressed in the osteoblast lineage, whereas lmx1ba was featured in the MES_3 joint cell lineage (Fig. 6F). Their central roles in the regulatory network were underscored by their high centrality scores in MES_5 and MES_3, respectively (Supplemental Fig. S11B). In silico perturbations of mef2cb and lmx1ba in MES cells highlighted their crucial role, as the disruption of each TF gene led to the derangement of the entire osteoblast lineage (Supplemental Fig. S11C). Previous studies have shown mef2cb’s importance in zebrafish craniofacial development and heart development (Hinits et al. 2012; Adrião et al. 2023). Mef2 regulates a Runx2 enhancer for osteoblast-specific expression and controls chondrocyte hypertrophy and bone development in mice (Arnold et al. 2007; Kawane et al. 2014). In contrast, overexpression of Lmx1b negatively regulates osteoblast differentiation and bone formation, and its absence can lead to a regeneration outgrowth failure in mouse digit tip (Cesario et al. 2018; Kim et al. 2022; Castilla-Ibeas et al. 2023). These findings suggest that mef2cb and lmx1ba might play important roles in lineage specification within osteoblast MES population.
Discussion
Unlike mammals, zebrafish exhibit remarkable regenerative capabilities, serving as a unique model for exploring the regeneration of various tissues and organs, such as the heart, hair cells, spinal cord, retina, and fin. Numerous studies have discovered crucial genes responsive to injury and regeneration, as well as have characterized the activation of these genes following injury. Several cis-regulatory elements, known as tissue regeneration enhancer elements (TREEs), regeneration-responsive elements/enhancers (RREs), and tissue regeneration silencer elements (TRSEs) (Kang et al. 2016; Lee et al. 2020; Thompson et al. 2020; Wang et al. 2020a; Cao et al. 2022; Harris 2022; Jimenez et al. 2022; Sun et al. 2022; Cigliola et al. 2023; Ando et al. 2024), have been recognized for their roles in activating or repressing regeneration-associated genes in various tissues postinjury. For instance, the LEN enhancer upstream of the lepb gene is active during zebrafish fin and heart regeneration, although distinct regions of LEN are activated in different tissue (Kang et al. 2016). The enhancers fgf20aE48, mdkaE20, and cx43E24 exhibit spatially distinctive activities (Thompson et al. 2020). These discoveries raise questions about which gene expression programs are activated or repressed in different cell types, what cis-regulatory elements are linked to gene expression, and which key TFs regulate gene expression programs through these cis-elements.
Employing the paired single-cell transcriptome and chromatin accessibility profiling method, we consistently identified major cell types in both preinjury and postinjury fin tissues. In line with our previous report (Hou et al. 2020), the major cell populations are three layers of epithelial cells: EM cells, HEM cells, and MES cells. Our current study also captured ENDO cells, PGM cells, METs, and two unannotated cell clusters. METs are a newly characterized type of immune cell in zebrafish that do not derive from hematopoiesis. The marker gene expression of MET in our data set aligns with the previous studies (Alemany et al. 2018; Lin et al. 2020; Zhao et al. 2023; Zhou et al. 2023). Our data set also provides the single-cell level chromatin accessibility of MET for the first time. EM cells, METs, and the unannotated clusters expressed the gene cldnh at high levels. The unannotated clusters exhibited a unique marker gene expression similar to that of intestinal tuft cells and lateral line neuromasts, respectively, both of which play roles in chemosensation and immune response (Froehlicher et al. 2009; Schneider et al. 2019). This suggests the presence of potentially uncharacterized cell types in zebrafish fin and requires further exploration.
In dominant cell types, gene expression is dynamic during regeneration, displaying a pattern consisting of an initial downregulation of genes related to processes such as antiproliferation and of rapid upregulation of genes related to protein catabolic and ATP metabolic processes. Subsequently activated genes are mainly involved in development and morphogenesis programs. Notably, several processes related to development and morphogenesis were shared between MES dynamic module 1 and dynamic module 3. This suggests that downregulation of these processes at early stages of regeneration enables cells to dedifferentiate and re-enter the cell cycle, whereas the reactivation at later stages of regeneration promotes redifferentiation and outgrowth.
In contrast to these dynamic gene expression patterns, chromatin accessibility shows marked differences between pre- and postinjury states but remains relatively stable throughout regeneration. Aggregated ATAC-seq signals reveal that RRRs gain accessibility at or before 1 dpa, indicating that chromatin accessibility changes are early and immediate responses to injury. This early openness of RRRs gradually reduces throughout all the regenerative time points we profiled, but upregulation of adjacent genes happens gradually and in a coordinated fashion, particularly in the MES. This finding suggests a decoupling between chromatin accessibility and gene expression dynamics, in which early chromatin openness may prime gene expression programs necessary for regeneration. Specifically, the early and pronounced chromatin openness at the initial stages of regeneration might facilitate the activation of gene expression programs associated with regeneration. This phenomenon of modality decoupling, in which changes in chromatin accessibility precede those in gene expression, aligns with patterns observed in developmental processes, particularly during lineage commitment (Ma et al. 2020). However, the specific regulatory mechanisms driving these early chromatin changes in regeneration remain to be fully characterized. Based on existing literature, we hypothesize several potential mechanisms—(1) histone modifiers, such as histone acetyltransferases (HATs) (Bannister and Kouzarides 2011); (2) pioneer TFs, such as members of the AP-1 family (Beisaw et al. 2020; Wolf et al. 2023); and (3) chromatin remodelers, such as the SWI/SNF complex (Alver et al. 2017; Wolf et al. 2023)—might play key roles in facilitating early chromatin accessibility by repositioning nucleosomes. Future studies, including experiments that profile chromatin dynamics at earlier regeneration time points and employ targeted genetic and biochemical approaches, will be necessary to test these hypotheses and identify the precise regulatory mechanisms involved in early chromatin remodeling during fin regeneration. Such findings suggest a complex interplay between chromatin accessibility and gene expression, highlighting the need for further investigation into the mechanisms driving these processes in tissue regeneration. In BE cells and MES cells, these evolutionarily conserved RRRs are located near genes enriched in development, morphogenesis, and regeneration processes. We validated multiple cell-type-shared/specific opening RRRs for their enhancer activity using in vivo enhancer reporter assays and confirmed that different enhancers of the same gene can be activated in distinct cell types and tissue positions. Moreover, we validated three HEM RRRs with enhancer activity, which contain motif footprints of TFs associated with hematopoiesis and immune response.
Through the integration of various analytical approaches, including peak-to-gene linkage, TF–target inference, correlation of gene expression, and analysis of TF motif footprints, we have constructed GRNs for major cell types across stages. Consistent with prior research, we found that TFs belonging to the AP-1 and ETS families are crucial across the major cell types. We also observed lineage-specific TFs that target genes involved in lineage-specific processes. This network serves as valuable blueprint for further regulatory mechanism research in the tissue regeneration area. The MES population can be split into two main lineages, that is, the fibroblast MES cells and osteoblast MES cells, and the osteoblast lineage further differentiates into joint cells and osteocytes. In silico perturbation (Kamimoto et al. 2023) of key TFs identified in these lineages results in corresponding lineage disruptions. Given the multifaceted functions of these key TFs in developmental processes, conditional knockouts or knockdowns are necessary for further exploration of their specific roles in zebrafish fin regeneration. Overall, our data set, along with the identification of RREs and key TFs, provides a valuable resource for studying zebrafish fin regeneration.
Methods
Fish husbandry and procedures
All zebrafish were used in accordance with protocols 20-0435 and 24-0038 approved by the Washington University institutional animal care and use committee (IACUC). Wild-type TU, AB, and Tg(sp7:EGFP) strains were maintained under standard husbandry in the Washington University fish facility, with system water temperature at 28.5°C and a day–night cycle controlled as 14 h light/10 h dark. For fin amputation, we anesthetized 1-year-old fish with 0.16 g/L MS-222 in system water and then removed the distal half of their caudal fin with sterilized razor blades. The fish were then sent back to a circulating water system for recovery. For all samples, we first amputated the distal half of the caudal fin. For the 0 dpa samples, we collected the first two segments of the fin rays adjacent to the amputation sites immediately after amputation. For the 1, 2, 4, and 6 dpa samples, we collected the regenerated caudal fin tissue adjacent to the amputation sites at specified time points. Each time point included two biological replicates (Supplemental Table S1A). Tissues were collected in 1 mL 1× PBS on ice.
Nuclei preparation and single-nucleus multiome sequencing
Fresh tissues were treated with 1 mL 1× TrypLE Express enzyme (Gibco) expression with rotation for ∼30 min at 30.5°C. Isolation status was checked under a microscope every 15 min after gentle mixing. Once the cells were well dissociated, they were centrifuged at 500g for 3 min at 4°C to remove supernatants. One milliliter of 1× cold PBS buffer was used to wash cells, followed by centrifuging at 500g for 3 min at 4°C. For the 0 dpa sample, supernatants were discarded, and the remaining fin tissue was further dissociated into a single-cell suspension with 750 µL of 0.5 mg/mL Liberase DL (Roche) in enzyme-free cell dissociation buffer (Gibco) at 30.5°C for 15 min with gentle agitation. Liberase DL contains highly purified collagenase I and collagenase II and facilitates cell dissociation from the intact fin tissue. After Liberase dissociation, samples were centrifuged at 500g for 3 min at 4°C and supernatants were discarded, and then they were washed with 1 mL precold 1× PBS and supernatants were discarded again. After enzyme dissociation, cells were filtered through a 30 µm or 40 µm strainer followed by centrifugation at 500g for 3 min at 4°C and were washed in 500 µL sort buffer once. Then, cells were resuspended in 300 µL DNase solution with pipetting mix five times and incubated on ice for 5 min. After adding 1 mL 1× cold PBS with 0.04% BSA, samples were then centrifuged at 500g for 3 min at 4°C. This wash step was repeated for two more times. Then, cells were resuspended in 0.1× lysis buffer with gentle pipetting and then incubated on ice for 3 min. One milliliter of wash buffer was added, and tubes were gently inverted three to four times. Then, samples were centrifuged at 300g for 3 min at 4°C, and supernatants were removed. This wash step was repeated two more times followed by checking nuclei status under a microscope. After the last round centrifugation, 10 µL of supernatant was left in the tube, and then 50 µL of 1× diluted nuclei buffer was added, followed by centrifugation at 300g for 3 min at 4°C. After removing supernatants, samples were resuspended with 20 µL of 1× diluted nuclei buffer. Trypan blue was used to count nuclei. The final nuclei concentration was 2000–2500 nuclei/µL. The chromium next GEM single-cell multiome ATAC + gene expression reagent bundle from 10x Genomics was used for library preparation. Sequencing was performed using NovaSeq 6000 (Illumina).
Single-cell analysis
Preprocessing
The sequenced files were processed by default Cell Ranger ARC (v2.0.0). The reads were aligned to genome danRer11, and feature counting was based on Ensembl release 104 (cellranger-arc count). Cell Ranger ARC processed RNA cell–gene matrices were converted to Seurat objects using Seurat (v4.0.3) (Butler et al. 2018), and the ATAC fragment files were converted to Seurat objects using Signac (v1.6.0) (Stuart et al. 2021). R 4.1.1 was used (R Core Team 2021). Low-quality cells were then removed if they had low RNA counts (nCount_RNA < 1000), high RNA counts (nCount_RNA > 50,000; 6 dpa replicate 1: nCount_RNA > 500,000), high RNA mitochondrial reads percentage (percent.mt > 10), low ATAC counts (nCount_ATAC < 1000), high ATAC counts (nCount_ATAC > 200,000; 6 dpa replicate 2: nCount_ATAC > 500,000), high ATAC mitochondrial reads percentage (pct.mt.atac > 10), high nucleosome signal (mononucleosomal/nucleosome-free reads ratio, nucleosome signal > 2), and low TSS enrichment score (TSS.enrichment < 2).
Processing of RNA expression data
After filtering low-quality cells, RNA matrix was normalized using SCTransform (Hafemeister and Satija 2019) with total RNA counts, mitochondrial gene ratio, and cell cycle scores (CC. Difference) regressed out. The “RunPCA” function with default parameters was used to perform linear dimensional reduction. All samples were then integrated using the reciprocal PCA method.
Processing of DNA accessibility data
For scATAC-seq data, after low-quality cell filtering, peaks were called by sample using MACS2 (Zhang et al. 2008) in the Signac “CallPeak” function with the following parameters: -g 1.4 × 109 -f BEDPE ‐‐keep-dup all -B ‐‐SPMR ‐‐nomodel ‐‐extsize 200 ‐‐shift -100 -q 0.05 ‐‐call-summits. Then, peaks from all samples were merged using the “reduce” function. Peaks with length above 10,000 and below 20, peaks in nonstandard chromosomes, and peaks overlapping with zebrafish blacklist (Yang et al. 2020) were removed. Then, the union peak set was used to create the Signac “peak assay” for each sample. For normalization, feature selection, and linear dimensional reduction, the “RunTFIDF,” “FindTopFeatures,” and “RunSVD” with default parameters in Signac (v1.6.0) were sequentially performed. Harmony (Korsunsky et al. 2019) was then used to integrate all samples. After clustering with LSI components two to 40 and a resolution of 0.3, two clusters of cells with extremely high RNA raw counts and ATAC fragment counts were further removed before renormalization, reselection of features, and further linear dimensional reduction.
Multimodal clustering
We computed a joint neighbor graph using the weighted nearest neighbor (WNN) method in Seurat v4 (Hao et al. 2021) to jointly analyze both DNA accessibility and gene expression. We performed graph-based clustering on PCA components one to 45 and LSI components two to 40 followed by “RunUMAP” and “FindClusters” with a resolution of 0.2. Cell types were annotated based on DEGs and known markers with supporting literature evidence. We then performed peak calling again using MACS2 on the identified cell types.
DEGs and DARs
All DEGs were assessed with the “Seurat::FindMarkers” function for transcripts detected in at least 10% of cells and a log2 fold change threshold of 0.25. Bonferroni-adjusted P-values were used to determine significance with a threshold of P.adj < 0.05. For DARs, Seurat (v4.4.0) was used, and all DARs were assessed with the “FindMarkers” function utilizing a logistic regression model for peaks detected in at least 10% of cells and a log2 fold change threshold of 1.5. Bonferroni-adjusted P-values were used to determine significance with a threshold of P.adj < 0.05.
Computing peak-to-gene links
The “Signac::LinkPeaks” function was used to calculate linkage scores for each peak–gene pair. The detailed analysis steps are described by Stuart et al. (2021). Briefly, Pearson correlation coefficients between gene expression and accessibility of peaks within 100 kb from each gene TSS were calculated. Expected correlation coefficients for each peak were calculated based on its GC content, accessibility, and length and were used to compute a Z-score and P-value. Significant links were filtered using default parameters suggested by the developer, including a P-value cutoff of 0.05, a correlation coefficient cutoff of 0.05, and the requirement that both the gene and the linked open peak be present in at least 10 cells.
ChromVAR motif analysis
ChromVAR (Schep et al. 2017) analysis was performed in Signac (v1.6.0) to determine the global TF activity in each cell. The JASPER2022 motif database (Castro-Mondragon et al. 2022), BSgenome.Drerio.UCSC.danRer11, and the cell-by-peak matrix were initially used as input to add motif information to the Seurat object using the “Signac::AddMotifs” function. The “Signac::RunChromVAR” function was used to create a Z-scored cell-by-motif activity matrix stored in the “chromvar” assay. To identify motifs with differential activity across compared groups, we applied the “Seurat::FindMarkers” function, specifying “mean.fxn = rowMeans and min.pct = 0.2.”
GRN construction by stage within cell types
We constructed GRNs following methods described in previous studies (Lyu et al. 2021, 2023). The approach includes six steps:
-
TF-motif correlation calculation. The Pearson's correlation between the TF gene expression and chromVAR (v1.12.0) motif activity score was calculated by cell type by time point. Only correlations greater than 0.05 or less than −0.05 were kept.
-
Cis-regulatory element identification. A union set of cell-type-specific called peaks were annotated based on their genomic location: (1) promoter (±1 kb around TSS), (2) gene body region, and (3) intergenic region. Then, the “Signac::LinkPeaks” function was used to calculate peak–gene correlation (links). Each cell type was further subset to preinjury (0 dpa) and regenerating stage (1/2/4/6 dpa) cells for peak–gene correlation calculation, with “distance = 100000, P-value_cutoff = 0.05, and score_cutoff = 0.05” parameters specified.
-
TF binding sites prediction. We used the motifmatchr (v1.12.0) package (“matchMotifs”) to identify matching motifs within ATAC peaks, with a P-value cutoff of 5 × 10−5. Input motifs were based on the JAPSPAR 2022 database (Castro-Mondragon et al. 2022). Footprint signals for each stage and cell type were calculated using the same method as the original approach. BAM files used for footprint signal calculation were prepared as mentioned in the Genome Browser Track View section below. TOBIAS (v0.14.0) (Bentsen et al. 2020) was used for producing bias-corrected Tn5 signal in bigWig file format from BAM files. TF binding sites were retained based on the criteria: NR + NL − 2 × NC > 0.1, where NR is the signal of the right region next to the motif, NL is the signal of the left region, and NC is the signal of the motif binding region.
-
TF–target correlation calculation. The “grnboost2” function from arboret package (Aibar et al. 2017) was used to infer the importance of TF–gene pairs. The top 10% of pairs with the highest scores was retained. Each cell type was further separated by time points. For each time point, a cell-by-gene expression matrix was extracted and used along with a TF gene list converted from the JASPAR 2022 as input for the grnboost2 analysis. Pearson's correlation was also calculated for each TF–gene pair.
-
TF-peak–target link construction. Information from the previous steps was integrated to establish connections between TFs, peaks, and target genes.
-
TF ranking by centrality. TFs were ranked by degree centrality in the network using the “degree_centrality” function from the igraph package. Centrality scores were normalized to the maximum observed in the network.
Identification of lineage-specific key regulators
MES cells were initially subset from whole Seurat object. “RunPCA,” “FindNeighbors,” “FindClusters,” and “RunUMAP” were performed for the MES subset with dimension one to 20 and a resolution as 0.6 to identify subclusters. Pseudotime lineages for fibroblast MES and osteoblast MES were inferred using SlingShot (v1.8.0) (Street et al. 2018). The Seurat object was then converted to CellOracle object. All steps were followed in the CellOracle (v0.11.0) (Kamimoto et al. 2023) standard tutorials with default parameters for GRN construction for each subclusters and in silico TF perturbation.
GO enrichment analysis
GO enrichment analyses were performed using the R package clusterProfiler (Yu et al. 2012) with the “enrichGO” function; Metascape (v.3.5) (Zhou et al. 2019) with D. rerio as input and analysis species and default settings was used.
GREAT analysis
GREAT (v3.0.0) (McLean et al. 2010) was used to annotate RRRs. RRRs were converted from the danRer11 to danRer7 using the liftOver tool (Hinrichs et al. 2006). Then, the danRer7 coordinates of RRRs were used as input for the GREAT analysis. Distance settings included a proximal range defined as 5 kb upstream and 1 kb downstream, with distal regions up to 100 kb.
Conservation score calculations
Conservation scores for RRRs and shuffled genomic regions were calculated using phastCons vertebrate eight-way (Siepel et al. 2005) and phyloP vertebrate eight-way (Pollard et al. 2010). Shuffled genomic regions were generated using the BEDTools package (Quinlan and Hall 2010). RRRs and shuffled regions were converted from the danRer11 to the danRer7 assembly using liftOver.
Genome browser track view
The WashU Epigenome Browser tool (Li et al. 2022) and “Signac::CoveragePlot” function were used to display track views. For generating WashU Epigenome Browser tracks, sinto (https://timoast.github.io/sinto/) was used to split BAM files into pseudobulk groups. SAMtools (Danecek et al. 2021) was used to merge the splitted BAM files. “bamCoverage” in the deepTools package (Ramírez et al. 2016) was used to convert BAM file to bigWig format. RNA tracks were normalized by CPM, and ATAC tracks were normalized by RPKM, with a bin size of 10 and an effective genome size of 1.4 × 109.
In vivo activity validation of RREs
The modified zebrafish enhancer detection (ZEDtw) vector backbone was the same as described in a previous study (Lee et al. 2020). Candidate enhancer elements and negative control sequences were PCR-amplified from zebrafish genomic DNA using primers listed in Supplemental Table S4. The amplified elements were then cloned into ZEDtw vector by the Gateway in vitro recombination system, as described previously (Bessa et al. 2009). Purified plasmids were injected into wild-type AB zebrafish embryos at the one-cell stage. All mCherry + F0 embryos were raised up to adulthood. For some enhancer elements, F1 stable lines were also established by outcrossing F0 with wild-type AB zebrafish. Once the F0 or F1 zebrafish reached adulthood, caudal fins were amputated. EGFP and mCherry expressions were monitored and photographed on the regenerating fin at 1/2/4/6 dpa.
Statistical information
To identify differential gene expression across cell groups, we applied the nonparametric Wilcoxon rank-sum test using Seurat. Differential accessibility of chromatin regions was analyzed using logistic regression in Seurat. P-values were adjusted using Bonferroni correction across all features. The significance of overlaps between RRR-nearest genes and RRGs within each cell type was evaluated using the hypergeometric test (stats::phyper in R), with “lower.tail” parameter set to “FALSE.” For gene expression comparisons in Figure 3C and Supplemental Figures S3B and S4, we used the Wilcoxon rank-sum test via the “rstatix::wilcox_test” function in R.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE261907. Aggregated snRNA-seq and snATAC-seq tracks, split by cell type and stage, can be visualized in the WashU Epigenome Browser (https://epigenomegateway.wustl.edu/browser/?genome=danRer11&hub=https://epigenome.wustl.edu/FinRegen_snMultiome/FinRegen_scMultiome_browser_track.json). All custom scripts used to perform the analysis in this study are available at GitHub (https://github.com/yjchen1201/FinRegen_scMultiome_pub) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Brian Stephens, Amelia Hummel, and other staff members in the fish facility of the Washington University in St. Louis for basic fish husbandry. We thank Jennifer Ponce, Andrew Emory, and other staff members at the McDonnell Genome Institute (MGI) and the Genome Technology Access Center (GTAC) at Washington University in St. Louis for generating and sequencing snRNA-seq and snATAC-seq libraries. This work is supported by the Children's Discovery Institute of Washington University and the St. Louis Children's Hospital through microgrants from the Zebrafish Models for Pediatric Research Services Cooperative (ZRSC). Y.C., Q.Z., X.X., and T.W. are also supported by National Institutes of Health (NIH) grants R01HG007175, R01AG078964, R01AG083568, U24NS132103, and U01HG013227. J.K. is supported by NIH grant R35GM137878, NIH grant R01HL151522, and University of Wisconsin Institute for Clinical and Translational Research (UW ICTR) pilot grant. K.S. is supported by Stem Cell and Regenerative Medicine Center Research Training Award.
Author contributions: Y.C., Y.H., and T.W. conceived the idea for this project. Y.C., Y.H., and T.W. designed the experiments. Y.H., Y.C., and X.X. performed the tissue collection and nuclei extraction. Y.C., Y.H., T.W., G.Z., and C.H. interpreted the data. Y.C., I.W., M.S., Q.Z., K.S., and J.K. performed the regeneration enhancer assay. T.W. and G.Z. supervised the work for this project. Y.C., Y.H., T.W., and G.Z. wrote the manuscript with input from all the other authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279372.124.
-
Freely available online through the Genome Research Open Access option.
- Received March 18, 2024.
- Accepted November 4, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








